
Abstract
Background:
Toll-like receptor 4 participates in the process of acute heart injury. The underlying mechanisms of its protection are multifactorial, but we hypothesized that toll-like receptor-mediated autophagy control plays a vital role. The purpose of this study was to clarify the effect of autophagy on cardiac fibrosis.
Methods:
Cardiac fibrosis was induced by subcutaneous isoproterenol (ISO) injection, and rapamycin was simultaneously administered orally for 14 days. Animal echocardiography was then used to evaluate the success of the cardiac fibrosis model, and the mice were killed after the echocardiography examination.
Results:
Toll-like receptor 4 knockout (TLR4 KO) mice had better heart function than did wild-type (WT) mice (P < .05). Rapamycin treatment reduced the left ventricular ejection fraction to 23.5% (P < .05), and the collagen volume fraction of the ISO and ISO plus rapamycin groups was 5.9% and 25.9%, respectively, in TLR4 KO mice. Compared with the WT mice, Beclin 1 and autophagy were downregulated in TLR4 KO mice (P < .05); however, the ISO plus rapamycin group had higher autophagy activity than did the ISO group in TLR4 KO mice (P < .05).
Conclusions:
Our results suggest that TLR4 KO-induced cardioprotection against ISO-induced cardiac fibrosis is associated with reduced autophagy induction. Cardiac fibroblast autophagy participates in its own activation. The moderate inhibition of autophagic activity may be a new strategy for treating cardiac fibrosis.
Introduction
There is increasing evidence for the benefit of treating fibrotic diseases with rapamycin, but some disputes remain on its treatment of cardiac fibrosis. 1 –3 Myofibroblasts are critical for tissue repair and fibrotic disease pathogenesis. The phenotype transition is still a puzzle. 4,5 Whether rapamycin can activate cardiac fibroblasts by stimulating autophagy remains uncertain. Therefore, we treated cardiac fibrosis with rapamycin in toll-like receptor 4 knockout (TLR4 KO) mice and observed its influences on cardiac fibroblasts.
Rapamycin is a typical autophagy tool drug, and it can stimulate cardiac cell autophagy. 6 The function of autophagy in promoting or ameliorating fibrosis is debatable. Moderate autophagy and excessive autophagy adversely contribute to this biological function. 7,8 Autophagy-dependent cardiac myocyte death has been demonstrated in myocardial infarct animal models; however, few studies have reported whether autophagy can influence cardiac fibroblast activation. We hypothesized that autophagy could promote fibroblast activation in both proliferation and differentiation.
In previous work, we demonstrated that TLR4 KO mice lack autophagy activity. 9,10 We used TLR4 KO mice as negative controls for inadequate autophagy. Toll-like receptor 4 participates in the process of acute heart injury. The underlying mechanisms of TLR4 KO-mediated protection are multifactorial, 11 –13 but we hypothesized that TLR4-controlled autophagy played a vital role. The purpose of this study was to clarify the effect of autophagy on cardiac fibrosis, focusing mainly on the autophagic activity of cardiac fibroblasts. 14 –16
Methods and Materials
Ethics Statement
All of the mice were maintained in the laboratory of Animal Experiments at Capital Medical University. The mice were given a standard diet. The investigation was approved by the Animal Care and Use Committee of Capital Medical University.
Animal and Treatments
Male TLR4 KO mice (C57BL/10ScN) were obtained from the Model Animal Research Center of Nanjing University, and male C57BL/6J mice (wild-type [WT] mice) were obtained from Vital River Laboratory Animal technology (Beijing, China). The average weight was 27 ± 2 g; no significant differences were identified among the WT and the TLR4 KO mice. We divided the mice into 4 groups: vehicle, isoproterenol (ISO; WT and TLR4 KO mice each constituted a group), and ISO plus rapamycin. The vehicle group was treated with saline. Cardiac fibrosis was induced by subcutaneous ISO injection daily (10 mg/kg/d for 3 days and 5 mg/kg/d for 11 days), and rapamycin (2.2 mg/kg/d for 14 days) was administered by oral gavage according to a published report. 17 After 14 days, animal echocardiography was used to preliminarily evaluate the success of the cardiac fibrosis model, and then the mice were killed.
Animal Echocardiography
Mice were anesthetized with intraperitoneal 4% chloral hydrate by weight, and the heart rate was controlled at 360 ± 30 beats per minute. We performed echocardiography 14 days after starting drug administration. Systolic and diastolic functions were evaluated individually by left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS), and the movement velocity of the mitral annulus (E’). 18
Morphological and Histological Evaluation
The hearts were rapidly excised, fixed with 4% paraformaldehyde, and embedded in paraffin for histopathological examination as previously described. Heart tissue sections (3-µm thick) were prepared and stained with hematoxylin and eosin (H&E) or Masson trichrome. Collagen deposition from 10 randomly chosen regions per tissue sample was analyzed at ×400 magnification. Collagen volume fractions were measured using Image-Pro Plus 6.0 software.
Confocal Microscopy
Heart tissue sections (5-μm thick) were prepared and stained with the indicated primary antibodies overnight at 4°C. The sections were washed twice, incubated with fluorochrome-labeled secondary antibodies (1:200) for 45 minutes, and washed 3 times after staining. Images were observed with a Leica SP2 confocal microscope (Leica Microsystems, Exton, Pennsylvania) and analyzed using the Leica confocal software. Autolysosomes were identified by LC3 and lysosome-associated membrane protein 1 (LAMP1) colocalization, 19 and cardiac myofibroblasts were identified by α-smooth muscle actin (α-SMA) and Vimentin colocalization. 4
Western Blotting
Proteins were extracted from heart tissue using radioimmunoprecipitation assay buffer with 1 mmol/L phenylmethylsulfonyl fluoride and protease inhibitor cocktail. Total proteins (40 µg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, Massachusetts). Membranes were incubated overnight with primary antibodies. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies, followed by signal detection with an enhanced chemiluminescence detection system (Amersham Bio-sciences, United Kingdom).
Semiquantitative Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, California) following the manufacturer’s instructions. Next, the RNA was reverse transcribed and amplified. Polymerase chain reaction was performed using a Mycycler thermal cycler and analyzed using 2% agarose gels. The following primer sequences were used: 5′-CATGTTCAGCTTTGTGGACCT-3′ and 5′-GCAGCTGACTTCAGGGATGT-3′ (collagen type I); 5′-TCCCCTGGAATCTGTGAATC-3′ and 5′-TGAGTCGAATTGGGGAGAAT-3′ (collagen type III); 5′-TTACCAACTGGGACGACATG-3′ and 5′-ATACAGGGACAGCACAGCCT-3′ (β actin). All of the values obtained were normalized to the β-actin values.
Electron Microscopy
Heart tissue samples were fixed in 3% glutaraldehyde, postfixed in 1% osmium tetroxide, rinsed in sodium phosphate buffer (pH 7.2), dehydrated, and embedded in Epon 812. Semithin sections stained with 2% toluidine blue-borax mixture were used for orientation by light microscopy. Ultrathin sections were picked up on grids, stained with 10% uranyl acetate and 1% lead citrate, and then examined with a JEM-1400 electron microscope (JEOL, Japan).
Statistical Analysis
The data were represented as the mean ± standard error, and Student t test was used for comparisons between the groups. Comparisons among 3 or more groups were analyzed using a 1-way analysis of variance; P values <.05 were considered to be statistically significant. All of the statistics were analyzed using the SPSS 16.0 software.
Results
Toll-Like Receptor 4 Knockout Protected Against Cardiac Dysfunction, While Rapamycin Treatment Counteracted the Protective Effect
We evaluated cardiac systolic and diastolic functions by echocardiography. Isoproterenol reduced the LVEF and LVFS in WT mice, but TLR4 KO mice were protected (P < .05). Rapamycin treatment reduced the LVFS to 23.5% (P < .05; Figure 1). Moreover, diastolic function was evaluated by measuring the movement velocity of the E’. The E’ of the ISO plus rapamycin group was reduced to 24.3 ± 5.8 mm/s (the value of the vehicle group was 31.5 ± 7.3 mm/s, P < .05). Compared with the WT mice, the TLR4 KO mice had better diastolic function when treated with ISO alone (27.1 ± 6.1 mm/s vs 30.2 ± 3.8 mm/s). Although we studied the E/E’ value (the ratio of mitral flow and mitral annulus), we did not consider it to be effective and did not include the results in our further analysis. Heart function was measured in mice with similar heart rates (360 ± 30 beats per minute, P < .05).
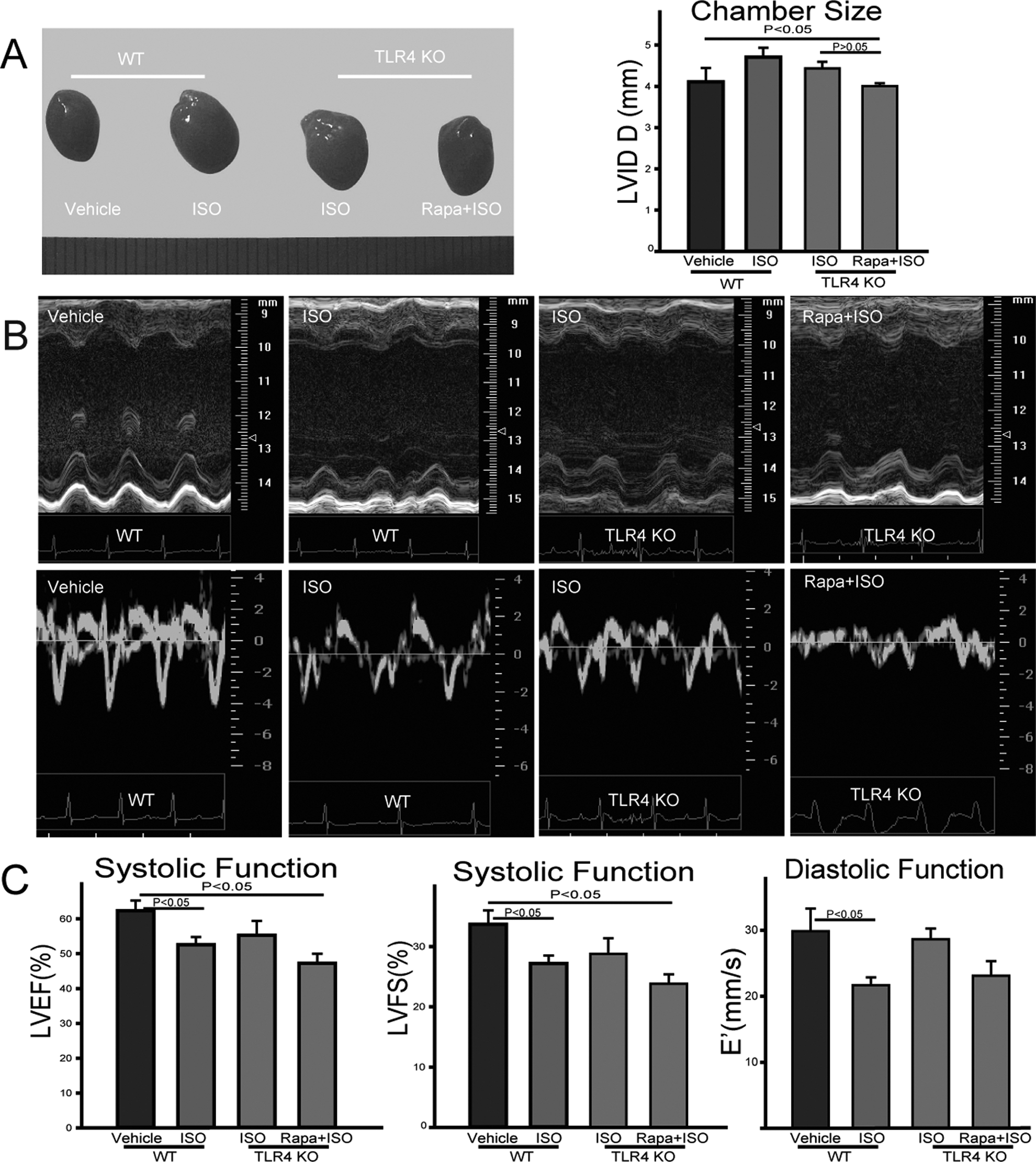
Rapamycin promoted isoproterenol (ISO)-induced cardiac dysfunction in toll-like receptor 4 knockout (TLR4 KO) mice. A, Heart morphology. Heart size in TLR4 KO mice was smaller than that of the wild-type (WT) mice when treated with ISO. B, Echocardiography of WT and KO mice. M-Mode echocardiography indicated that ISO treatment decreased the left ventricular fractional shortening (LVFS) and left ventricular ejection fraction (LVEF), but TLR4 KO mice were protected. Rapamycin treatment was the worst among the 4 groups, and the same results were obtained by measuring diastolic function. Diastolic function was evaluated by mitral annulus velocity using the tissue Doppler (N = 6-9 for each group). C, Systolic and diastolic functions were compared among the 4 groups. E’ represents the mitral annulus movement velocity.
Rapamycin Accelerated Cardiac Fibrosis in TLR4 KO Mice
The H&E and trichrome staining gave some clues about the cardiac fibrosis. Interestingly, there were different cardiac fibrosis phases. The cardiac fibrosis appeared following cardiac myocyte loss. The ISO plus rapamycin group had more myocyte loss and more severe fibrosis. The ISO caused cardiac fibrosis in both the WT and the KO mice. Rapamycin treatment exacerbated ISO-induced cardiac fibrosis. The collagen volume fraction of the ISO group in WT mice and the ISO plus rapamycin group was 20% and 25.9%, respectively (Figure 2). Compared with the WT mice, TLR4 KO mice were protected against ISO-induced cardiac fibrosis, but this protective effect was counteracted by rapamycin treatment. The collagen volume fraction of the 2 groups was 5.9% and 25.9%, respectively (P < .05).

Rapamycin accelerated cardiac fibrosis, while toll-like receptor 4 knockout (TLR4 KO) were protected. A, Massive myocyte necrosis and myocardial fibrosis were both confined to the subendocardium and midwall. B, Hematoxylin and eosin (H&E) staining demonstrates that both myocyte necrosis and fibrosis developed in the same myocardial regions. Where there was no myocyte loss, there was no fibrosis. Myocyte loss in the isoproterenol (ISO) plus rapamycin group was the most severe among the 4 groups. C, Masson’s trichrome staining was performed to evaluate collagen deposition in left ventricular (LV) tissue. Collagen volume fraction (CVF) was measured using the Image-Pro Plus 6.0 software. More collagen was deposited in rapamycin-treated mice (N = 6-9 for each group).
We eliminated the failed model according to the pathological results, and we analyzed the heart tissue by Western blot. The results supported the differentiation of cardiac fibroblasts and collagen deposition in the ISO plus rapamycin group. Phosphorylated SMAD3 (pSmad3) was upregulated, which is a key fibrogenesis factor. 20 α-Smooth muscle actin expression in the ISO plus rapamycin group was the highest among the 4 groups (P < .05); furthermore, the collagen type 3 messenger RNA levels were upregulated in the ISO plus rapamycin group (Figure 3).
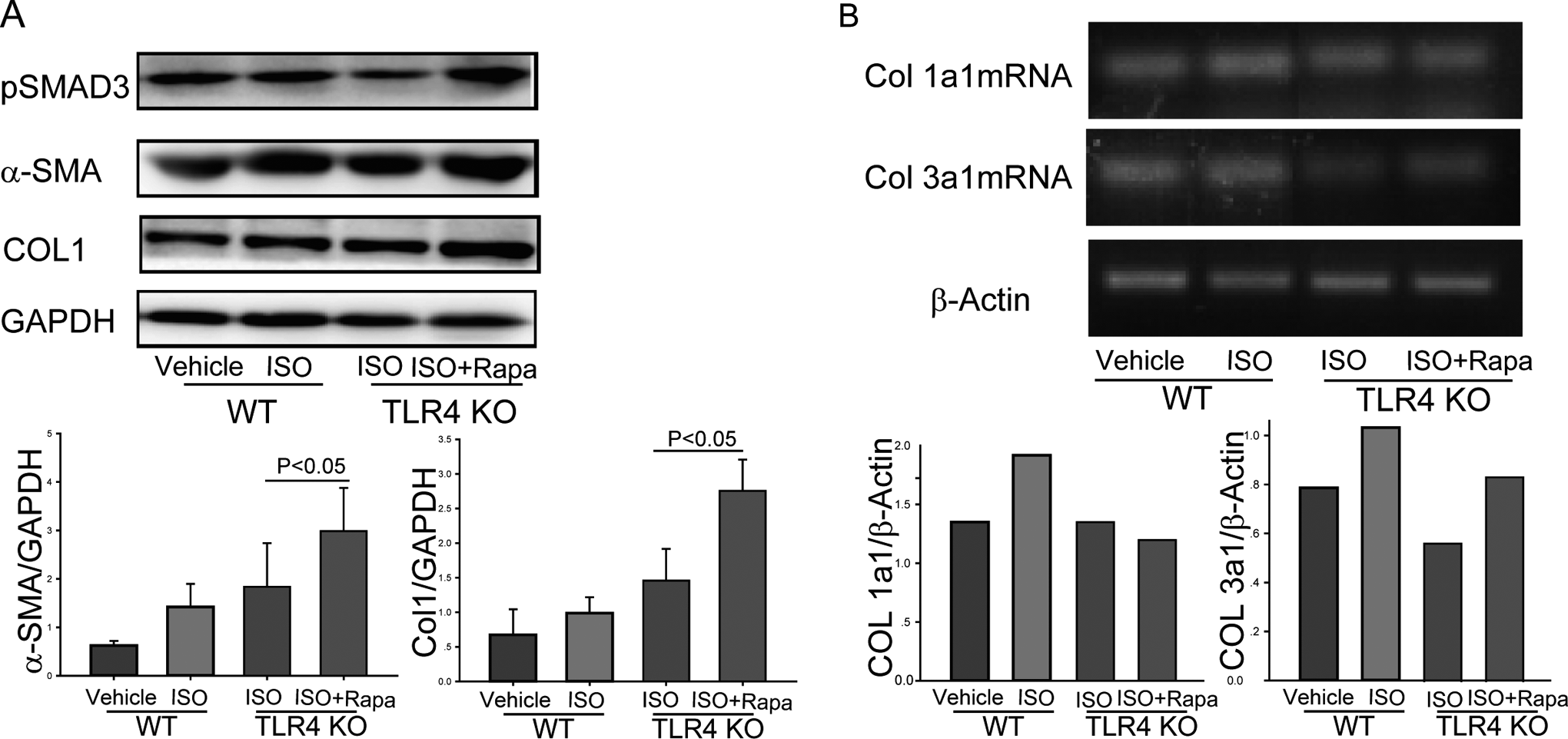
Smad3 phosphorylation promoted collagen expression, and rapamycin promoted fibrosis through the Smad3 signaling pathway. A, α-Smooth muscle actin (α-SMA) and pSmad3 expressions were downregulated in toll-like receptor 4 knockout (TLR4 KO) mice; rapamycin promoted α-SMA expression. More collagen was observed in the rapamycin treatment group. B, Collagen messenger RNA (mRNA) expression was downregulated in TLR4 KO mice, but rapamycin treatment promoted collagen type 3 mRNA expression with no statistical significance.
Beclin 1 Downregulation Provided Moderate Autophagy Activity
The pathology results gave a clue that cardiac fibroblasts accumulated in the same zone where the cardiac myocytes were lost. Thus, we hypothesized that there was communication between cardiac myocytes and cardiac fibroblasts, 21 and that TLR4 may be the receptor that entails the communication. The TLR4 can receive signals from dying and/or injured cardiac myocytes, and it controls autophagy through the protein Beclin 1. 22 Signaling proteins related to autophagy were analyzed to prove the hypothesis (Figure 4). Compared with WT mice, LC3 expression was decreased in the ISO-treated TLR4 KO mice. Interestingly, Beclin 1 expression was decreased. Thus, Beclin 1 is the vital mediator by which TLR4 controls autophagy activity. Moreover, rapamycin partially restored autophagy in a Beclin 1-independent manner (Figure 4).

Autophagy activity was analyzed by Western blot. Excessive autophagy activity was induced after treating with rapamycin, and insolvable p62 was aggregated in the rapamycin-treated group. Insolvable p62 is an important measurement to evaluate autophagy pathway efficacy. The toll-like receptor 4 controls cardiac cell autophagy through Beclin 1. Downregulated Beclin 1 provided moderate autophagy activity.
Cardiac Fibroblast Activation by Stimulating Autophagy
As mentioned earlier, it was unclear what cells accumulated in the focal pathological zone. We used the markers vimentin and α-SMA individually for fibroblasts and their activated phenotype. The accumulated cells were demonstrated to be activated myocardial fibroblasts (cardiac myofibroblasts) by confocal and electron microscopy (Figures 5 and 6). The cardiac fibroblast proliferation and phenotype transition were increased when mice were treated with ISO, especially in combination with rapamycin treatment. The most exciting finding was altered autophagy activity. We evaluated autophagy activity using LC3 and Lamp1 markers and their colocalization implicated autolysosome formation. Most accumulated myofibroblasts were full of autolysosomes in the ISO plus rapamycin group, while the ISO-treated KO group lacked autolysosomes. Cardiac myofibroblast activation was enhanced by the increased autolysosome formation.
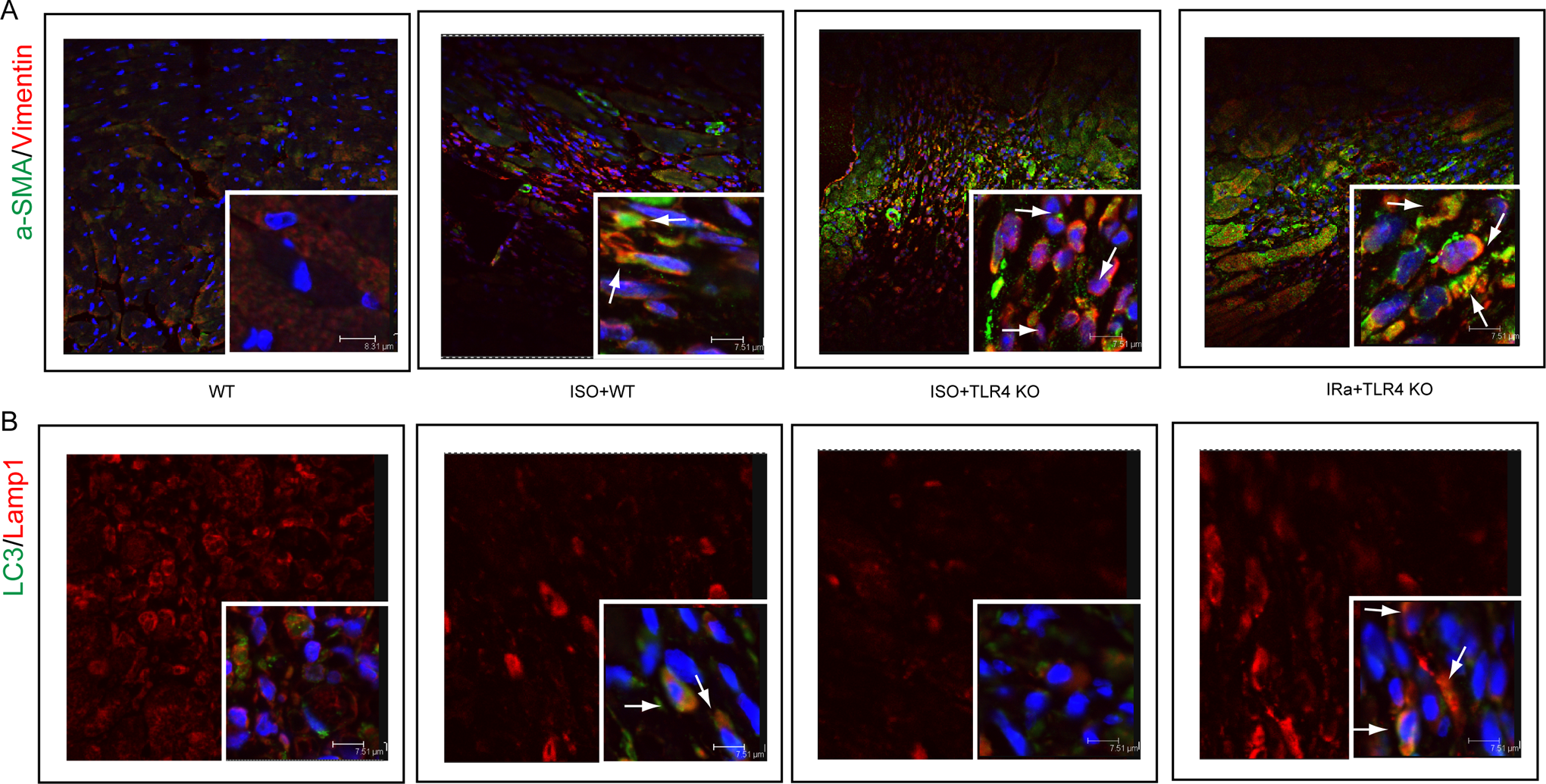
Accumulated myofibroblasts were full of autolysosomes in the isoproterenol (ISO) plus rapamycin treatment group. A, Autophagy enhanced cardiac fibroblast differentiation and proliferation. B, Most of the accumulated myofibroblasts were full of autolysosomes in the ISO plus rapamycin treatment group. Autolysosomes were observed indirectly by confocal microscopy through LC3 and lysosome-associated membrane protein 1 (LAMP1) colocalization. (LC3, green; LAMP1, red. The white arrowheads indicate colocalization of the 2 colors.)

Altered myocardial ultrastructure after isoproterenol injection. A, Normal myocardial heart ultrastructure. White arrows denote the mitochondria. B, Moderate cardiomyocyte injury by isoproterenol (ISO) injection. The myofibrils are intact while the mitochondria show severe swelling. C, Severe cardiomyocyte injury. Myofibril destruction (black arrow) and mitochondrial swelling or vacuolization (white arrow). D, Collagen type I (black arrow) and collagen type III (white arrow) in the intercellular space. Cardiac fibroblasts were the main cells in the fibrosis region. E, Autolysosomes in the myocytes (black arrow). F, Autolysosomes in the cardiac fibroblasts (black arrow). Cardiac fibroblast proliferation can be observed in the fibrosis region.
Because of the pathology results, we examined the heart tissue further (Figure 4). The results were surprising because excessive autophagy activity was observed in the rapamycin treatment group, and there was accumulated p62 in the same group. Beclin 1 was decreased in the TLR4 KO group, and rapamycin stimulated autophagy through a Beclin 1-independent pathway.
Discussion
In the present study, we demonstrated that the TLR4 KO-induced cardioprotection against ISO was associated with reduced autophagy induction and that enhancing autophagy counteracted this protection. We also found that the TLR4-mediated, Beclin 1-dependent autophagy pathway may be pivotal in cardiac fibrosis pathogenesis.
We were fascinated that the toll-like receptor can recognize endogenous pathogens, which are released by dying and/or injured cells such as high-mobility group box 1 and heat-shock protein 70. Thus, we have adopted the ISO-induced replacement fibrosis model. The dose we used caused a myocardial infarction. Both cardiac myocyte necrosis and fibrosis developed in the same myocardial regions, and the pathological zones were confined to the subendocardium and midwall. Loss of cardiac myocytes induced cardiac fibroblast proliferation, which was also demonstrated by Benjamin in 1989. 23 Although the reason remains unclear, TLR4-controlled autophagy may provide an explanation. As noted by Mann, the innate immune system proves a short-term adaptive response to tissue injury, but the beneficial effects may be lost if innate immune signaling becomes sustained and/or excessive. 24
Autophagy was induced after the severe heart injury. During the inflammatory phase, myofibril destruction and mitochondrial swelling or vacuolization as well as autophagy-dependent apoptosis occurred in the cardiac myocytes. During the proliferative phase, autophagy promoted fibroblast activation. Thus, autophagy was delicately regulated by the organism so we still have a long way to go to harness this phenomenon.
Excessive autophagy promoted autophagy-dependent cardiac myocyte apoptosis. The most plausible reason for the loss of myocytes was cardiac myocyte death. 25 We observed more cardiac myocyte loss in the ISO plus rapamycin group, even loss of the whole heart midwall. Meanwhile, excessive autophagy was induced by ISO plus rapamycin treatment. Thus, it is reasonable to presume that the massive cardiac myocyte losses were caused by excessive autophagy activity (Figure 6). All of these findings indicate that excessive autophagy activity can lead to cardiac myocyte death, 26 while moderate autophagy can maintain metabolic functions. Two mechanisms regarding autophagy-dependent apoptosis have been noted by Maiuri et al. 27 First, autophagy may constitute a mechanism through which the cell that is destined to die initiates its catabolism, thereby accelerating cell disappearance. Second, autophagy may also help maintain optimal, high adenosine triphosphate levels, which may facilitate the apoptotic process. Further evidence has been established to support myocyte loss via autophagy-dependent apoptosis. 28,29
Cardiac fibroblast autophagy promoted cardiac fibroblast proliferation and differentiation. Autolysosomes were obviously increased in cardiac myofibroblasts in the fibrosis zone using colocalization techniques; moreover, cardiac fibroblast differentiation was observed in the same pathological zone. However, few autolysosomes were observed in the ISO-treated TLR4KO mice. As mentioned earlier, Beclin 1 downregulation may be the reason why few autolysosomes were found in the KO mice. Interestingly, rapamycin can restore the autophagy activity in KO mice, which may explain the aggravated cardiac fibrosis in the ISO plus rapamycin group. Thus, cardiac fibroblast autophagy participated in self-differentiation and self-proliferation. The same biological behavior was also confirmed by Thoen et al, 30 who noted that autophagic flux was increased with hepatic stellate cell activation. What is changed by the recycling function of autophagy? How does the autolysosome promote cell proliferation and differentiation? Further studies are still needed to answer these questions, but we support the view that differentiation and proliferation events require cellular self-digestion, cellular housekeeping, and/or cellular recycling to meet high nutritional and energy demands. 31 Autophagy can satisfy all of these demands, which is surely the best choice that cells can select.
Conclusion
Our results suggest that TLR4 KO-induced cardioprotection against ISO-induced cardiac fibrosis is associated with reduced autophagy induction. Cardiac fibroblast autophagy participates in its own activation. Moderately inhibiting autophagy activity may be a new strategy for treating cardiac fibrosis.
Footnotes
Acknowledgments
We thank Dr Bin Xu for revising the article.
Authors’ Contribution
R. Dong contributed to conception and design of the study, with substantial contribution to data acquisition, analysis and interpretation of the data, and drafting of the manuscript; Z. Wang substantially contributed to data acquisition; C. Zhao substantially contributed to data acquisition; H. Gu substantially contributed to data acquisition; Z. Hu contributed to analysis and interpretation of the data; J. Xie substantially contribution to data acquisition; Y. Wu contributed to conception and design of the study, analysis and interpretation of the data, and critical revision of the manuscript for intellectual content.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the funding from the “215” high-level personnel construction from Beijing Municipal Health Bureau, 215 Healthy and Technical Project (grant no. 2011-3-003).