
Abstract
Periodontitis, an inflammatory disease initiated by Gram-negative bacteria such as Porphyromonas gingivalis (Pg), is considered as a risk factor for rheumatoid arthritis (RA). Our study aimed to determine the effect of Pg and its LPS on the expression of peptidyl arginine deiminase isotypes (PADs) in human primary chondrocytes (HC). HCs were infected with Pg and activated by its LPS (LPS-Pg). The mRNA expression levels of human PADs (1, 2, 3, 4 and 6) and bacterial enzyme (PADPg) were quantified by RT-qPCR. Cellular extracts served to measure the enzymatic activities of PADs and PADPg and to visualize the profiles of citrullinated proteins/peptides by Western blotting. Our data showed significant inhibitions of mRNA expressions of human PAD-2, PAD-3 and PAD-4 during infection of HC with live Pg. Activation of HC by LPS-Pg increased mRNA expressions of human PAD-2 and PAD-3. The PADPg enzymatic activity was significantly increased in only infected HC. Analysis of citrullinated proteins/peptides profiles revealed the occurrence of low molecular bands only in cellular extracts from HC infected with Pg. Our data showed that Pg and its LPS differentially regulate the expression of PADs in human chondrocytes and that Pg favors the apparition of new citrullinated proteins/peptides.
Introduction
Rheumatoid arthritis (RA) is a common systemic autoimmune disease characterized by synovial hyperplasia, inflammatory cell recruitment and destruction of the joint architecture due to intra-articular fibrin deposition. 1 This disease share many common features with periodontitis (PD), a chronic inflammatory disease initiated by bacteria affecting gingiva, periodontal ligament, cementum and alveolar bone. 2 The two diseases have in common many risk factors and immunogenic features, especially HLA-DR4, as well as the same local immune response amplified by recruiting inflammatory cells to the target tissue (periodontium or synovial membrane).3,4 These cellular and humoral immune reactions contribute to the pathogenesis of RA and PD that ultimately lead to the destruction of adjacent bone.5,6 Despite differences in initiating etiological mechanisms, evidence now emerging from numerous clinical and epidemiological studies suggests an association between RA and PD. 7 Indeed, subjects with RA are at increasing risk of developing PD. 8 Among the common features of RA and PD, low-grade infections are involved in the pathogenesis of these diseases. Porphyromonas gingivalis (Pg) is a Gram-negative anaerobe bacterium involved, with other bacteria, in the onset of chronic PD and may be also in RA, as DNA from Pg was found in synovial fluids and plasma samples from RA patients. 8 A unique feature of RA is the presence of specific auto-Abs in sera from patients, with anti-citrullinated protein Abs being the most present, which make them useful diagnostic markers of the disease.9,10 These anti-citrullinated protein Abs (ACPAs) are produced in the synovial fluid of inflammatory joints and give false signals to the immune system to generate Abs against these proteins, among them fibrin, vimentin and fibrinogen. 11 Citrullination of proteins is achieved by a group of enzymes, peptidyl arginine deiminases (PADs), that generate a non-essential amino acid, citrulline, by de-imination of arginine residues on polypeptidic chains. 12 In humans, a family of five PADs enzymes (PAD1, 2, 3, 4 and 6) has been described, but little is known on their respective ability to specifically citrullinate each of the protein markers of RA. 13 In addition to human PADs, a bacterial PAD (PADPg), a unique feature of Pg, has been identified. 14 Although not completely homologous to the five human PADs counterparts, this PADPg is able to citrullinate both bacterial and mammalian proteins, but with different specificities. 15 The ability of Pg to express a specific PAD suggests that infection with this organism could impact both the onset of RA and the progression of the disease by facilitating the occurrence of citrullinated auto-Ags. 16 Until now, no clear evidence implicates the bacterial PAD in the generation of ACPAs in vivo. Although the precise role of PADPg remains to be clarified in RA, recent data suggest an important role for this enzyme in the pathogenicity of PD. Indeed, experiments performed in vitro with purified PADPg indicated that the enzyme could distort the crosstalk between the epithelium and epidermal growth factor (EGF) after citrullination of the C-terminal arginine of purified EGF, thus hindering its ability to stimulate epidermal cell proliferation. 17 Whether such an in vitro effect occurs physiologically in tissue damage and delayed healing within Pg-infected periodontium is presently unknown. Regarding RA, little is known on how Pg might be involved in the pathogenesis of the disease. 18 A few reports on primary human chondrocytes (HCs) highlighted a possible role in apoptosis that might contribute to the joint damage observed during the progression of RA. 19 Similarly, in experimental arthritis, concomitant PD caused by oral infection with Pg enhanced articular bone loss. 20 As citrullination is a widespread phenomenon in normal physiology and the inflammation process, it is important to clarify which among the human PADs could be involved in the formation of citrullinated auto-Ags in RA. Furthermore, considering the possible involvement of Pg in the etiology of RA, our working hypothesis was to investigate if Pg could interfere with the expression of human PADs in HCs especially, by modifying the expression of human PADs with consequences in the production of anti-citrullinated Ags/proteins. This hypothesis should reinforce the idea that Pg or one of its virulence factors, i.e. the LPS, could be considered as a potent molecular link between PD and RA.
Material and methods
Bacterial culture
Pg strain 33277 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and was cultivated anaerobically in brain–heart infusion medium supplemented with menadiol (0.5 mg/ml) and hemin (5 µg/ml) (all products obtained from Sigma, St. Louis, MO, USA). The culture was grown at 37℃ until an OD600 of 1.0 was reached. Concentration of bacteria was determined optically at OD600 (1 OD = 5 × 108 bacteria/ml) as previously described. 21 When using heat-killed Pg, bacteria were heated at 85℃ for 10 min before the beginning of the experiment. Bacterial inactivation was confirmed by determination of the absence of colony-forming units (CFU/ml) after anaerobic cultivation.
Human primary chondrocyte culture
Human primary chondrocytes (HC) were obtained from PromoCell (Heidelberg, Germany) and were isolated from normal human articular cartilage from knee and hip joints. HC were cultured in a ready-to-use chondrocyte grown medium supplemented with FCS and antibiotics (penicillin 100 units/ml; streptomycin, 100 mg/ml). Cells were cultured at 37℃ in humidified air with 5% CO2. In all the experiments, HC used were obtained between passages 3–5. All the media and antibiotics were obtained from PromoCell.
Infection of HC with P. gingivalis
The day before infection, HC were plated at 2 × 105 cells/plate in the complete HC medium without antibiotics. At the day of infection, cells were washed twice with PBS and infected for 2 h with Pg or with heat-killed Pg at a MOI of 10 and 100 bacteria/cell in the HC medium without antibiotics. Uninfected HC served as controls. The cells were co-cultured with Pg and with heat-killed Pg during 2 h in humidified air with 5% CO2. HC were then washed at least twice with sterile PBS to eliminate non-adsorbed bacteria. Total RNA and cellular extracts were prepared.
Activation of HC by LPS purified from P. gingivalis (LPS-Pg)
Commercial ultrapure LPS-Pg was obtained from InvivoGen (San Diego, CA, USA). LPS-Pg was dissolved in ultra-pure water at a final concentration of 1 mg/ml and conserved at –80℃ by aliquots. HC were plated at 2 × 105 cells/plate in the complete HC medium. The day after, medium was changed, cells washed twice with PBS and 1 µg/ml purified LPS-Pg was added for 2 h and for 24 h in the complete HC culture medium. Plates without LPS-Pg served as controls. Total RNA and cellular extracts were prepared at each time-point of the time course activation.
Preliminary experiments
Preliminary experiments for infection and activation of HC were also performed with a different strain of Pg (strain W381) and with its purified LPS ranging from 0 µg/ml to 5 µg/ml. All these experiments were performed in the same conditions as mentioned above. As the results obtained were similar to those with the ATCC strain, only results obtained with this ATCC strain are presented.
Quantitative RT-PCR (RT-qPCR)
Total RNA was isolated using the High Pure RNA Isolation kit (Roche Applied Science, Meylan, France) according to the manufacturer’s instructions. RNA quality and concentration were determined spectrophotometrically using a NanoDrop apparatus (Fisher Scientific, Illkirch, France). One µg total RNA was used as template for cDNA synthesis using the iScript Reverse Transcriptase Supermix (Bio-Rad, Hercules, CA, USA) in conditions described by the manufacturer. A negative control was performed using total RNA with no enzyme. Validated PCR primers pairs for the human genes β-actin, PAD-1, PAD-2, PAD-3, PAD-4 and PAD-6 (Quantitect Primer Assay) were purchased from Qiagen (Courtaboeuf, France). The primer pair for the bacterial PADPg (forward primer: 5’-CCT TGGGGGTACTGCATCGTG-3’ and the reverse primer: 5’-ACCTCGATGCCGTCGCCCTCTCG-3’) was obtained synthetized from Fisher Scientific.
For quantitative RT-qPCR, equal amounts of cDNA (200 ng) were employed using iTaqTm Universal SyB®GreenSupermix (Bio-Rad) with primer concentrations and conditions indicated by the manufacturer. An iCycler IQ with iCycler IQ software (version 3.1; Bio-Rad) was used with the auto-calculated cycle threshold (CT) selected. β-Actin was selected as a stable reference gene under the experimental conditions of this study. The CT value of each gene was normalized to β-actin using the comparative 2 −ΔΔCT method.
Preparation of cellular extracts
Media were removed and cells were washed twice with ice-cold PBS, collected by scraping in 5 ml ice-cold PBS and harvested by centrifugation at 1000 g for 5 min at 4℃. Supernatants were aspirated and cells were lysed for 10 min at 4℃ in 200 µl RIPA buffer (20 mM Tris-HCl, pH 7.4, 120 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, 50 mM NaF, 0.5 mM Na3VO4, 1 mM phenylmethylsulphonyl fluoride and 2 µg/ml of each aprotinin, leupeptin and pepstatin). All mentioned compounds were purchased from Sigma. Lysates were clarified by centrifugation for 10 min at 13,000 g at 4℃ and supernatants (cellular extracts) were collected. The protein concentration was determined with the Bio-Rad DC assay.
Western immunoblotting
Five µg proteins from cellular extracts were used for SDS-PAGE electrophoresis as described previously. 21 Proteins were transferred by electroblotting onto nitrocellulose membranes and blots were blocked for 2 h at 4℃ in TBS with 5% BSA. A primary anti-citrulline Ab (Cat: ab100932) purchased from Euromedex (Souffelweyersheim, France) was used overnight at 4℃ under gentle agitation in the blocking buffer at a dilution recommended by the manufacturer. The anti-citrulline Ab does not react with free citrulline and specifically recognizes intra-peptidic citrulline independently of the amino-acid sequence. We also used an anti-GAPDH polyclonal Ab (Cat: 10494-1-AP; Proteintech Europe, Manchester, UK) as an internal control to verify equal loading of proteins in all cellular extracts. Ag–Ab binding was detected using HRP-conjugated species-specific secondary Ab for 2 h at room temperature followed by several washes with TBS containing 0.05% Tween 20. Enhanced chemiluminescence substrates for HRP (Euromedex) were applied and signals acquired using the Image Lab Software version 3.0 (Bio-Rad).
Quantification of human and bacterial PAD enzymatic activities
The total human PAD activity was measured in cellular extracts with the Ab-based assay using a protocol adapted for use in microtiter plates. 22 Five µg proteins from cellular extracts were used, and the assay quantification was performed using as substrate N-α-benzoyl-l-arginine ethyl ester (BAEE) (Sigma) and the anti-citrulline Ab in a following step. The calibration curve was established using peptidyl arginine deiminase type-4 isolated from rabbit skeletal muscle and furnished by Sigma. Assays were performed in triplicate and wells without added cellular extract served to measure the internal background of the reaction. Absorbance was measured at 450 nm using the microplate reader Multiskan FC (Fisher Scientific). One unit of human PAD activity was defined as the amount of enzyme that produces 1 µmol N-α-benzoyl-citrulline ethyl ester from BAEE per h at 55℃ at pH 7.2. Results were expressed relative to the control assay.
The presence of endogenous citrulline and bacterial PADPg activity were determined in cellular extracts using a modified method by Boyde and Rahmatullah, 23 which was based on chemical modification of the citrulline side chain and colorimetric detection of the derivatives. Five µg proteins in cellular extracts were used and assays were performed in triplicate using as substrate 10 nM of N-Ac-l-Arg-OH (Sigma). Owing to the presence of citrullinated proteins/peptides and free citrulline generated by arginine deiminases, a group of distinct enzymes that only acts on free arginine residues and different from human PADs and bacterial PADPg, we measured the base level of citrullination and of PADPg activity by adding PBS instead of cellular extracts for the reaction. Human PADs are inactive against C-terminal arginine residues and do not deiminate arginine residues in the substrate N-Ac-l-Arg-OH. At the end of the reaction, absorbance at 535 nm was measured using the microplate reader Multiskan FC and the obtained OD was converted to micromoles of citrulline using a calibration standard curve prepared using free l-citrulline purchased from Sigma at 1 to 100 nmol per well. The base citrulline level was subtracted from the read-out with the substrate and the resulting value represented PADPg activity in the sample. One enzymatic unit was defined as the production of 1 µmol citrulline in 1 h at 55℃. Results were expressed relative to the control assay.
Statistical analysis
Statistical analyses were performed using the Student's t-test. A P-value <0.05 was considered statistically significant. The reported data are the means of at least three separate experiments performed under similar conditions and results were expressed as means ± SD.
Results
Infection of HCs with Pg inhibited mRNA expression of human PADs
HCs were infected during 2 h with live Pg at two different MOI (of 10 and 100) and with heat-killed Pg at a MOI of 100. Total RNA was extracted and mRNA expression levels of β-actin, PAD-1, PAD-2, PAD-3, PAD-4 and PAD-6, as well as the bacterial PAD (PADPg) were measured by RT-qPCR. As normalized to β-actin, our experiments showed that relative expressions of PAD-2, PAD-3 and PAD-4 were significantly down-regulated after infection with Pg at a MOI of 100 (5.5-fold, 3.2-fold and 3.7 fold, respectively) (Figure 1a). These inhibition effects were Pg dose-dependent as an infection of HC performed with Pg at a MOI of 10 reduced the measured effects. Although detected in all experiments, the mRNA expression level of PAD-3 was weakly expressed as compared with those of PAD-2 and PAD-4, whereas mRNA expression of PAD-1 and PAD-6 was not detectable. In HC infected with heat-killed Pg, no significant variations of the expression of mRNAs encoding for all human PADs were observed, suggesting that the down-regulated effects observed with live Pg might originate from a heat-labile component shared with the lived pathogen or from a rapidly expressed cellular/bacterial factor acting on the transcription rate of the tested genes (Figure 1a). As a control, the expression of mRNA encoding for bacterial PADPg was detected only in infected HC, suggesting that the pathogen might penetrate cells to be active.
Expression of mRNAs encoding for human PADs in HC infected with Pg (a) and activated by LPS-Pg (b). (a) HC were infected for 2 h with Pg at MOI of 10 and 100 and with heat-killed Pg (Ina) at a MOI of 100. Uninfected cells served as control. (b) HC were activated for 2 h and for 24 h by 1 µg/ml of LPS-Pg. Non-stimulated cells served as controls. Total RNAs were prepared and levels of mRNAs encoding for the five human PADs (PAD-1; PAD-2; PAD-3; PAD-4 and PAD-6) and the bacterial PADPg were determined by RT-qPCR. The mRNA expressions of PAD-1 and PAD-6 were not detected. All results were presented as the quantity relative to β-actin as a reference gene. The significance of differences (*) between a test mRNA and control HC was analyzed using a Student’s t-test (P < 0.0005). All experiments were repeated three times and results expressed as mean ± SD.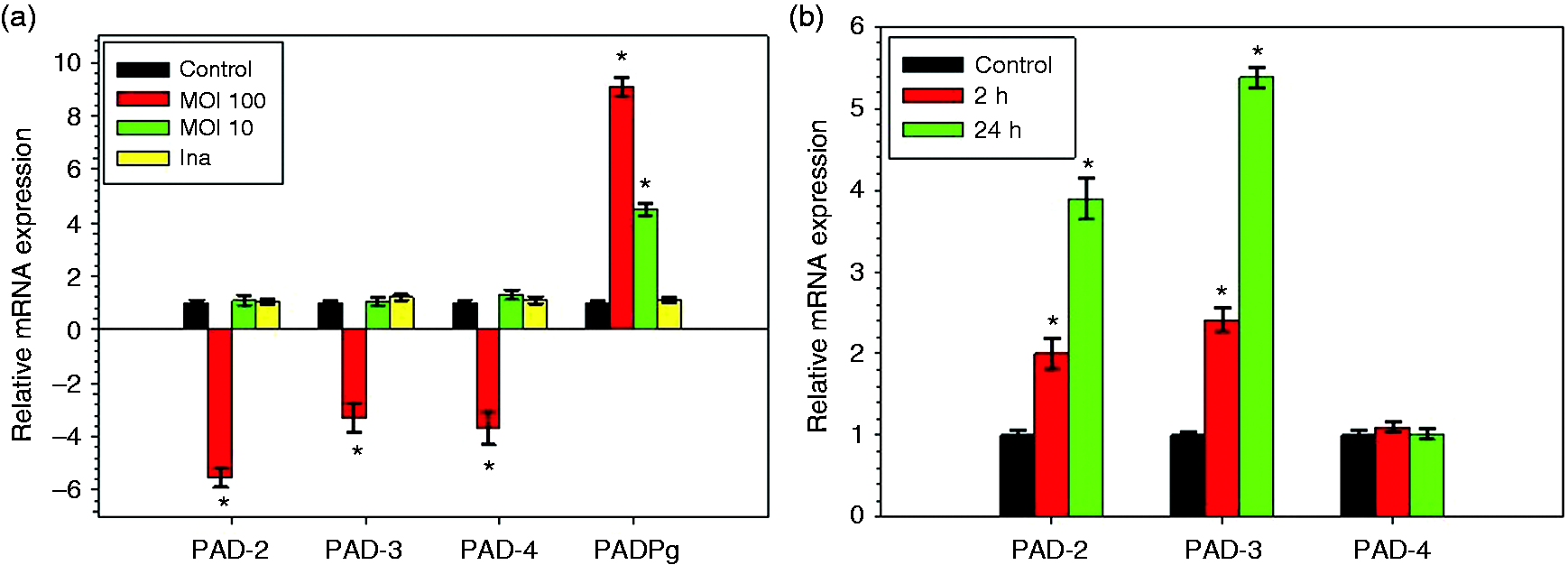
Effect of LPS-Pg on mRNA expression of human PADs
Among all the components and virulence factors expressed by Pg, we tested whether its purified LPS, a potent pro-inflammatory mediator, led to similar down-regulated effects observed with the live pathogen. HC were stimulated for 2 h and 24 h by 1 µg/ml LPS-Pg and mRNA expression levels of human PADs were determined by RT-qPCR. During the time-course activation of HC, and compared with the corresponding non-stimulated control cells, expressions of mRNAs encoding for PAD-2 and PAD-3 were significantly increased after 2 h of activation to reach a maximum at 24 h (3.9-fold and 5.4-fold for PAD-2 and PAD-3, respectively) (Figure 1b). No significant variation of expressions of mRNA encoding for PAD-4 was detected at any time of the HC activation process and no detection of mRNAs encoding for PAD-1, PAD-6 and PADPg were observed.
Variations of the enzymatic activity of bacterial and human PADs in infected and activated HCs
We quantified both human and bacterial PAD enzymatic activities in cellular extracts obtained from HC infected with Pg and activated by LPS-Pg. During the infection of HC, we measured a Pg dose-dependent and significant increase of the bacterial PADPg enzymatic activity, from 2.1-fold at a MOI of 10 to 4.2-fold at a MOI of 100 (Figure 2a). As expected, PADPg enzymatic activity was not detectable in cellular extracts of HC infected with the heat-killed bacterium. By measuring human PAD activity, a significant decrease (2.3-fold) was only observed in cellular extracts from HC infected with Pg at a MOI of 100, as compared with control cellular extracts (Figure 2a). This decrease was not observed with heat-killed Pg, suggesting that the inhibition of human PAD enzymatic activity might originate either from a heat-labile component(s) shared by Pg or from synthesized bacterial factor(s) acting on PAD mRNAs or proteins (Figure 2a).
Modulation of PADPg and human PADs enzymatic activities in HC infected with Pg (a) and activated by LPS-Pg (b). (a) HC were infected for 2 h with Pg at a MOI of 10 and 100 and with heat-killed Pg (Ina). Uninfected cells served as controls. (b) HC were activated for 2 h and for 24 h by purified LPS-Pg at a final concentration of 1 µg/ml. Non-activated cells served as controls. Cellular extracts were prepared and PADPg and human PAD enzymatic activities were determined. The significance of differences (*) between an assay and control HC was analyzed with a Student’s t-test (P < 0.0005). All experiments were repeated at least three times and results expressed as mean ± SD.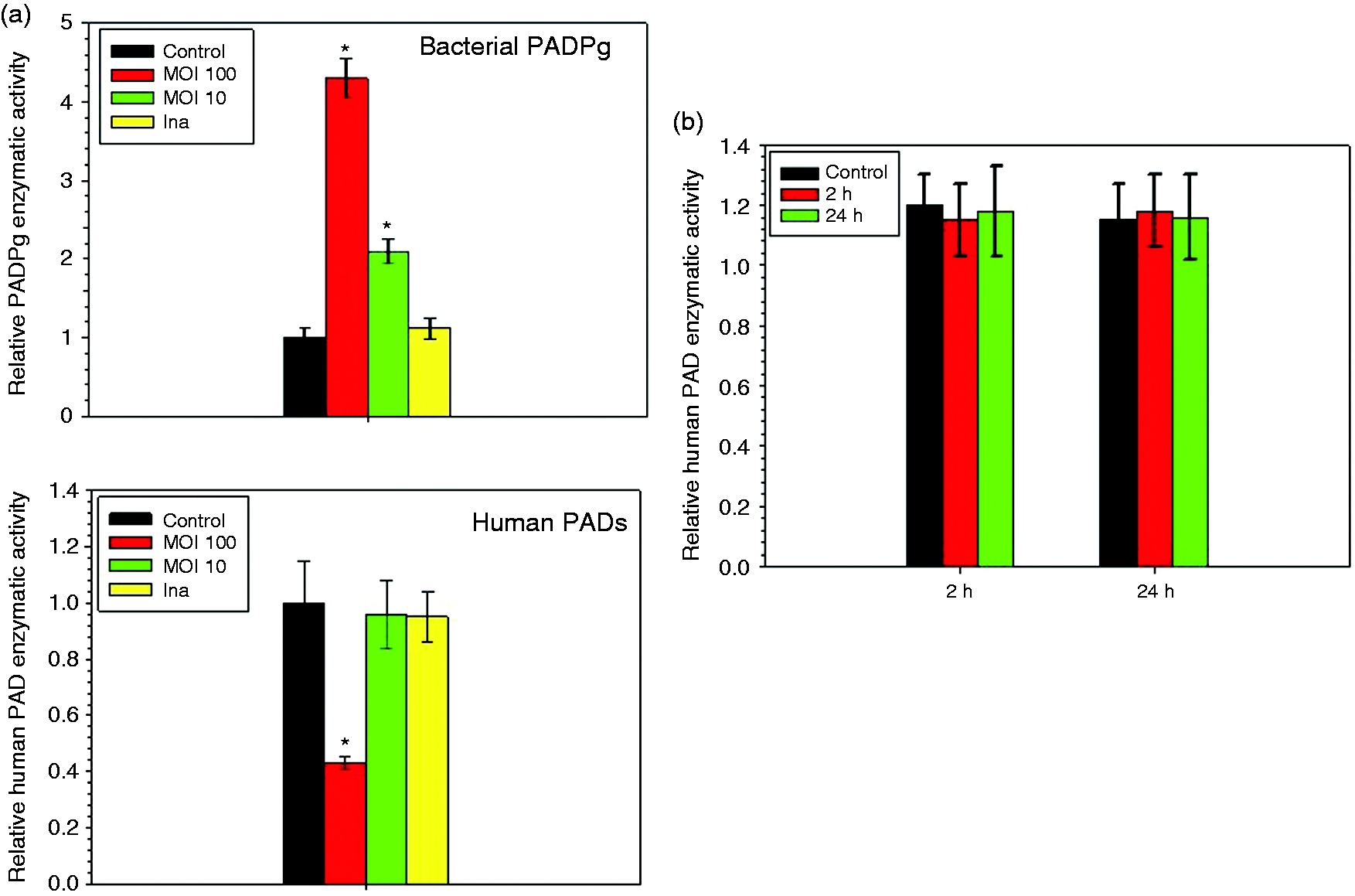
In HC activated by LPS-Pg, no change in the human PAD enzymatic activity was measured as compared with corresponding controls at any time point of the stimulation process (Figure 2b). Indeed, increasing expressions of mRNAs encoding for PAD-2 and PAD-3 previously measured during the activation process were not reflected at the level of PAD enzymatic activity. As expected, no detection of PADPg enzymatic activity was observed (data not shown).
Profiles of citrullinated peptides/proteins in cellular extracts from HC infected with Pg and activated by LPS-Pg
We determined if citrullination profiles of peptides/proteins were modified during infection and activation of HC. Cells were infected during 2 h with Pg and heat-killed Pg, and the cellular extracts were analyzed by Western blotting using an anti-citrulline Ab. As compared with peptides/proteins profiles obtained with cellular extracts from uninfected HC controls, we noticed the apparition of new citrullinated bands with molecular masses ranging from 15 kDa to 45 kDa (Figure 3a). These bands were not present during infection performed with heat-killed Pg and were poorly visible during infection performed with Pg at a MOI of 10.
Western blotting of citrullinated peptides in cellular extracts of HC infected with Pg (a) and activated with LPS-Pg (b). (a) HC were infected for 2 h with Pg at MOI of 10 (Pg 10) and 100 (Pg 100) and with heat-killedPg (Ina) at a MOI of 100. Uninfected cells served as controls (C). (B) HC were activated for 2 h and for 24 h by 1 µg/ml LPS-Pg. Non-stimulated cells served as controls (C). In both experiments, cellular extracts were prepared and analyzed by Western blotting using an anti-citrullinated Ab. Arrows indicate the citrullinated bands (MM ranging from 15 kDa to 45 kDa) appearing in cellular extracts of infected human chondrocytes as compared with the corresponding control. An anti-GAPDH polyclonal Ab served to verify equal loading of proteins between all wells.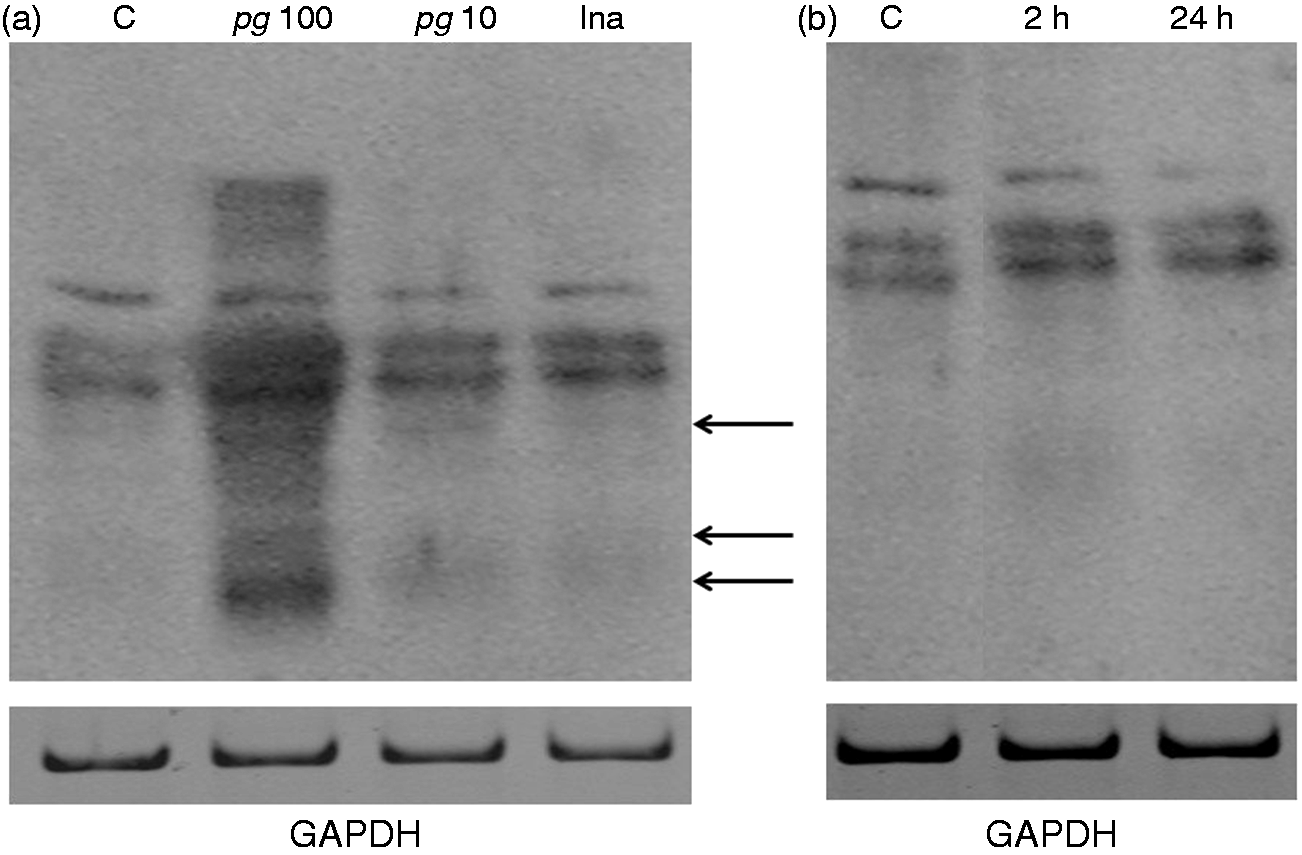
Analysis of cellular extracts of HC activated by LPS-Pg did not reveal any change of peptides/proteins profiles as compared to the control profile and at any time of the stimulation process (Figure 3b).
Discussion
RA is characterized by disease-specific ACPAs generated by cellular PADs. A bacterial PAD (PADPg) has also been described in Pg, but little is known on the effects of cellular PADs and PADPg in HCs, a cell-type producing cartilage. 24 Although recent data using this cell-type highlighted a possible effect of Pg during apoptosis, 25 none is specifically known on the implications of human PADs and PADPg. Our data indicated that mRNA expression levels of PAD-2, PAD-3 and PAD-4 were significantly decreased during infection of HC with live Pg. PAD-2 and PAD-4 expressions have been particularly studied in RA, and both enzymes, together with citrullinated proteins, have been found in synovial fluids and synovial membranes.26–28 PAD-4 has been demonstrated to be the candidate isotype that was most closely associated with RA, according to genetic and immunohistochemical analyses. 29 Additionally, expression of PAD-4 has been associated with RA in Asian, but not in Caucasian populations, and showed a more stable mRNA probably related to the presence of the disease-associated PADI4 haplotype.30,31 PAD-2 expression was also found in patients with RA and osteoarthritis (OA), particularly in close association with inflammation in RA synovial tissue. 32 Besides expression of mRNA encoding for PAD-2 and PAD-4, our data also indicated a weak but significant expression of PAD-3 in HCs. Expression of PAD-3 has only been reported in the upper layers of epidermis and in hair follicles. 33 However, non-expression of human PAD-3 has been reported in various blood-derived cells from healthy patients, notably in macrophages stimulated by LPS from Escherichia coli and in synovial tissue of patients with RA and OA. 27 In our experiments, the mRNA expression of PAD-3 was inconsistent with these clinical observations. This discrepancy might be explained by the fact that the biological samples evaluated by these authors (synovial fluids, synovial tissues, blood, etc.) were heterogeneous in terms of cell-type composition (monocytes, macrophages, synoviocytes, etc.) and did not include HCs. Moreover, our cellular model has been elaborated in vitro and did not take into account the inflammatory process that may affect mRNA expressions of human PADs in tissues.
During infection of HC, significant Pg dose-dependent decreases of mRNA expressions encoding for human PAD-2, PAD-3 and PAD-4 were consistent with a significant decrease of human PAD enzymatic activity only observed with a high-dose of Pg (MOI of 100). In contrast to its human counterparts, the bacterial PADPg enzymatic activity increased and might indicate a potential role for this enzyme during the infection process. Indeed, PADPg might explain the apparition of new produced citrullinated peptides/proteins observed by Western blotting during infection of HC. Although the ability of PADPg to citrullinate cellular/bacterial proteins has already been demonstrated, in particular for fibrinogen and α-enolase, 15 our data did not provide evidence that PADPg contributed directly to citrullination of new peptides/proteins appearing during infection. Nevertheless, we have to bear in mind that these citrullinated peptides/proteins were not present when infection was performed with heat-killed Pg, maybe as a result of the inactivation of PADPg by heating. 14 Enzymes involved in citrullination of these peptides/proteins remain to be identified but we cannot rule out the hypothesis that human PADs could be involved in citrullination of these peptides/proteins. However, such a hypothesis was difficult to consider as significant down-regulation of both human PAD enzymatic activity and corresponding mRNAs were observed during infection of HC.
As Pg possess many virulent factors, including PADPg, we investigated if its purified LPS could be involved in the down-regulation of PAD mRNA expressions initiated by the whole bacteria. Indeed, during activation of HC by LPS-Pg, we observed significant increases of mRNA expression levels of PAD-2 and PAD-3. Despite these increasing expressions, PAD-2 and PAD-3 did not promote the stimulation of total human PAD enzymatic activity and the emergence of new citrullinated peptides/proteins. How expressions of these two human PADs contribute to the action of LPS-Pg in HCs is presently unknown.
Our data demonstrated that Pg was able to initiate the emergence of new citrullinated peptides/proteins in HCs. Whether such citrullinated peptides/proteins included ACPAs remains to be elucidated but our data add further support for a potential causal role of Pg in the etiology of RA. Pg might contribute to establish the joint damage observed in RA pathogenesis and to breach immune tolerance in this disease. 34 To complete this hypothesis, further experiments are needed using either a PADPg-deficient Pg strain to infect HC or the purified PADPg to stimulate HCs and to verify the citrullinated status of auto-Ags.
Footnotes
Acknowledgments
We thank Professor Jacques Eric Gottenberg (Department of Rheumatology, Hôpitaux Universitaires de Strasbourg, France), Professor Olivier Huck and Professor Jean Luc Davideau (both from the Periodontology Department, Faculty of Dental Research, Strasbourg, France) for their continuous interest in our work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors would like to thank the INSERM, the University of Strasbourg and the Dental Faculty of Strasbourg for the financial support.