
Abstract
The type III histone deacetylase SIRT1 has recently emerged as a critical immune regulator by suppressing T-cell immunity and macrophage activation during inflammation, but its mechanism in regulating inflammatory response in macrophages remains unclear. Here we show that the expression of SIRT1 in macrophage cells decreased following the release of inflammation cytokines when the cells were stimulated with LPS. IRF8, an important regulator in monocyte differentiation and macrophage polarization, showed the opposite trend, with SIRT1 expression levels increasing after the cells treated with LPS. Co-immunoprecipitation and immunofluorescence experiments showed that SIRT1 could not only interact with IRF8, but also deacetylate it. LPS treatment had no effect on the expression of IRF8 in macrophage cells in which sirt1 was specifically deleted. Our results show that IRF8 may be the target of histone deacetylase SIRT1 to regulate the inflammation in the macrophage cells.
Introduction
Severe sepsis, or septic shock, represents one of the oldest problems in medicine. Septic shock is caused by severe infection of invading microbes that either produce endotoxin (Gram-negative) bacteria or functional analogous molecules (Gram-positive bacteria or fungi). 1 Endotoxins (outer membrane LPS) represent the major component of these membranes in most Gram-negative bacteria. In Gram-negative sepsis, toxic LPS induces profound activation of, for example, macrophages and the production of potent pro-inflammatory cytokines such as TNF-α, IL-1 and IL-6. 2 These pro-inflammatory cytokines act on immune cells, resulting in multi-organ failure that affects the liver, kidneys and central nervous system. Macrophages are a heterogeneous population of immune cells that are essential for the initiation and resolution of pathogen- or tissue damage-induced inflammation. 3 They demonstrate considerable plasticity, which allows them to respond, and physiology in response to cytokines and microbial signals. 4 These changes can give rise to a population of cells with distinct functions that are phenotypically characterized by the production of pro- and anti-inflammatory cytokines. 5
The Sirt2 gene was first discovered in yeast as a transcription repressor. Its mammalian orthologues, the sirtuins, are a family comprising seven members: SIRT1– SIRT7. Additional evidence suggests that SIRT1 represses inflammatory signaling in multiple tissues and cell types, including macrophages. 6 Caloric restriction increases SIRT1 protein levels in peritoneal macrophages and suppresses the production of pro-inflammatory mediators. 7 Likewise, siRNA-mediated SIRT1 knockdown in RAW264.7 cells increases TNF-α secretion; pharmacological activation of SIRT1 suppresses cytokine release from stimulated macrophages;8,9 myeloid-specific SIRT1 deletion increases macrophage infiltration and pro-inflammatory cytokine production. 10 Taken together, these findings suggest a close link between SIRT1 and macrophage activity in inflammation. However, the mechanism whereby SIRT1 regulates macrophages in response to sepsis is unclear.
IFN regulatory factor 8 (IRF8) is a member of the IRF family, whose expression is restricted to the hematopoietic system. 11 In contrast to a more common dichotomy between transcription factors involved in development and transcription factors involved in environmental responses, 12 IRF8 is required not only for macrophage differentiation, but also for a stimulus-induced expression of some critical immune response genes for IL12p40 and IFN-β. 13 Notch-RBP-J and TLRs are integrated at the level of synthesis of IRF8 protein where heterologous signaling pathways can regulate the TLR-induced pro-inflammatory polarization of macrophages. 14
In this study, we showed that the expression of SIRT1 is decreased in sepsis; SIRT1 functions as a negative regulator of pro-inflammatory cytokines. At the molecular level, SIRT1 interacts with IRF8 and inhibits IRF8 acetylation. Myeloid-specific Sirt1 gene deletion inhibits LPS-induced IRF8 expression. Thus, SIRT1 appears to restrain pro-inflammatory processes by interacting with IRF8.
Materials and methods
Animals, isolation and culture of macrophages
Male C57BL/6 mice (20–24 g, 6–8 wk) were used in the experiments, as well as myeloid-specific SIRT1 knockout (lysMcre Cre+ SIRT1flox/flox) mice with a C57BL/6 background and WT (lysMcre Cre- SIRT1flox/flox) mice.
The bone marrow in the femurs and tibias was exposed and flushed with RPMI 1640 medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated FBS (Gibco), 100 U/ml penicillin and 100 mg/ml streptomycin (Beyotime, Shanghai, China). Medium containing bone marrow cells was centrifuged for 5 min at 400 g.
The method used to isolate peritoneal macrophages from mice has been described previously. 15 Briefly, mice were killed, and 5 ml sterile PBS was injected into the abdomen. The fluid was withdrawn, centrifuged, the cell pellet re-suspended and cultured in DMEM (Gibco) containing 10% FBS, 100 IU/ml penicillin and 100 mg/ml streptomycin, and maintained at 37℃ in a humidified incubator with 5% CO2. The cultured medium was exchanged with new medium after 12 h.
The bone marrow cells were cultured for 3 d. The immortal mouse macrophage cell line RAW264.7 was obtained from American Type Culture Collection (Livingstone, MT, USA). Cells were cultured in DMEM containing 10% FBS, 100 IU/ml penicillin and 100 mg/ml streptomycin, and maintained at 37℃ in a humidified incubator with 5% CO2. RAW264.7 cells were collected by scraping, then centrifuged at 400 g for 5 min, and re-suspended in serum-free DMEM. Cells were seeded in six-well plates (approximately 3.0 × 104 cells/cm2) before further treatments.
Western blotting and immunoprecipitation
Abs to SIRT1 and acetyl lysine, and anti-GAPDH Ab were purchased from Abcam (Cambridge, MA, USA), and anti-IRF8 Ab from Proteintech (Chicago, IL, USA). Protein bands were analyzed using ImageJ densitometry analysis and the fold expression was indicated as the relative protein level. For the protein–protein interaction study, macrophages were washed with PBS once and then lysed in cold RIPA buffer (Beyotime) for 10 min at 4℃. Cellular lysates were subjected to immunoprecipitation with IgG or anti-Sirt or anti-IRF8 Abs in a buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 0.1% Nonidet P (NP)-40, 1 mM EDTA, 1 mM PMSF and a protease inhibitor mixture (Roche, Indianapolis, IN, USA). After overnight incubation at 4℃, protein A/G-agarose beads (Santa Cruz, CA, USA) were added to the immunoprecipitation and incubated for 1 h at 4℃. The agarose beads were then washed extensively three times before being subjected to Western blot analysis.
Quantitative real-time PCR
Total RNA was extracted from RAW264.7 cells by using RisoPlus (TaKaRa, Shiga, Japan) according to the manufacturer’s protocol. Obtained RNA was reversely transcribed into cDNA using the PrimeScriptTM RT Master Mix Kit (Perfect Real Time, TaKaRa). The primer sequences for each gene were as follows: SIRT1: Sense, 5′-CAGACCCTCAAGCCATGTTTGATA-3′; Anti-sense, 5′-TTGGATTCCTGCAACCTGCTC-3′. TNF-α: Sense, 5′-CGTCAGCCGATTTGCTATCT-3′; Anti-sense, 5′-CGGACTCCGCAAAGTCTAAG-3′. IL-1β: Sense, 5′-GCCCATCCTCTGTGACTCAT-3′; Anti-sense, 5′-AGGCCACAGGTATTTTGTCG-3′. GAPDH: Sense, 5′-TGTGTCCGTCGTGGATCTGA-3′; Anti-sense, 5′-TTGCTGTTGAAGTCGCAGGAG-3′. IL10: Sense, 5′-GCCAGAGCCACATGCTCCTA-3′; Anti-sense, 5′-GATAAGGCTTGGCAACCCAAGTAA-3′. IRF8: Sense, 5′-TGACACCAACCAGTTCATCCGAGA-3′; Anti-sense, 5′-CACCAGAATGAGTTTGGAGCGCAA-3′. iNOS: Sense, 5′-CAAGCACATTTGGGAATGGAGA-3′; Anti-sense, 5′- CAGAACTGAGGGTACATGCTGGAG-3′. Arg1: Sense, 5′- AGCTCTGGGAATCTGATGG-3′; Anti-sense, 5′- ATTACACGATGTCTTTGGCAGATA-3′; IL12a: Sense, 5′CTCCATCGCTTCTCTCATATTC-3′, Anti-sense, 5′AGTTCTTGCTCTTCTGCTAAC3′.
ELISA
Supernatants from macrophage cultures were harvested at the indicated time points. Mouse TNF-α and mouse IL-1β were detected using an ELISA kit (RayBio®, Norcross, GA, USA and NeoBioscience, Shenzhen, China, respectively) according to the manufacturer’s instructions. Concentration was calculated by regression analysis of a standard curve.
Lentivirus infection
Lentiviruses expressing mouse siRNA targeted Sirt1 were constructed by life technology (Invitrogen, Carlsbad, CA, USA). We cultured macrophage cells in DMEM supplemented with 10% FBS. For knockdown experiments, we infected the cells with various lentiviruses for 6 h in cultured media with 2% FBS. The medium was then replaced by fresh DMEM supplemented with 10% FBS for another 48 h. Cells were stimulated with LPS.
Immunofluorescence staining
Macrophage cells stimulated with or without LPS were fixed using 4% paraformaldehyde for 20 min and stained for IRF8 (rabbit) or SIRT1 (mouse). DAPI was used for nuclear staining. Expression and localization of IRF8 and SIRT1 was analyzed using an Olympus IX71 light microscope (Olympus, Tokyo, Japan).
Statistical analysis
All experiments were performed at least three times using different batches of cells. Results are presented as mean ± SEM. Statistical analysis of the data was performed using SPSS 17.0 software (IBM, Armonk, NY, USA) by one-way ANOVA with least significant difference or S-N-K’s post-hoc analysis. Values of P < 0.05 were considered to be statistically significant.
Results
TLR-induced inflammatory cytokine expression in macrophages
To investigate whether LPS could induce the expression of inflammatory cytokines in macrophages, we separated mice peritoneal macrophages. Cells were stimulated with 1 µg/ml LPS,14,16 and cytokine expression was measured by RT-PCR or ELISA. IL-1β (Figure 1a) and TNF-α (Figure 1b) mRNA were increased at 2 h after LPS stimulation, and continued to increase until 12 h. There were no significant differences at 24 h, although the mRNA level was higher compared with that at 0 h. The release of IL-1β (Figure 1g) and TNF-α (Figure 1h) were up-regulated at 6 h post LPS-treatment which continued to increase until 24 h. iNOS (Figure 1c) and IL-12 a (Figure 1d) mRNA were up-regulated and increased at 6 h post-LPS treatment, and the iNOS mRNA level continued to increase until 24 h. Furthermore, we tested the expression of anti-inflammation factors Arg1 and IL-10. The results showed that the expression level of Arg1 was down-regulated at 6 h and continued to decrease until 24 h (Figure 1e). The IL-10 mRNA level was significantly down-regulated at 24 h (Figure 1f). The results in the RAW 264.7, a macrophage-like cell line, showed a similar trend (data not shown). These results suggested that the macrophage status was effected by TLR ligands.
Assessment of the expression levels of pro-inflammatory cytokines in peritoneal macrophages treated with 1 µg/ml LPS. (a–f) Peritoneal macrophages were exposed to 1 µg/ml LPS and cultured at the indicated time points. The mRNA levels of (a) IL-1β, (b) TNF-α, (c) iNOS, (d) IL-12 a, (e) Arg1 and (f) IL-10 (f) were assessed by real-time PCR. The protein level of (g) IL-1β and (h) TNF-α were examined by ELISA. Each data point is the mean ± SEM of three experiments and normalized against corresponding GAPDH mRNA level with the value at 0 h arbitrarily set as 1. *P < 0.05.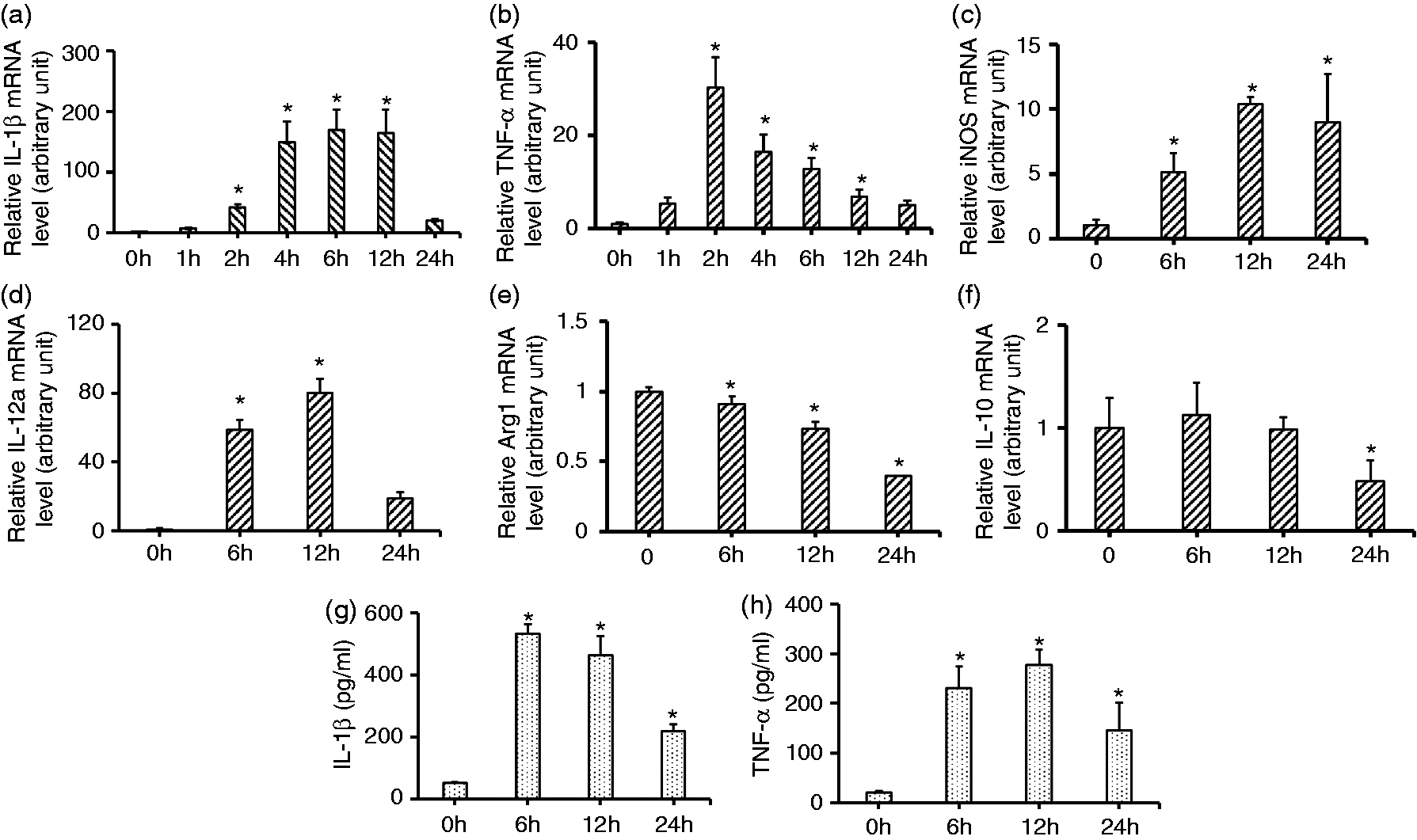
SIRT1 suppresses inflammation cytokines in macrophages
Recent studies discovered that SIRT1 plays an important role in immunity response.
10
To understand the role of SIRT1 in LPS-induced cell signaling processes, we firstly determined the time course for the expression of steady-state SIRT1 in macrophages in vitro, in response to LPS. The SIRT1 protein expression level was detected by Western blot. Treatment of RAW264.7 cells with LPS resulted in a substantial decrease in steady-state SIRT1 protein expression level (at 12 h and until 24 h; Figure 2a). Furthermore, peritoneal macrophages also showed decreased levels of SIRT1 expression when stimulated with LPS for 12 h, and until 24 h (Figure 2b). We then analyzed the mRNA level of the sirt1 gene. The results indicated that stimulation of RAW264.7 cell lines (Figure 2c) and peritoneal macrophages (Figure 2d) with LPS significantly decreased sirt1 mRNA levels at 12 h, and until 24 h. Then we examined whether activation of SIRT1 inhibited pro-inflammatory cytokines in the peritoneal macrophages. The cells were pre-treated with SRT1720 for 6 h, a SIRT1 chemical activator, and then stimulated with LPS for 12 h. P53 as the target of SIRT1 reflects the enzyme activity of SIRT1.
17
Firstly, acetylation of p53 was examined which was decreased when the cells were exposed to SRT1720. This suggested that SRT1720 could activate SIRT1 (Figure 2e). Secondly, we observed that the mRNA levels of pro-inflammatory IL-1β (Figure 2f) and TNF-α (Figure 2g) were decreased compared with the cells without SRT1720 pre-treatment. The trend in RAW264.7 cells was similar (data not shown). These results indicated that SIRT1 could mediate the inflammation response in macrophages.
SIRT1 mRNA and protein expressions in macrophages in response to endotoxin. (a) RAW 264.7 or (b) peritoneal macrophage cells were treated with LPS (1 µg/ml) at different time points. Various cell lysates (30 mg protein) were prepared and subjected to Western blot analysis by using Abs specific for SIRT1 and GAPDH. RT-PCR assessing the change in SIRT1 mRNA level after treatment of (c) RAW264.7 cells or (d) peritoneal macrophage cells with LPS (1 µg/ml) at the indicated time points. (e) Peritoneal macrophage cells lysates were subjected to Western blot analysis by using Abs specific for acetyl/Lys379-p53 and p53. Peritoneal macrophage cells were stimulated with 1 µg/ml LPS after pre-treatment with 1uM SRT1720 for 6 h. The mRNA levels of (f) IL-1β and (g) TNF-α were assessed by RT-PCR. Each data point is the mean ± SEM of three experiments and normalized against corresponding GAPDH level. *P < 0.05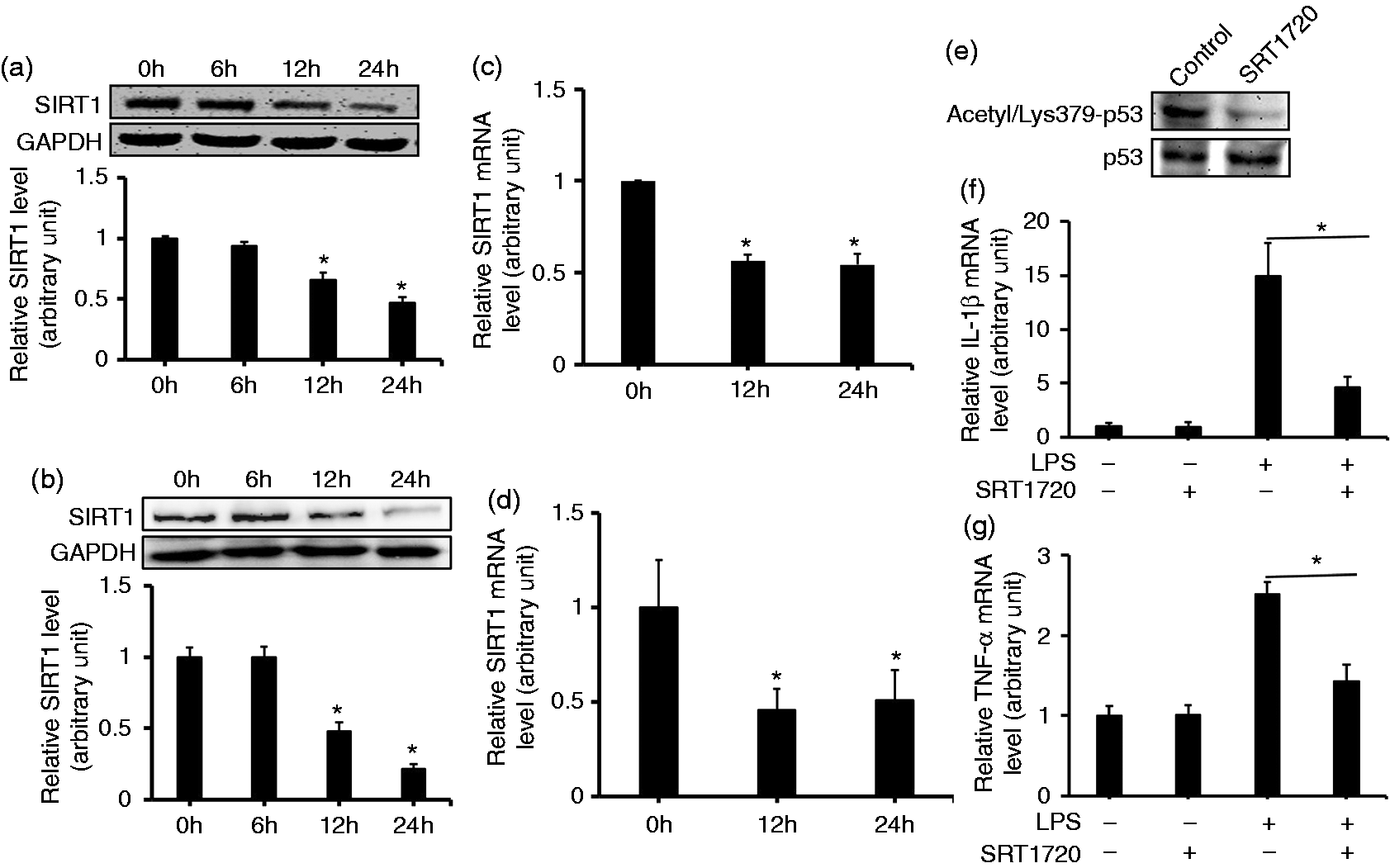
TLR ligand induces IRF8 expression in macrophages
Some members of the IRF family have been shown to be induced in macrophages by pathogenic components,
18
which raised the possibility that the recognition of molecular PAMPs by PRRs might be responsible for their expression. As anticipated, elevated levels of irf8 mRNA were observed in LPS-stimulated RAW264.7 cells (Figure 3a) and the peritoneal macrophages (Figure 3b) compared with those in control cells at 12 h. Levels remained elevated even at 24 h. When protein levels were examined in RAW264.7 cells, a higher IRF8 expression occurred together with the mRNA level (Figure 3c). Furthermore, peritoneal macrophages showed increased levels of IRF8 expression when stimulated by LPS (Figure 3d). Additionally, immunofluorescence imaging in the RAW264.7 cells showed the induction of IRF8 (Figure 3e). Together, these results indicated that IRF8 expression is controlled by TLR signaling.
IRF8 expression level in macrophages in response to LPS treatment. RT-PCR assessed the change in SIRT1 mRNA levels after treatment of (a) RAW264.7 cells or (b) peritoneal macrophage cells with LPS (1 µg/ml) at indicated time points. (c) RAW 264.7 cells or (d) peritoneal macrophage cells were treated with LPS (1 µg/ml) at the indicated time points. Various cell lysates (30 mg protein) were prepared and subjected to Western blot analysis by using Abs specific for IRF8 and GAPDH. (e) The immunofluorescence images show the protein expression of IRF8 after treatment of RAW264.7 cells with LPS (1 µg/ml) for 24 h. Each data point is the mean ± SEM of three experiments and normalized against corresponding GAPDH level. *P < 0.05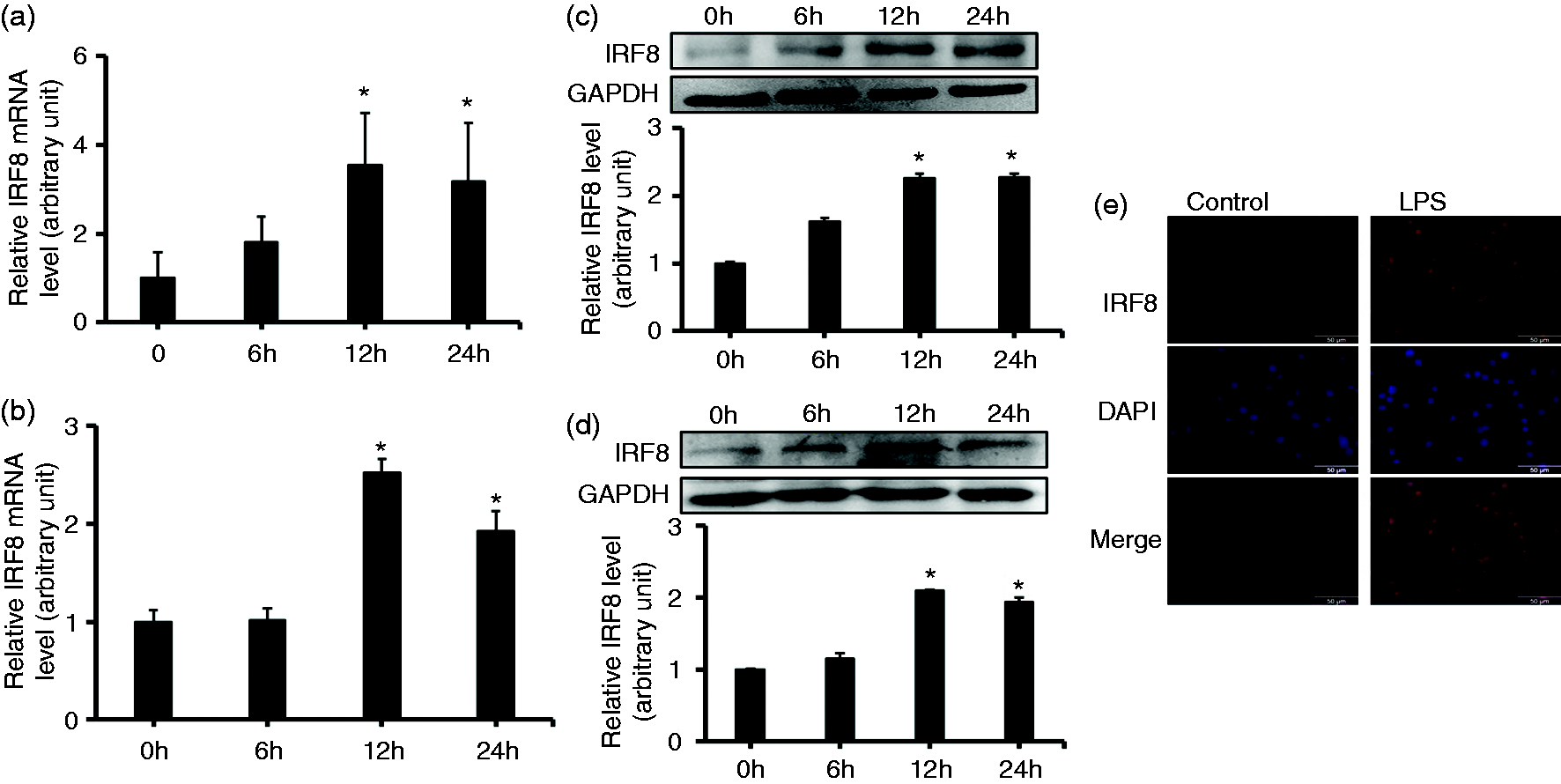
SIRT1 interact with IRF8 in macrophages
As a deacetylase, SIRT1 may suppress pro-inflammatory cytokine production by deacetylation of the transcription factor IRF8 that mediates macrophage polarization. To test this hypothesis, the interaction of SIRT1 with IRF8 was analyzed. Firstly, the localization of SIRT1 and IRF8 was investigated. Immunofluorescence imaging of the RAW264.7 cells showed that both proteins co-localize in the nucleus with some amount also present in the cytosol (Figure 4a). To further verify the localization, the nucleus and cytosol proteins were separated with a kit. The results proved that both proteins were expressed in the nucleus and cytosol (Figure 4b). These results suggested that the two proteins interact. So, to confirm SIRT1 interaction with IRF8, whole extracts of RAW264.7 cells were collected, and the interaction of endogenous SIRT1 with IRF8 was determined by co-immunoprecipitation and Western blotting. The IRF8 band was detected in the anti-SIRT1 immunoprecipitation (Figure 4c). In summary, these results indicated that SIRT1 interacted with IRF8 in macrophages.
SIRT1 interacts with IRF8. (a) Immunofluorescence images identified the localization of SIRT1 and IRF8 in RAW264.7 cells. Green: SIRT1; red: IRF8; blue: DPAI; orange: merger of green and red. (b) Nuclear and cytoplasm extracts of RAW264.7 cells are prepared for Western blot analysis by using Abs specific for SIRT1, IRF8, GAPDH and histone H3. (c) The interaction of SIRT1 with IRF8 was determined by co-immunoprecipitation with anti-SIRT1 Ab and Western blotting with anti-IRF8 Ab. The expression of SIRT1 in the whole-cell lysates was confirmed by Western blotting (input).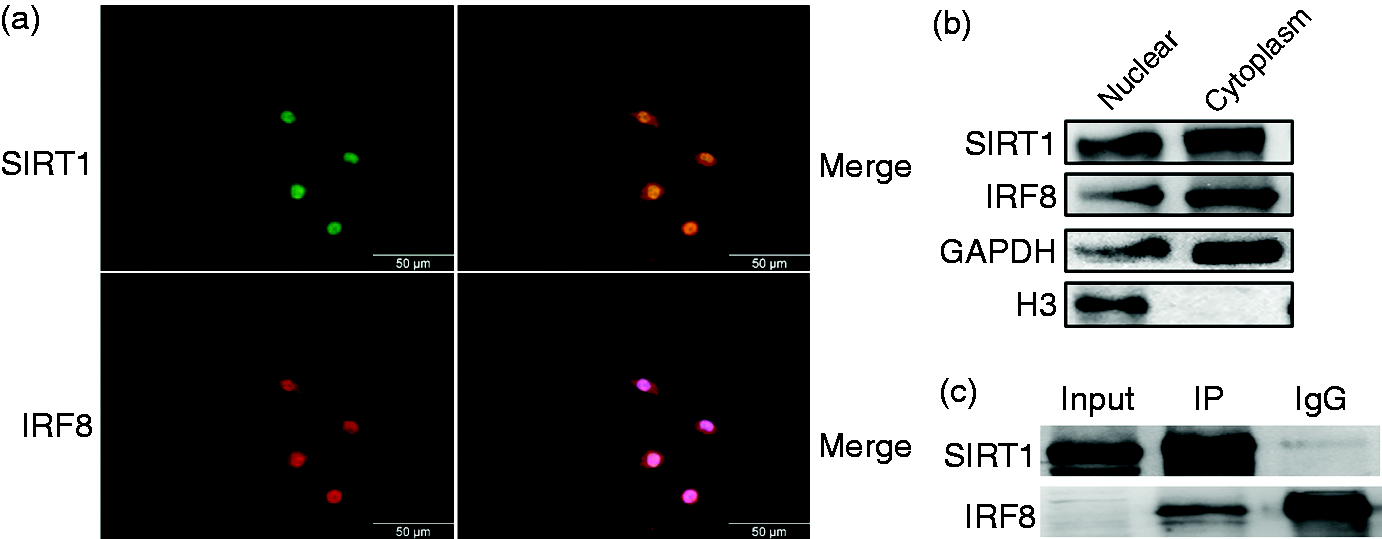
IRF8 expression is regulated by SIRT1
As shown above, SIRT1 interacted with IRF8; thus, it might be involved in LPS-induced IRF8 gene expression. To test this hypothesis, peritoneal macrophages from sirt1 conditional knock-out mice were utilized. Western blotting results showed that SIRT1 protein was not expressed in these peritoneal macrophages or in bone marrow monocytes (Figure 5a). The peritoneal macrophages from wild type and sirt1 knock-out mice were exposed to LPS at different times, and IRF8 expression levels were analyzed by Western blotting. The results showed that LPS stimulation had no effect on IRF8 expression at the different time points. However, IRF8 expression was increased by LPS treatment of peritoneal macrophages from wild type mice at 12 h and until 24 h (Figure 5b). Additionally, we transduced the RAW264.7 cells with lentivirus encoding sirt1 specific siRNA. Number 2, number 3 and number 4 lentivirus siRNA significantly inhibited the expression of SIRT1 (Figure 5c). The cells were transduced with number 4 lentivirus siRNA following treatment with LPS for 12 h. No significant change in the expression of IRF8 was observed (Figure 5d). These results suggested that SIRT1 mediated LPS-elicited IRF8 expression.
The effects of SIRT1 on IRF8 protein expression. (a) Peritoneal macrophage (PM) and Bone marrow monocyte (BMM). from Sirt1+/+ and Sirt1–/– mice were acquired. Cell lysates (30 mg protein) were prepared and subjected to Western blot analysis by using Abs specific for SIRT1 and GAPDH. (b) Peritoneal macrophage cells from wild type mice and Sirt1–/– mice were treated with LPS (1 µg/ml) at the indicated time points. Various cell lysates (30 mg protein) were prepared and subjected to Western blot analysis by using Abs specific for IRF8 and GAPDH. (c) RT-PCR assessing the change in SIRT1 mRNA level after treatment of RAW264.7 cells with lentivirus-targeted SIRT1. (d) Cells were pre-infected with lentivirus-4 for 48 h, then exposed to LPS (1 µg/ml) for 12 h. Cell lysates (30 mg protein) were prepared and subjected to Western blot analysis by using Abs specific for IRF8 and GAPDH. Each data point is the mean ± SEM of three experiments and normalized against corresponding GAPDH level. *P < 0.05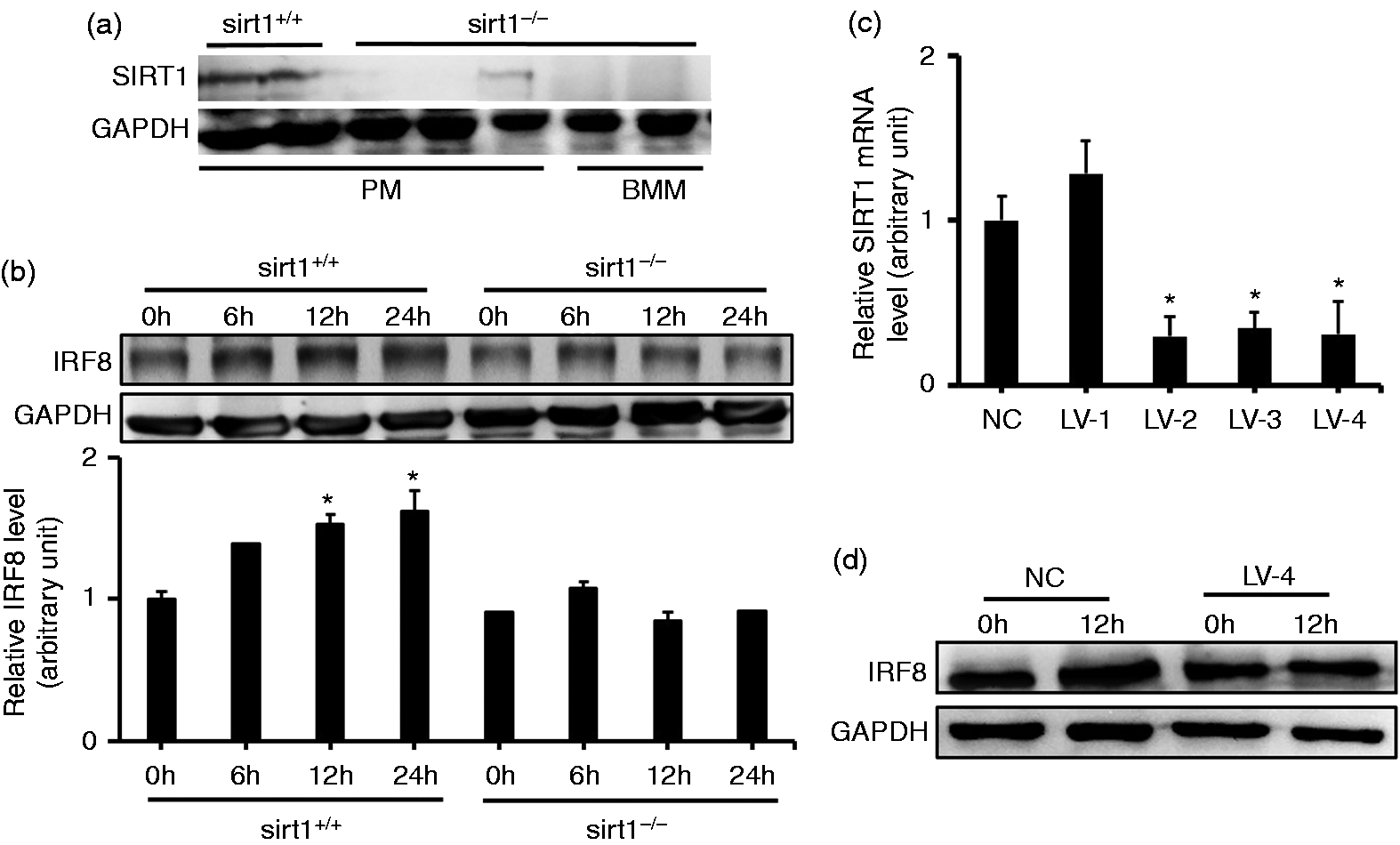
SIRT1 deacetylates IRF8 in macrophages
Acetylation of IRF family transcription factors by acetyl-transferase p300, cAMP-response element-binding protein and p300/CBP-associated factor has been shown to be critical for their functions.19,20 As SIRT1 is a deacetylase, we asked the question of whether SIRT1 regulates IRF8 acetylation. RAW264.7 cells were pre-treated with the SIRT1 inhibitor EX527 for 6 h, and the whole-cell extracts were collected for co-immunoprecipitation. The acetyl-Lys band was detected in the anti-IRF8 immunoprecipitation; and the acetyl-Lys increased in the EX527 pre-treated cells compared with that from cells without pre-treatment (Figure 6). Therefore, SIRT1 deacetylates IRF8 in macrophages.
SIRT1 deacetylates IRF8. RAW264.7 cells were treated with 10 µM EX527 for 6 h and the cell lysates were prepared. IRF8 acetylation was determined by immunoprecipitation of IRF8 with anti-IRF8 Ab and Western blotting with anti-acetyl-lysine Ab.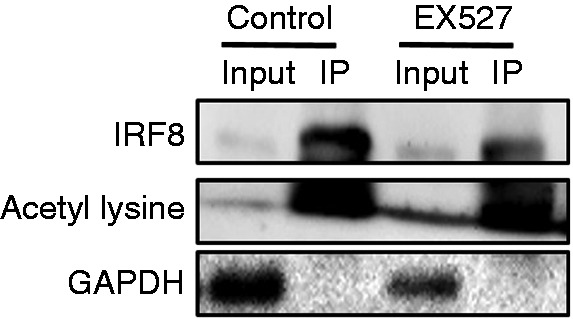
Discussion
Since the discovery of sirtuins as longevity determination genes in yeast over a decade ago, much effort has been spent in elucidating the molecular mechanism and physiological functions of their mammalian homologs. Recent data pointed towards an association between SIRT1 and the transcription factor NF-κB, which renders this signaling pathway an intriguing target for the treatment of chronic inflammatory disease.21,22 SIRT1 has been proven to be a critical immune regulator by suppressing macrophage functions. 23 In our study, we used an in vitro sepsis model to explore the role of SIRT1 in the regulation of the inflammatory response. We demonstrated that macrophages stimulated with LPS had higher mRNA levels of pro-inflammatory cytokines, such as TNF-α, IL-1β, iNOS and IL-12 a. Moreover, the release of TNF-α and IL-1β were increased. Conversely, the mRNA levels of anti-inflammatory factors, such as IL-10 and Arg1, were inhibited. We also found that SIRT1 expression was down-regulated in macrophages exposed to LPS. When the cells were pre-treated with SIRT1 activator, the mRNA levels of pro-inflammatory cytokines were decreased. These results suggest that SIRT1 plays an important role in controlling the cytokine production in sepsis.
The IRF family consists of nine members (IRF1–IRF9) in mammals and exerts essential regulatory functions in the immune system,24,25 participating in divergent and overlapping molecular programs. IRF8 expression is known to be transcriptionally inducible by IFN-γ.26,27 Here we found that LPS induced rapid expression of IRF8 protein in RAW264.7 cells and peritoneal macrophages. We also confirmed that stimulation with LPS resulted in higher expression of IRF8 mRNA.
Accumulated evidence suggests that acetylation of IRF family transcription factors is required for their optimal transcriptional activity. 28 Acetylation of diverse domains of IRF members results in a conformational change and thus influences their DNA-binding activities. 29 Our current data revealed that SIRT1 and IRF8 co-localize in both nucleus and cytoplasm, as shown by immunofluorescence and Western blot. The co-localization of the two proteins may be the prerequisite of their interaction. Also it was shown that SIRT1 interacted with IRF8 in macrophages. Previous studies have revealed that SIRT1 can modulate the IRF1 acetylation status in dendritic cells.30,31 SIRT1 inhibits IL-27p28 subunit expression by suppressing IRF1 transcriptional activity. Our data showed that LPS stimulation had no effect on the expression of IRF8 in SIRT1-deleted peritoneal macrophages. When SIRT1 expression was interfered with in the RAW264.7 cells with lentivirus, there was no change in IR8 expression due to LPS treatment. These results may suggest that IRF8 expression was regulated by SIRT1. However, further investigations are necessary to confirm how SIRT1 regulates the IRF8 expression in macrophages. Lys-78 of helix α3 within the DNA binding domain was the major acetylation site for both IRFs. 32 We found that a SIRT1 inhibitor could increase the acetylation of IRF8 in RAW264.7 cells. However, it is necessary to verify the effects of acetylation status on IRF8 function.
In summary, these results indicate that LPS down-regulates SIRT1 expression and increases IRF8 expression level; SIRT1 interacts with and regulates the acetylation status of IRF8. Therefore, IRF8 may point to new mechanism of macrophage inflammatory response and to a potential target for the development of a sepsis therapy.
Footnotes
Acknowledgements
We thank Chaowu Tang (Department of Burns and Cutaneous Surgery, Xijing Hospital) for his kind technical support in this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We thank the Natural Science Foundation of China (grant no: 81530064) and Xijing Hospital Natural Science Funds for Distinguished Young Scientists (grant no: XJZT15M16) for financial support.