
Abstract
Neutrophil (polymorphonuclear leukocyte) activation with release of granule contents plays an important role in the pathogenesis of acute lung injury, prompting clinical trials of inhibitors of neutrophil elastase. Despite mounting evidence for neutrophil-mediated host tissue damage in a variety of disease processes, mechanisms regulating azurophilic granule exocytosis at the plasma membrane, and thus release of elastase and other proteases, are poorly characterized. We hypothesized that azurophilic granule exocytosis would be enhanced under priming conditions similar to those seen during acute inflammatory events and during chronic inflammatory disease, and selected the cytokine TNF-α to model this in vitro. Neutrophils stimulated with TNF-α alone elicited intracellular reactive oxygen species (ROS) generation and mobilization of secretory vesicles, specific, and gelatinase granules. p38 and ERK1/2 MAPK were involved in these components of priming. TNF-α priming alone did not mobilize azurophilic granules to the cell surface, but did markedly increase elastase release into the extracellular space in response to secondary stimulation with N-formyl-Met-Leu-Phe (fMLF). Priming of fMLF-stimulated elastase release was further augmented in the absence of NADPH oxidase-derived ROS. Our findings provide a mechanism for host tissue damage during neutrophil-mediated inflammation and suggest a novel anti-inflammatory role for the NADPH oxidase.
Introduction
Aberrant or prolonged neutrophil (polymorphonuclear leukocyte or PMN) activation is well recognized as a cause of host-tissue damage in a range of local and systemic inflammatory conditions. Among PMN granules, the azurophilic or primary granules possess the most toxic contents including a number of proteases. Two azurophilic granule proteins, elastase and cathepsin G, have specific targets for host injury in the lung via degradation of the surfactant proteins A and D as previously described.1,2 Moreover, there are extensive data supporting a causative role for neutrophil activation including release of granule contents in the pathogenesis of acute lung injury (ALI).3,4 In animal models of ALI, neutrophil elastase has also been demonstrated to be a mediator of tissue injury, 5 with blockade or deletion of this protease eliciting improved outcomes.6,7 Clinically, this has led to a number of clinical trials using an inhibitor of human neutrophil elastase to treat patients with acute respiratory distress syndrome and ALI.8,9 Despite this appreciation of the potential host toxicity stemming from inappropriate activation of PMN, there is limited understanding of the mechanisms governing mobilization of azurophilic granules to the cell surface. Much of our early understanding of granule mobilization came from studies of PMN phagocytosis. 10 More recent studies using physiologically relevant soluble stimuli show that granule exocytosis at the plasma membrane may function in a mechanistically distinct fashion from delivery to the phagosome.
Neutrophil priming or pre-activation has been extensively investigated in the laboratory using both in vitro analyses in human PMN and using murine models of inflammation. A growing literature has demonstrated that priming is not a laboratory artifact, but occurs in vivo with primed neutrophils identified in the circulation of a number of diverse patient populations following systemic insults. Most of the in vivo data have focused on acute inflammatory events including sepsis, ALI, hemorrhagic shock, and other severe trauma.11,12 However, there are also descriptions of neutrophil priming during chronic inflammatory disease, including rheumatoid arthritis and inflammatory bowel disease.13–16 Exploration of TNF-α as a priming agent seems particularly relevant given that circulating TNF-α levels are elevated in patients with these diseases and that anti-TNF-α therapy is a mainstay of treatment. 17 Although many investigators have studied priming by TNF-α, our laboratory has specifically focused on stimulation with physiologic levels of TNF-α that mimic those found in the serum during acute and chronic inflammation. 18 For example, in inflammatory bowel disease, serum levels ranging from 0 to 150 pg/ml have been reported. 15 We have demonstrated PMN priming in response to TNF-α at levels as low as 1 pg/ml, 18 and that responses vary significantly between low and high TNF-α concentrations. TNF-α levels in the lungs during ALI events are not known, but are likely greater than that seen during chronic inflammation.
Current understanding suggests that PMN do not release cytolytic granule contents after interaction with only the priming stimulus, but instead display enhanced readiness for response to a subsequent stimulus and progression to full activation. Under infectious conditions, this process would appear to be a pathophysiologically appropriate mechanism to enhance neutrophil interactions with pathogenic bacteria after entry of PMN into an infected tissue. However, there are a number of sterile chronic inflammatory conditions where the presence of primed PMN may result in significant host tissue damage with no obvious benefit to the host. Particularly of concern is the potential destructive effect of azurophilic granule release based on the proteolytic contents. Although it has recently been demonstrated that azurophilic granule release can be altered by endotoxin priming, 19 there is limited evidence regarding the effects of low-level TNF-α stimulation on priming of azurophilic granule mobilization, 20 which would be highly relevant to inflammatory disease pathogenesis.
In the current study we focus on the priming of azurophilic granule exocytosis in response to TNF-α. We provide evidence that TNF-α at pathophysiologically relevant concentrations primes neutrophils for enhanced azurophilic granule exocytosis in response to a secondary stimulus. To better understand the mechanisms controlling this priming response, we investigated the role of MAPK signaling and NADPH oxidase-derived reactive oxygen species (ROS) signaling. Although we have previously demonstrated that TNF-α priming elicits activation both of p38 and ERK1/2 MAPKs, neither is involved in the priming of elastase release. Moreover, the NADPH oxidase appears to serve as a repressor of azurophilic granule release under both basal and priming conditions. This anti-inflammatory signaling role for ROS provides a novel mechanism for maintaining homeostasis and has significant implication for the treatment of patients both with acute and chronic inflammation.
Materials and methods
Materials
Hanks Balanced Salt Solution (HBSS) was purchased from BioWhittaker (Walkersville, MD, USA), Dulbecco’s Phosphate-Buffered Saline (DPBS) from Mediatech (Manassas, VA, USA), N-Formyl-Met-Leu-Phe (fMLF) from Fisher Scientific (Pittsburgh, PA, USA), dextran from Pharmacosmos (Holbaek, Denmark), Ficoll-Paque from GE Healthcare (Piscataway, NJ, USA), HSA from Talecris Biotherapeutics (Durham, NC, USA), normal goat serum and 7-amino-4-methylcoumarin (AMC) from MP Biomedicals (Solon, OH, USA), OxyBurst® Green H2HFF BSA (OxyBurst) and Amplex® UltraRed from Molecular Probes (Eugene, OR, USA), paraformaldehyde from Electron Microscopy Sciences (Hatfield, PA, USA), and MAPK inhibitors from EMD Millipore (Billerica, MA, USA). Abs included active CD11b from BioLegend (San Diego, CA, USA), and CD63 from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA, USA). CD66b, lactoferrin and biotinylated lactoferrin Abs, and lactoferrin standard were purchased from Abcam (Cambridge, MA, USA). Recombinant TNF-α, avidin-HRP, matrix metallopeptidase 9 (MMP-9) and biotinylated MMP-9 Abs, and recombinant human MMP-9 were purchased from R&D Systems (Minneapolis, MN, USA). Fluorescently conjugated secondary Abs were from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Additional reagents were obtained from Sigma (St. Louis, MO, USA). All buffers and reagents were strictly endotoxin free.
Human PMN purification
Human PMN were isolated according to standard techniques from heparin anti-coagulated venous blood from healthy consenting adults and individuals with X-linked chronic granulomatous disease (CGD) following written informed consent and in accordance with a protocol approved by the Institutional Review Board for Human Subjects at the University of Iowa and the University of Texas Southwestern Medical Center. PMN were isolated using dextran sedimentation and Hypaque-Ficoll density-gradient separation followed by hypotonic lysis of erythrocytes as previously described. 21
Analysis of cell surface protein expression by flow cytometry
PMN were analyzed using a FACScan or FACScalibur flow cytometer from BD Biosciences (Franklin Lakes, NJ, USA). For assessment of surface expression of active CD11b, CD66b, and CD63, PMN were incubated in HBSS containing 1% HSA and 0.1% dextrose ± TNF-α, as specified. Following stimulation, cells were centrifuged, resuspended in blocking buffer (DPBS with 2% nonfat dry milk and 4% normal goat serum), and incubated on ice for 20 min. Primary Abs were added after blocking and incubated on ice for one hour. PMN were centrifuged and resuspended in fluorescein isothiocyanate (FITC)-conjugated secondary Ab diluted 1:1000 and incubated on ice for 30 min.
Analysis of lactoferrin and gelatinase release by ELISA
Freshly isolated PMN were diluted in HBSS containing 1% HSA and 0.1% dextrose. A total of 4.5 × 105 PMN were added per well of a 96-well microplate and stimulated with TNF-α or no agonist, as specified, in a 37℃/5% CO2 incubator for 60 min. In some experiments, fMLF (1 µM) was added as a secondary stimulus after a 30-min priming period with TNF-α. After stimulation, supernatant was collected from the wells and centrifuged to remove cells. The supernatant was collected and stored at –20℃ until ELISA was performed. A 96-well NUNC MaxiSorp microplate from Thermo Fisher Scientific (Rochester, NY, USA) was coated with 8 µg/ml MMP-9 or 2 µg/ml lactoferrin Ab in carbonate buffer (100 mM NaHCO3, 34 mM Na2CO3, pH 9.6) and incubated at room temperature (22℃) on a rotator for 16h. Wells were washed three times in wash buffer (0.05% Tween 20 in DPBS), then blocked with DPBS containing 1% BSA and 5% sucrose for 1h. Samples and standards, diluted in assay diluent (0.1% BSA in DPBS), were applied and allowed to incubate for 2h. Next, 0.4 µg/ml biotinylated MMP-9 or 0.5 µg/ml biotinylated lactoferrin Ab was added and allowed to incubate for 2h. Avidin-HRP was added to the wells and allowed to incubate for 20 min. Color development occurred during incubation with tetramethylbenzidine for one to 3 min and was stopped by addition of 0.5 M H2SO4. All steps were carried out at room temperature, and the plate was washed three times with wash buffer between all steps.
Elastase activity
Freshly isolated PMN were diluted in PICM-G Buffer (10 mM sodium phosphate buffer with 2.7 mM KCl, 138 mM NaCl, 0.6 mM CaCl2, 1.0 mM MgCl2 and 0.1% dextrose) containing AMC (20 µM) and added to a 96-well microplate as described above. For priming experiments, fMLF (1 µM) was injected at specified time points following treatment with TNF-α or no agonist, as specified. PMN were also exposed to dihydrocytochalasin B (DHCB) (2.5 µg/ml), a microfilament-disrupting agent, prior to stimulation with fMLF as a measurement of maximum elastase release from the azurophilic granules. In some experiments, diphenyleneiodonium (DPI) (50 μM) was used to inhibit the NADPH oxidase. Fifty µM DPI was chosen after preliminary studies demonstrated > 99% inhibition of superoxide generation in response to phorbol-12-myristate-13-acetate as measured by reduction of ferricytochrome c, whereas 10 μM DPI inhibited approximately 95% of superoxide. The FLUOstar Omega was used to measure fluorescence at an excitation of 360 ± 20 nm and emission of 450 ± 30 nm with readings every 60s. Elicited elastase release was expressed as the change in fluorescence over a specified time period.
MAPK signaling
In some experiments, inhibitors of p38 MAPK (SB 203580) or ERK1/2 (PD 98059) signaling were used as previously described. 18 The p38 MAPK inhibitor or a control inhibitor (SB 202474), both at 10 µM, was added to PMN at specified time points before or following treatment with TNF-α (1 ng/ml) or no agonist. In separate experiments, as described, the ERK1/2 inhibitor was added at 100 µM prior to or following treatment with TNF-α.
Measurement of intracellular ROS by flow cytometry
Freshly isolated PMN were stimulated with TNF-α or no agonist in suspension at 37℃ in the presence of OxyBurst (100 µg/ml), as specified, to detect ROS present in endocytic compartments. Immediately following the specified time points, the cells were placed on ice and analyzed by flow cytometry.
Measurement of NOX2 activity by lucigenin-enhanced chemiluminescence (LUC-CL) and Amplex UltraRed
LUC-CL and Amplex UltraRed assays of NADPH oxidase 2 (NOX2) activity were performed in a 96-well microplate using the FLUOstar Omega. Freshly isolated PMN were diluted in HBSS containing 1% HSA and 0.1% dextrose. A total of 4.5 × 105 PMN (LUC-CL) or 1.0 × 106 PMN (Amplex UltraRed) were added per well in the presence of lucigenin (100 µM) or Amplex UltraRed (50 µM) and HRP in excess as determined experimentally. For priming experiments, fMLF (1 µM) was injected at specified time points following treatment with TNF-α (1 ng/ml) or no agonist. NOX2 activity was expressed as relative light units (LUC-CL) or relative fluorescence units (Amplex UltraRed) using a kinetic assay with readings every minute for 60 min unless otherwise stated. In some Amplex UltraRed experiments, the µM H2O2 generated was determined by extrapolation from a standard curve.
Statistical analysis
Results are expressed as means ± standard error of the mean. Statistical comparisons were performed by analysis of variance (ANOVA) with Tukey’s correction for multiple comparisons or Student’s t-test. A probability of p ≤ 0.05 was considered significant. n represents the number of experiments.
Results
TNF-α elicits cell surface activation and mobilization of specific and tertiary, but not azurophilic granules
We have previously demonstrated concentration-dependent increases in surface CD11b and gp91phox levels following exposure to TNF-α.
18
However, these two proteins are expressed both in secretory vesicles and specific or secondary granules, therefore, we expanded our analyses to include more specific markers of cell activation and exocytosis. Using an Ab against an activation epitope of CD11b, we studied active CD11b levels and demonstrated very marked concentration-dependent increases in active CD11b in response to 30-min exposure to TNF-α (Figure 1(a)). Longer exposure (60 min) preserved the concentration-dependent findings but did not lead to enhanced cell activation (data not shown), supporting our selection of the 30-min priming time point for the majority of our studies. To specifically explore granule exocytosis in response to TNF-α priming, we measured PMN surface expression of CD66b and CD63, markers of specific granules and azurophilic granules, respectively. CD66b expression increased in a concentration-dependent manner when cells were treated with TNF-α for 30 min (Figure 1(b)), and there was no further increase at 60 min (data not shown). There was no effect of TNF-α on CD63 membrane expression indicating no azurophilic granule exocytosis (Figure 1(c)).
TNF-α elicits activation of cell surface proteins and mobilization of specific and gelatinase granules in a concentration-dependent fashion. Activation of the β2-integrin CD11b and specific and gelatinase (tertiary) granule exocytosis occurs in response to TNF-α as measured by concentration-dependent increases in cell surface levels of active CD11b (a) and CD66b (b). Neither azurophilic granules nor secondary granules are mobilized by TNF-α alone, as evidenced by no change in CD63 (c) surface expression or lactoferrin release (d). Tertiary granule exocytosis, as measured by gelatinase (e) release, is elicited by TNF-α alone. TNF-α alone did not elicit secretion of elastase, as measured by a fluorometric activity assay (f), n = 5–9, *P < 0.05, ***P < 0.001 as compared to no agonist.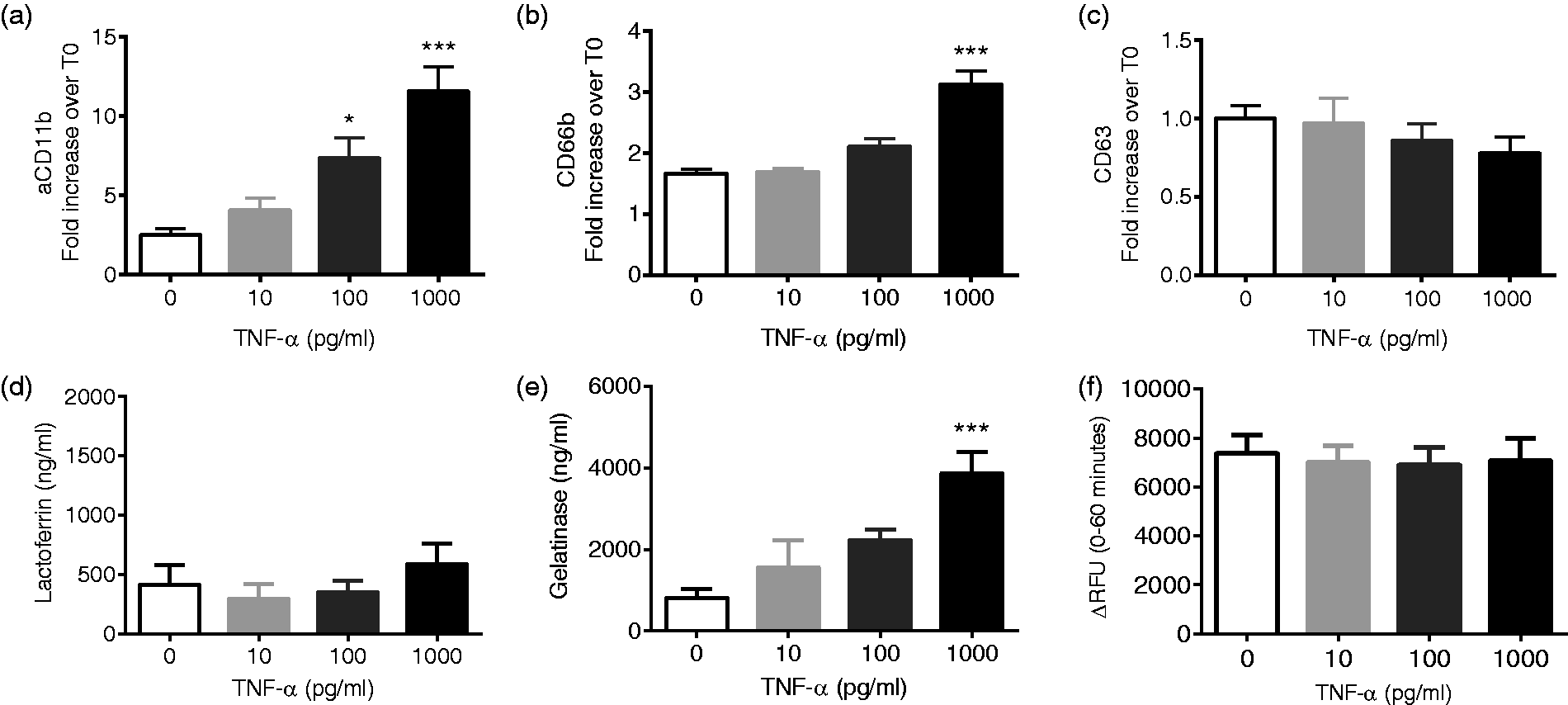
To complement these flow cytometry-based assays, we measured the release of soluble granule contents to the extracellular compartment. Exposure to TNF-α alone did not elicit lactoferrin release from the cell (Figure 1(d)), whereas tertiary granule exocytosis, as determined by gelatinase release, was more readily stimulated by TNF-α alone (Figure 1(e)). Consistent with our observations using flow cytometry, exposure to TNF-α alone at concentrations from 10 pg/ml to 1 ng/ml did not elicit elastase release above levels noted under control conditions (Figure 1(f)), suggesting that priming concentrations of TNF-α do not elicit azurophilic or primary granule exocytosis.
p38 MAPK and ERK1/2 differentially involved in TNF-α priming
We and others have demonstrated a requirement for p38 MAPK activity in TNF-α priming of the PMN respiratory burst,22–24 but specific mechanisms of p38 MAPK involvement have not been fully defined. Based on our previous data demonstrating that TNF-α priming leads to rapid phosphorylation of ERK1/2 and p38 MAPK, but not c-Jun N-terminal kinase (JNK),
18
we sought to further characterize the role of ERK1/2 and p38 MAPK in CD11b activation and granule exocytosis. Inhibition of p38 MAPK markedly diminished TNF-α-mediated activation of CD11b, as well as the mobilization of CD66b to the cell surface (Figure 2(a) and (b)) with no alteration in CD63 surface levels, as expected, since there is no change in these levels in response to TNF-α (Figure 2(c)). Although inhibition of ERK1/2 diminished TNF-α activation of CD11b (Figure 2(d)), there was no change in the surface levels of CD66b or CD63 (Figure 2(e) and (f)). These results support previous findings24,25 suggesting that p38 MAPK, but not ERK1/2, is specifically required for secondary granule exocytosis.
p38 and ERK 1/2 MAPK involved in cell activation and exocytosis elicited by TNF-α priming. p38 MAPK inhibition (added concurrently with TNF-α) significantly diminished CD11b activation (a) and CD66b upregulation (b), whereas there was no impact on CD63 cell surface levels (c) in cells treated with TNF-α for 30 min. ERK 1/2 inhibition similarly diminished CD11b activation (d), with no effect on CD66b or CD63 cell surface levels ((e) and (f)). n = 6, *P < 0.05.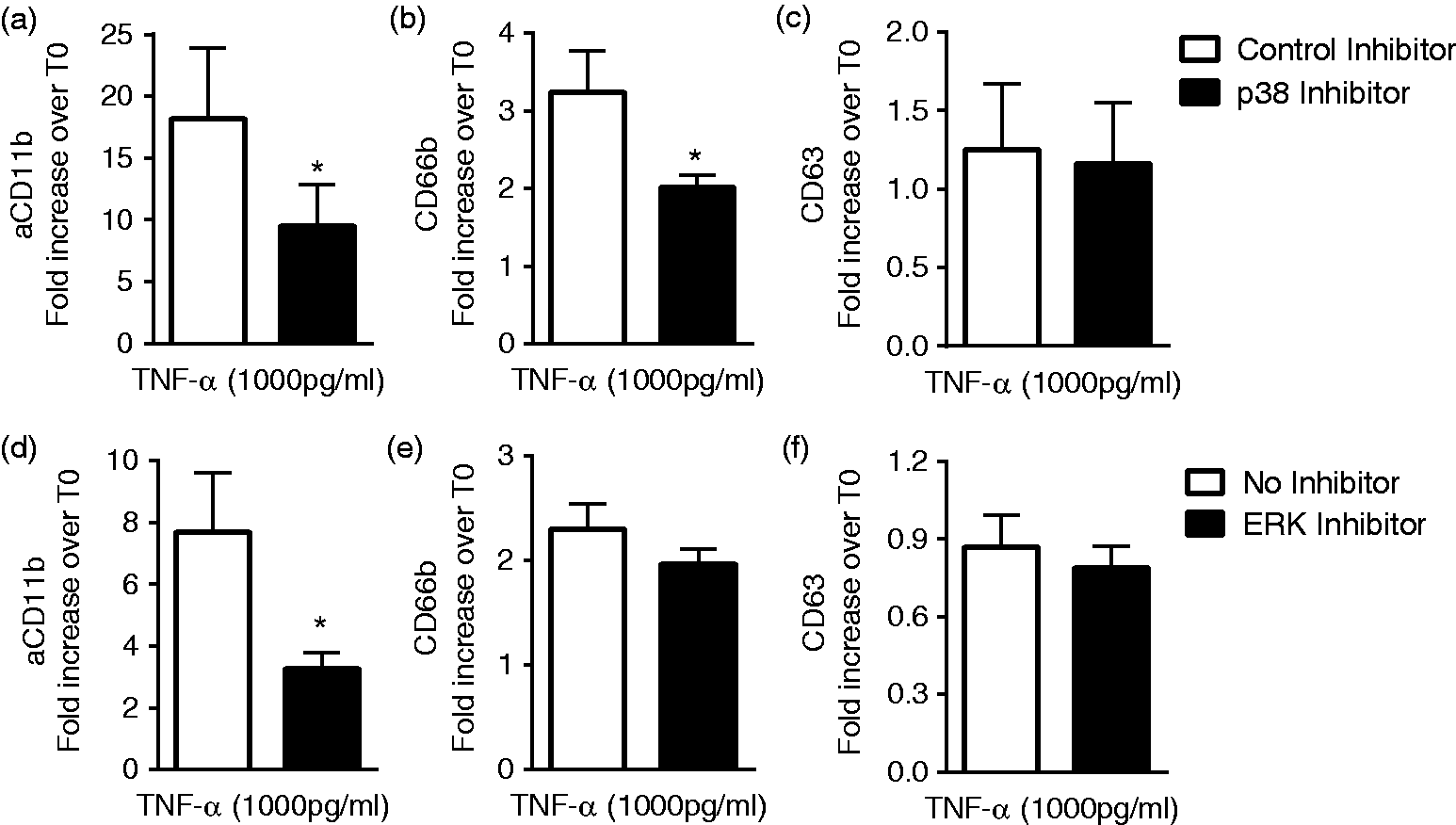
TNF-α primes granule release in response to a subsequent stimulus
Based on the current understanding of neutrophil priming as a state of pre-activation, it was not surprising that exposure to TNF-α alone had no significant effect on CD63 cell surface levels. Given the critical role that neutrophils are proposed to play in the pathogenesis of a number of chronic inflammatory diseases13,26 we postulated that exposure to low-level TNF-α might elicit priming of azurophilic granule release in response to a subsequent stimulus. To explore this hypothesis and also study the impact of priming on secondary and tertiary granule release in response to secondary stimuli, we used ELISA to measure extracellular lactoferrin and gelatinase, and a fluorometric activity assay to study the release of active elastase by TNF-α primed PMN. Although fMLF did not elicit release of lactoferrin in unstimulated cells, TNF-α priming led to markedly enhanced release of lactoferrin in response to subsequent stimulation with fMLF (Figure 3(a)). In addition, TNF-α alone elicits gelatinase release from PMN primarily. There was no synergistic increase in gelatinase release with TNF-α priming followed by fMLF (Figure 3(b)). As expected, TNF-α alone did not lead to significant elastase release in the 90-min priming assay when compared to no agonist conditions. Although there was a trend toward increased release of elastase with fMLF alone, this was not statistically significant (Figure 3(c)). Pre-incubation with TNF-α primed PMN for significantly increased elastase release in response to subsequent fMLF stimulation as compared to elastase secretion in response to fMLF alone (Figure 3(c)). These data demonstrate a novel component of the TNF-α priming phenotype with significant implications for host tissue damage.
TNF-α primes specific granule exocytosis and elastase release elicited by fMLF. Incubation with TNF-α markedly primes PMN for enhanced lactoferrin release in response to subsequent stimulation with fMLF (a). TNF-α alone elicits significant gelatinase release above baseline, and there is no synergistic effect of TNF-α plus fMLF (b). fMLF alone displayed a trend toward enhanced mobilization of elastase, but 30 min of priming with TNF-α markedly enhances elastase release (c), n = 9–12, *P < 0.05, **P < 0.01, ***P < 0.001.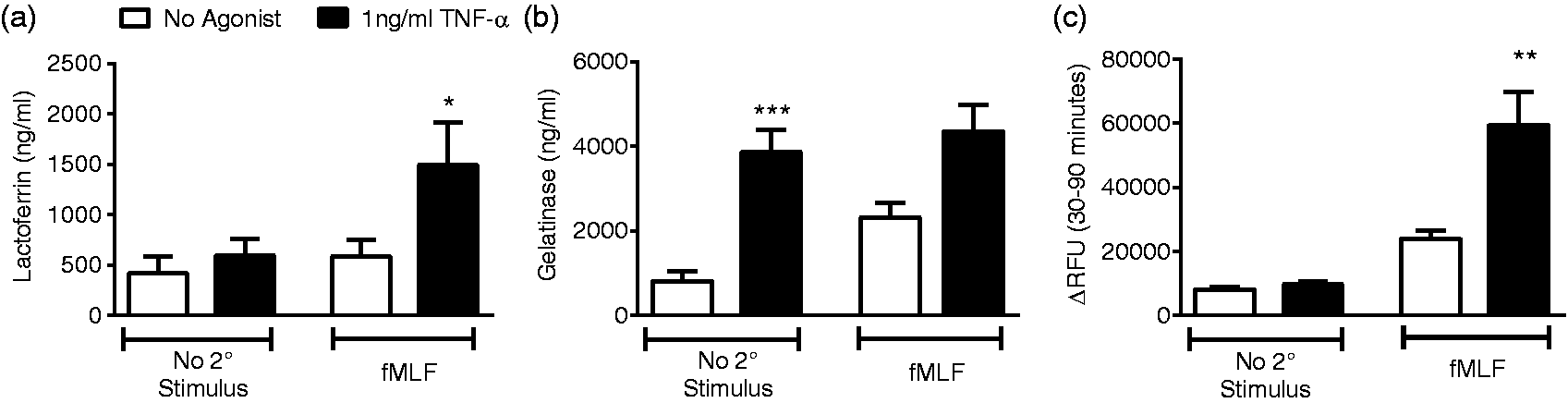
TNF-α elicits endosomal ROS production
We next sought to better characterize the signaling upstream of TNF-α priming of elastase release. We have previously demonstrated a requirement for NADPH oxidase-derived ROS in the generation of the primed state,18,27 and our previous studies of TNF-α priming suggested a significant oxygen-dependent component of priming of the respiratory burst.
18
In addition, we have recently demonstrated that endotoxin elicits low-level NADPH oxidase-generated intracellular endosomal ROS in human PMN.
28
We theorized that signaling ROS, generated by the NADPH oxidase, might have a role in elastase exocytosis. We sought to determine whether PMN stimulation by low-level TNF-α generated endosomal NADPH oxidase activity as we had previously demonstrated in endotoxin-stimulated PMN. Using flow cytometry-based detection to specifically measure intracellular ROS generation, we demonstrated a time- and concentration-dependent acquisition of intracellular fluorescence. TNF-α at a concentration of 1 ng/ml elicited a significant increase in measured intracellular ROS levels with 30- and 60-min incubations (Figure 4(a)). In response to TNF-α (1 ng/ml), a significantly increased percentage of PMN within the whole cell population developed OxyBurst positivity following 30- and 60-min incubations (Figure 4(b)). The probe used for these studies is conjugated to BSA and thus is not freely diffusible, providing a measure of endosomal ROS generation. Considered in combination, these data suggest intracellular endosomal ROS generation occurs in response to TNF-α priming.
TNF-α priming elicits direct intracellular endosomal ROS production. Intracellular ROS were measured using the OxyBurst Green-BSA probe. TNF-α (1 ng/ml) elicited time-dependent increases in intracellular fluorescence (OxyBurst GMI) (a). Incubation with 1 ng/ml TNF-α elicited a significant increase in the percentage of OxyBurst-positive cells (b), n = 6, *P < 0.05, **P < 0.01, ***P < 0.001 compared to no agonist.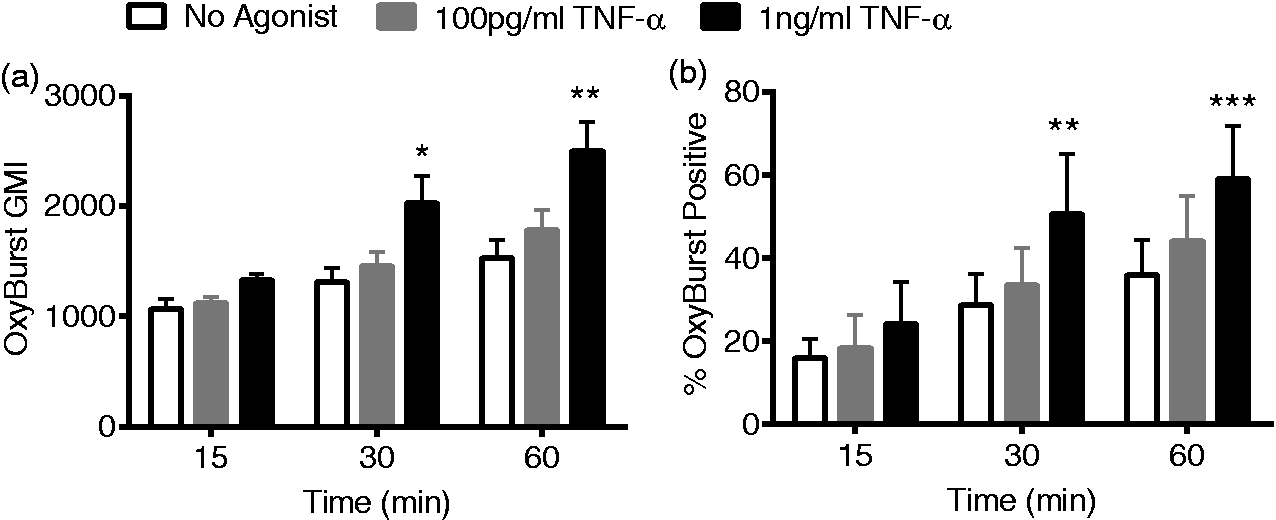
p38 MAPK activity is required for direct ROS generation in response to TNF-α
Next, we sought to determine the relationship between these endosomal ROS, p38 MAPK and TNF-α priming. We have previously reported on two distinct phases of ROS generation during PMN priming: the initial low-level direct ROS generated in response to the priming stimulus, followed by the primed ROS generated in response to fMLF or other secondary stimulus.18,27 More specifically, it has been demonstrated that p38 MAPK phosphorylates a specific residue on p47phox, and that this event is necessary for priming of the respiratory burst in response to TNF-α.29,30 However, this investigation did not distinguish between these two phases of NADPH oxidase activity. We postulated that p38 activity was necessary for the direct signaling of ROS generation following the priming stimulus alone, rather than for the primed ROS generation following the secondary stimulus, and investigated this hypothesis using two separate assays for ROS generation. Using timed addition of the p38 MAPK inhibitor, there was time-dependent inhibition of direct ROS generation elicited by TNF-α (Figure 5(a)–(c)). Addition of the p38 MAPK inhibitor at 15 min immediately abrogated the direct ROS generation with a 67% reduction in the peak ROS production (Figure 5(b)), as measured by LUC-CL. The control inhibitor did not have any effect on direct ROS generation as compared to no inhibitor in these analyses. Primed PMN NADPH oxidase activity in response to the secondary stimulus fMLF was assayed using both LUC-CL (Figure 5(d)–(f)) and Amplex UltraRed (Figure 5(g)–(i)). The extent of inhibition of the primed NADPH oxidase activity reflected the degree of inhibition of direct ROS activity, with much greater inhibition following addition of the inhibitor at 0 min (Figure 5(d) and (g)), some impairment with addition of the inhibitor at 15 min (Figure 5(e) and (h)), and no effect of the inhibitor added after direct ROS generation was complete (Figure 5(f) and (i)). The control inhibitor did elicit some reduction in ROS generated as compared to no inhibitor control using Amplex UltraRed, thus comparisons were made of control inhibitor versus p38 inhibitor. H2O2 generation was quantitated using Amplex UltraRed assay. Addition of the p38 inhibitor at 15 min elicited a 26.4% reduction in the primed burst after fMLF. As the p38 inhibitor is permeable and rapidly acting, these data suggest that the previously reported
30
p38 phosphorylation of p47phox occurs in the initial phase of direct ROS activation, but is dispensable for the primed respiratory burst. In addition, we studied the impact of ERK1/2 inhibition on these two phases of ROS production during TNF-α priming. We demonstrated that ERK1/2 inhibition led to a small but significant decrease both in ROS generated by the priming stimulus alone, and in the primed respiratory burst after secondary stimulation with fMLF. In contrast to our data using p38 inhibition, the magnitude of reduction in the direct ROS had no impact on the primed burst (Supplemental Figure 1).
p38 MAPK is required for direct ROS production in response to TNF-α. ((a)–(c)) Direct ROS generation elicited by TNF-α alone is blocked in a time-dependent fashion by addition of the p38 MAPK inhibitor, SB203580. When added concurrently with TNF-α, all direct ROS is blocked, n = 7 (a) with partial inhibition when added at 15 min, n = 4 (b). (c) Inhibitor added at 30 min after peak ROS production is reached has no effect, n = 6. ((d)–(f)) Inhibition of the primed respiratory burst in response to subsequent stimulation with fMLF is correlated with the degree of direct ROS inhibition seen in (a)–(c). Significant reduction in the peak ROS generation occurs with time 0, n = 5 (d) or time 15 min, n = 4 (e), whereas inhibitor added just prior to fMLF had no effect (f), n = 5. ((a)–(f), lucigenin-enhanced chemiluminescence). ((g)–(i)) Using Amplex UltraRed as an additional complimentary assay, addition of the inhibitor at 0 or 15 min similarly elicited a significant reduction in H2O2 generated in response to fMLF, n = 7, *P < 0.05, **P < 0.01, ***P < 0.001.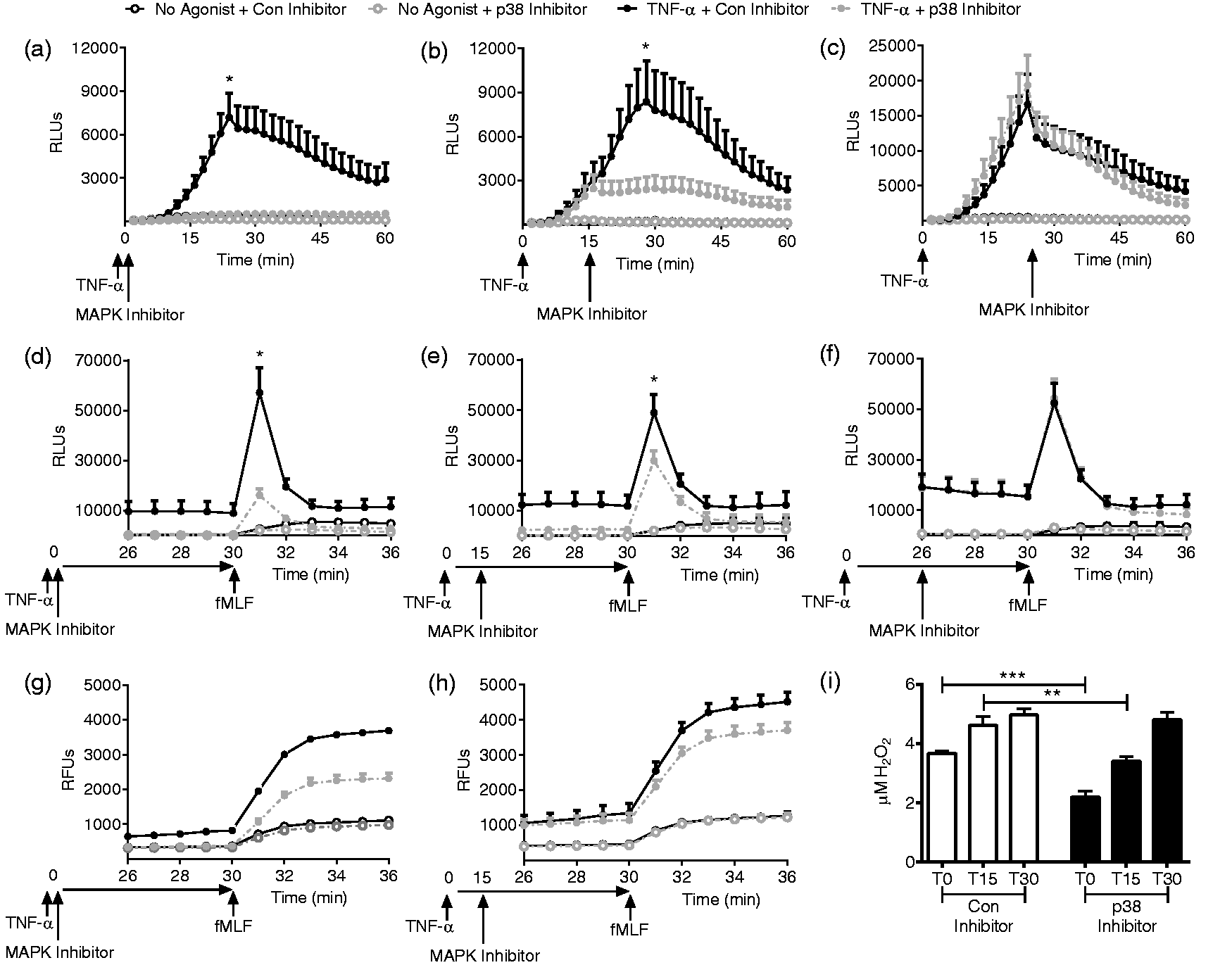
p38 MAPK not required for priming of fMLF-stimulated elastase release
Based on our data demonstrating that TNF-α priming of the respiratory burst is p38 MAPK and ROS dependent, we next investigated the involvement of these mechanisms in priming of azurophilic granule exocytosis. Preliminary experiments demonstrated no effect of the control p38 MAPK inhibitor on elastase release as compared to no inhibitor (data not shown). Under TNF-α priming conditions, p38 MAPK inhibition did not alter baseline (no agonist) or TNF-α elicited elastase release (data not shown). p38 MAPK inhibition had no effect on TNF-α priming of fMLF-mediated release (Figure 6(a)). Similarly, ERK1/2 inhibition had no effect on TNF-α priming of fMLF-mediated release (Figure 6(b)). This suggests that neither p38 MAPK nor ERK1/2 are required for the priming of azurophilic granule exocytosis.
Neither p38 nor ERK 1/2 MAPKs are required for TNF-α priming of elastase release. Inhibition of p38 MAPK (a) or ERK 1/2 (b) had no effect on fMLF-mediated elastase release following priming with TNF-α. n = 6.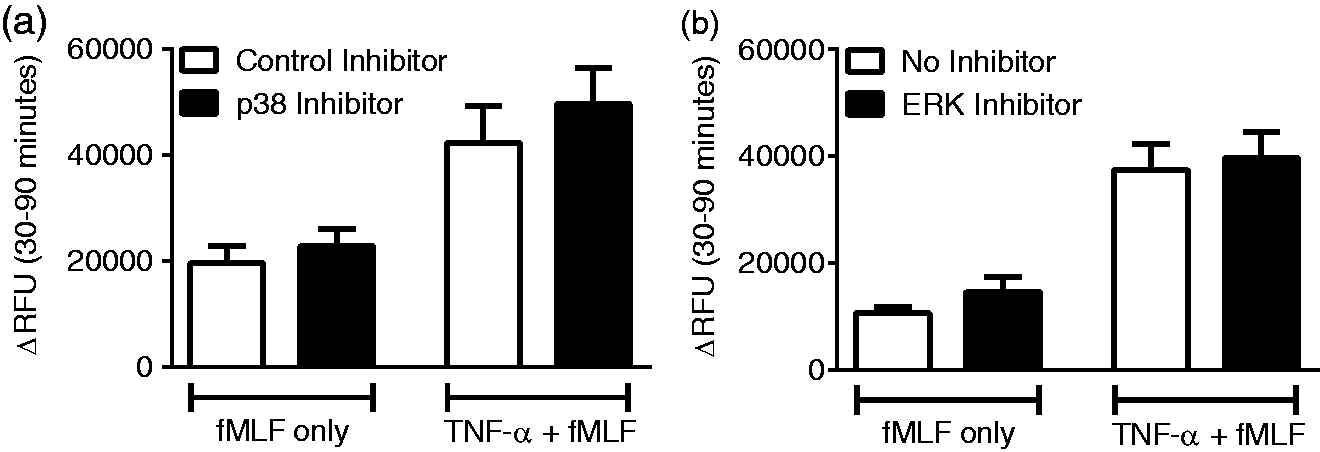
NADPH oxidase-dependent signaling involved in priming of elastase release
Although p38 MAPK activation allows the TNF-α mediated generation of endosomal ROS, it appears that p38 MAPK has no effect on the priming of elastase release, thus we predicted that NOX2-derived ROS would not be involved in the priming of elastase release. Unexpectedly, we noted a trend toward increased basal elastase release in unstimulated PMN lacking NOX2 function due to pharmacologic inhibition with DPI or due to genetic deficiency of NADPH oxidase function (PMN from patients with CGD (CGD-PMN)). CGD-PMN also displayed significantly enhanced release of elastase in response to TNF-α alone (Figure 7(a)). fMLF-stimulated release as a sole agonist was not altered in the absence of NOX2 function. TNF-α priming of fMLF-mediated release was significantly augmented by NOX2 inhibition or absence of function (Figure 7(b)). As expected, there were no differences in total measureable elastase activity in the absence of NOX2 activity under maximal release conditions (Figure 7(c)). Taken together, these data suggest that ROS signaling causes repression of elastase release under resting conditions, but this appears to occur in a p38 MAPK-independent manner.
NADPH oxidase-dependent signaling limits TNF-α priming of elastase release. CGD-PMN demonstrate enhanced elastase release in response to TNF-α alone (a). Priming of fMLF-mediated elastase release is enhanced in the absence of NADPH oxidase activity (either using DPI for inhibition, or CGD-PMN) (b). There were no differences in total measureable elastase release elicited in response to DHCB and fMLF in the setting of NADPH oxidase inhibition or genetic deficiency (c), n = 8–13 (n = 4 CGD), *P < 0.05, **P < 0.01 compared to control PMN.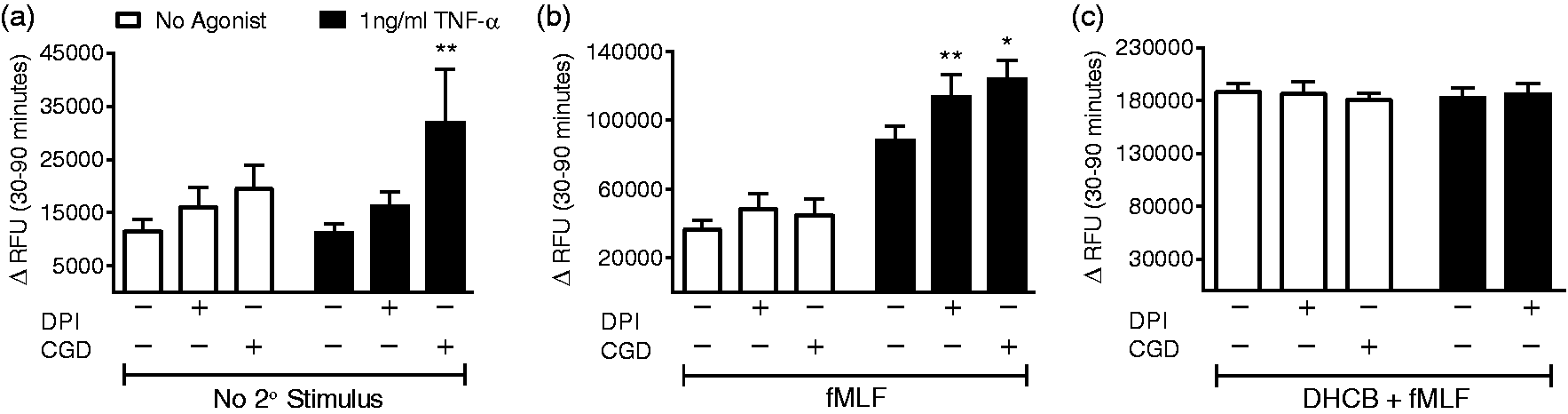
Discussion
As the most abundant circulating white blood cell and a critical component of microbial host defense, neutrophils are clearly recognized for their role in innate immunity. Over the last two decades, however, there has been an increasing understanding that PMN also mediate inflammatory tissue injury in a broad variety of disease states. Exuberant neutrophil activation with granule release, formation of neutrophil extracellular traps (NETs), and impaired neutrophil clearance are all contributing mechanisms in non-resolving inflammation.31–33 Neutrophilic inflammation is recognized as a key feature of ALI, chronic obstructive pulmonary disease, inflammatory bowel disease, and rheumatoid arthritis with elastase and other neutrophil granule products demonstrated in tissue specimens.14,34–36 As our understanding of PMN involvement in these conditions expands, it is critical to more specifically define the continuum of PMN activation from priming or pre-activation to “full” activation to apoptosis and cell death. The host-derived inflammatory cytokine TNF-α plays a pivotal role in the pathogenesis of many of these disease states, demonstrating the relevance of a clear understanding of TNF-α priming of PMN. Although many laboratory studies have justified higher concentrations of TNF-α (5–100 ng/ml) to model acute sepsis, these concentrations greatly exceed what has been documented in most inflammatory states. 37 Our laboratory has primarily focused our analyses on lower concentrations of TNF-α pertinent to ALI and chronic disease states including inflammatory bowel disease and rheumatoid arthritis.15,16,38
We have previously demonstrated that priming of the respiratory burst of PMN by TNF-α is achieved by exposure to low levels of the cytokine and occurs in an oxygen-dependent fashion. 18 In the current study we significantly extend those conclusions and expand our understanding of what phenotypic changes are encompassed by PMN priming. Our results show that low-level TNF-α priming markedly enhances the capability of the PMN to release both lactoferrin and elastase in response to subsequent stimulation. We have focused significantly on azurophilic granules as the priming of azurophilic granule exocytosis could be responsible for significant collateral damage by neutrophils that exit into inflamed tissue spaces. Although earlier studies demonstrated TNF-α priming of azurophilic granules using IL-820 and microcrystals 39 as secondary stimuli, these studies employed either supra-physiologic doses of TNF-α or PMN pre-treated with cytochalasin B, an actin destabilizer that leads to enhanced azurophilic granule exocytosis.
Subsequently, we sought to determine the mechanism underlying priming of azurophilic granule exocytosis. Our results in the current study, considered in combination with our previously published work, 18 suggest a complicated interaction between NADPH oxidase-derived signaling ROS and priming. The most extensively studied phenotype of neutrophil priming is the enhancement of the respiratory burst in response to a subsequent stimulus. Under the current paradigm, a priming stimulus induces an intracellular signaling pathway that leads to phosphorylation of Ser345 on p47phox, one of the required cytosolic subunits of the NAPDH oxidase multi-component enzyme complex. This phosphorylation event is mediated by p38 MAPK in response to TNF-α priming, whereas it is mediated by ERK1/2 in response to granulocyte-macrophage colony-stimulating factor (GM-CSF), and is required for the primed respiratory burst in response to a subsequent stimulus. 30 The overlap in signaling in response to two distinct cytokines has been proposed as a potential target for control of inflammation under sterile conditions. In the current study, we extend previous understanding of this process by dividing NADPH oxidase activation into two distinct stages. We have previously shown18,28 that priming stimuli directly elicit low level ROS production during the priming period, and here we demonstrate that these ROS are generated into intracellular endosomes. We have provided evidence that these initial ROS are required for subsequent enhancement of NADPH oxidase activation in response to the second stimulus. Here, we demonstrate that p38 MAPK activity is specifically required during the initial generation of low-level signaling ROS, but is dispensable during the secondary activation. Moreover, the priming effect of these “direct” ROS are not binary, but rather incremental, as partial abrogation by timed addition of p38 MAPK inhibitors partially reduces the subsequent primed burst. We propose that p38 phosphorylation of p47phox occurs on a subset of fully assembled NADPH oxidase complexes thus leading to endosomal ROS production, and the subsequent mobilization and assembly of additional NADPH oxidase complexes at the plasma membrane. 40 Considered in combination, these data suggest that activation of the NADPH oxidase for the generation of signaling ROS occurs independently of the signaling that activates the enzyme complex at the plasma membrane. This is a novel concept as it relates to endosomal signaling ROS; however, others have demonstrated that control of the NADPH complex at the plasma membrane and phagosomal membrane can occur independently of one another. 41
Mobilization of intracellular protein stores to the cell surface has also been investigated as a measure of cell priming, often focused on gp91phox or CD11b. 42 In resting neutrophils, these two proteins are expressed in three distinct locations: the plasma membrane, secretory vesicles, and secondary or specific granules, thus enhanced cell surface levels following priming does not definitively identify a mobilized granule population. We have previously demonstrated that low-level TNF-α stimulation augments cell surface CD11b levels. 18 In the current study, we found that priming enhanced activation of cell surface CD11b, as measured by exposure of an activation-specific epitope. Moreover, we demonstrate that priming elicits some degree of secondary granule exocytosis quantitated by enhanced cell surface CD66b levels, whereas TNF-α alone does not mobilize azurophilic granules based on unaltered surface CD63 levels. However, in the setting of ongoing exocytosis and endocytosis, cell surface protein levels are unlikely to provide the most reliable information about release of soluble granule contents into the surrounding tissues.
Based on our interest in the potential for neutrophil priming to cause host tissue damage, we focused our investigation on priming of azurophilic granule release, as this subtype contains the most toxic and cytolytic contents. For our studies we measured release of active elastase in response to TNF-α followed by a secondary stimulus. Priming of neutrophils for 30 min with TNF-α (1 ng/ml) markedly enhanced elastase release in response to fMLF. The regulation of granule exocytosis is quite complex and involves a series of tightly controlled steps that are unique for each subtype of granule. Initial trafficking includes cytoskeletal reorganization with actin depolymerization at the cell cortex allowing movement of the granule toward the periphery, followed by spatially directed F-actin polymerization. 43 This actin remodeling process brings the granule/vesicle in close proximity to the plasma membrane, allowing tethering and docking at the appropriate sites, followed by membrane fusion. The interactions between granules and the plasma membrane are orchestrated by combinations of SNARE proteins that are unique to each type of granule. 44
Significant progress has been made in identifying relevant proteins involved in azurophilic granule exocytosis including the small GTPase Rab27 and its effector SNARE proteins. 45 Although most of the initial work on granule exocytosis was not performed in the setting of priming, it was recently demonstrated that endotoxin exposure did prime azurophilic granule release in response to a second stimulus. The mechanism for this “priming effect” involved restriction of the motility of a subset of azurophilic granules to an area near the plasma membrane, thus enhancing their potential for membrane docking and fusion. 19 The current study provides evidence for a novel inhibitory or repressive signaling role for NADPH oxidase-derived ROS in this process. Although ROS production and accompanying oxidant stress have generally been perceived as pro-inflammatory, and our previous work has demonstrated a pro-inflammatory role for ROS signaling during TNF-α priming, the concept of a balancing anti-inflammatory signaling role is not novel. We, and others, have demonstrated hyperinflammation under conditions where the NADPH oxidase is non-functional, both in murine studies46,47 and human analyses.48,49
In summary, we have provided conclusive evidence that azurophilic granule exocytosis does not occur in response to TNF-α alone, although exposure to this cytokine does mobilize a portion of secondary and tertiary granules with release of contents to the extracellular space. In addition, incubation with TNF-α does prime specific granules for enhanced exocytosis following a subsequent stimulus. Importantly, azurophilic granules are also altered by the priming exposure such that subsequent stimulation with fMLF leads to markedly enhanced release of elastase. The NADPH oxidase provides inhibitory signaling that limits the release of azurophilic granule release under priming conditions. This is in contrast to priming of the respiratory burst, where oxidant signaling is required. Generation of oxidants in response to TNF-α is dependent on p38 MAPK activity, but this MAPK signaling pathway does not appear to be involved in priming of azurophilic granule release. Spatial and temporal segregation of NADPH oxidase-derived oxidants appears to explain some of these divergent roles.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (NIH) (grant number 5R21AI109127) to JGM and NIH (grant number 5K12 HD-068369-05) to RMP.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.