
Abstract
A major aspect of pathology in otitis media (OM), the most common childhood bacterial disease, is hyperplasia of the middle ear mucosa. Activation of innate immune receptors during OM leads to the activation of NF-κB, a pleiotropic transcription factor involved both in inflammation and tissue growth. To explore the role of NF-κB in mucosal hyperplasia during OM, we evaluated the expression of genes involved in two modes of NF-κB activation during a complete episode of acute, bacterial OM in mice. We also determined the effects of inhibitors of each pathway on infection-stimulated mucosal growth in vitro. A majority of the genes that mediate both the canonical and the non-canonical pathways of NF-κB activation were regulated during OM, many with kinetics related to the time course of mucosal hyperplasia. Inhibition of either pathway reduced the growth of cultured mucosal explants in a dose-dependent manner. However, inhibition of the canonical pathway produced a greater effect, suggesting that this mode of NF-κB activation dominates mucosal hyperplasia during OM.
Introduction
Otitis media (OM) is the most common bacterial disease of childhood. It leads to more antibiotic prescriptions and surgery in children than any other condition.1,2 It also causes an estimated 28,000 deaths per year worldwide, primarily in the developing world, and half of the world’s burden of serious hearing loss. 3
While OM is a multifactorial disease, with contributions from Eustachian tube dysfunction and allergy, a primary cause is infection by Streptococcus pneumoniae, non-typeable Haemophilus influenzae (NTHi) or Morexella catharralis. Bacterial infection of the middle ear (ME) often occurs following a cold or upper respiratory viral infection. In most children, OM is acute, resolving within one week, a period too short for cognate immunity to be responsible for the clearance of infection, implicating innate immunity.
A hallmark of acute and chronic OM is proliferation of the mucosa lining the ME. Unlike other mucosae, that of the ME radically transforms in response to infection, from a monolayer of squamous epithelium to a full-thickness respiratory epithelium within a few days, as shown over time in animal models 4 and in mature disease in humans.5–8 This hyperplasia contributes significantly to pathogenesis, through the production of mucus and other bioactive secretions as well as by ME cavity volume reduction. The mechanisms that mediate this response are far from clear. However, studies with ME epithelial cell lines have implicated NF-κB in the response of epithelial cells to bacteria.9,10 This is supported by the fact that NF-κB is activated downstream from many innate immune receptors. 11
NF-κB, a dimeric transcription factor, is a pleiotropic regulator of many immune and inflammatory processes in response both to injury and infection.12,13 It can also promote cell proliferation and survival, mediated via cell cycle regulation 14 and stimulation by growth factors. 15 Activation of NF-κB proteins is tightly regulated, and inappropriate activation of the NF-κB signaling pathways has been linked to autoimmunity, chronic inflammation, and various cancers. 16
NF-κB signaling consists of a canonical or classical pathway and a non-canonical or alternative pathway of activation (Figure 1). The canonical pathway
17
can be initiated by activated growth factor receptors (GFRs) via class I phosphatidylinositide 3-kinases (PI3Ks) and AKTs, TNF-α via its cognate receptor (TNFR1), IL-1 via the IL1 receptor (IL1R) or innate immune receptors including the TLRs or NLRs.16–18 Canonical NF-κB activation relies on the phosphorylation of IκBα, mediated by a heterotrimer of IκB kinases (IKKs), IKKα, IKKβ and IKKγ. This is followed by IκB ubiquitination and cleavage of the p105 subunit (NF-κB1) of a RelA/p105 dimer, releasing RelA/p50 to enter the nucleus and activate expression of many genes related to inflammation and/or tissue growth. The latter process can be inhibited by IKKɛ or BCL3.
Diagrammatic representation of NF-κB signaling via the canonical (classical) and non-canonical (alternative) activation pathways.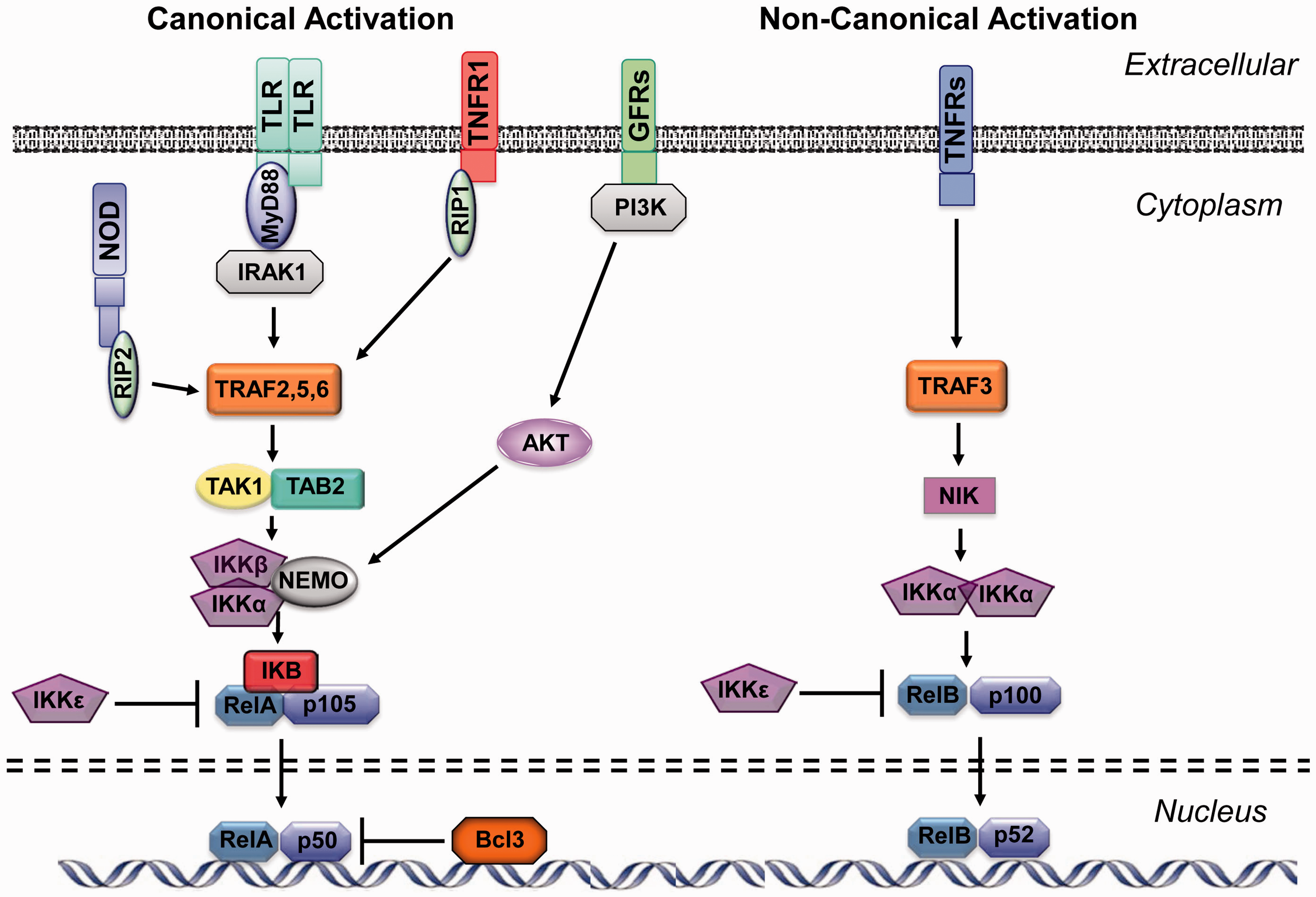
In the non-canonical pathway, 19 other members of the TNF receptor family, stimulated by various ligands, phosphorylate NF-κB-inducing protein (NIK). pNIK in turn activates an IKKα homodimer, independent of IKKβ and IKKγ, leading to phosphorylation and ubiquitination of the p100 (NF-κB2) member of p100/RelB dimers, followed by degradation of the inhibitory p100 C-terminus. This releases an active p52/RelB complex to enter the nucleus and induce the expression of genes related primarily, but not exclusively, to cognate immunity. This process can similarly be inhibited by IKKɛ.
The two pathways differ not only in the composition of their activation complexes but also in their downstream targets. Canonical activation preferentially regulates inflammatory, proliferation, cell survival, and innate immune genes. 17 The non-canonical pathway primarily regulates genes related to lymphoid organogenesis, B-cell maturation, and other aspects of adaptive immunity 16 that would be expected in chronic, but not acute, OM.
Failure of OM recovery has been associated with defects in innate immune receptors and adaptors, 20–23 differences in TNF-α expression, 24 and IL-1 processing by the inflammasome,25,26 while adaptive immunity plays a significant role in the prevention of re-infection. 27 In addition, as discussed above, mucosal proliferation plays a significant role in this disease. These findings suggest that NF-κB may serve as a molecular mechanism both for pathogenesis and recovery in OM.
The present study assessed the role of NF-κB signaling pathway in acute OM pathogenesis, and more specifically in mucosal proliferation. To determine whether NF-κB signaling is involved in ME mucosal hyperplasia, the expression of NF-κB signaling genes over the full course of an episode of acute, bacterial OM was assessed using gene arrays, 28 and compared temporarily to the growth of the mucosa. In addition, specific inhibitors of either the canonical or non-canonical NF-κB pathways were used in an in vitro model of infection-induced ME mucosal proliferation 29 to determine the relative contributions of each activation pathway. We hypothesized that many genes related to NF-κB signaling and both activation pathways would be regulated during OM, but that classical activation genes would be more closely related temporally to mucosal hyperplasia in acute OM. We further expected that inhibition of canonical NF-κB activation would reduce ME mucosal growth in culture. Since non-canonical NF-κB activation is primarily involved in adaptive immunity, we did not expect inhibition of this pathway to affect mucosal growth in culture, where cognate immunity is unlikely to be a significant factor.
Materials and methods
Animals
For gene arrays, 320 C57Bl/6/WB F1 hybrid mice 60–90 d of age were used. F1 hybrids were employed to avoid the recessive traits that are known to accumulate in inbred mouse strains. 30 For tissue culture, 24 Sprague-Dawley rats weighing 300 ∼ 350 g were used. Rats respond very similarly to mice following NTHi infection, 29 and provide substantially more tissue for in vitro studies, minimizing animal use. All procedures were conducted according to National Institutes of Health (NIH) standards for the care and use of animal subjects, and were approved by the Institutional Animal Care and Use Committee (IACUC) of the San Diego VA Medical Center.
OM induction
OM was induced by inoculation of strain 3655 NTHi into the ME cavities of mice or rats. This strain was originally isolated in 1979 from the ME of a child with OM in St. Louis, MO, USA. It has been used in many studies of experimental OM.20,29,31,32 OM was induced by injection through the tympanic bulla after a ventral surgical approach.22,29
OM transcriptome
ME mucosae were harvested from 40 deeply anesthetized mice at 0, 3, and 6 h, as well as 1, 2, 3, 5, and 7 d after inoculation of NTHi in saline. The tissue from 20 different mice was combined, to generate two independent samples. Separate groups of mice were injected with saline vehicle without NTHi, and otherwise identically treated. The tissue was homogenized in TRIzol™ (Invitrogen, Carlsbad, CA) and total RNA was extracted. Total RNA quality was assessed using the RNA 6000 Labchip Kit on the Agilent 2100 Bioanalyzer for the integrity of 18S and 28S ribosomal RNA. The mRNA was reverse transcribed using a T7-oligodT primer then in vitro transcribed using T7 RNA polymerase to generate biotinylated cRNA probes that were hybridized to Affymetrix MU430 2.0 microarrays. Duplicate arrays were hybridized for each time point, using RNA from pools of different mice to obtain independent biological replicates. Raw intensity data were median-normalized and statistical differences in gene transcript expression levels were evaluated using variance-modeled posterior inference (VAMPIRE). 33 This program uses a Bayesian approach to identify altered genes. Statistical analysis by VAMPIRE requires two distinct steps: (1) modeling of the error structure of sample groups and (2) significance testing with a priori-defined significance thresholds. VAMPIRE models the existing error structure to distinguish signal from noise and identify the coefficients of expression-dependent and expression-independent variance. These models are then used to identify microarray features that are differentially expressed between treatment groups. This method allows the use of small numbers of replicates to evaluate gene expression across a continuum of conditions, down to two or even one array per condition if (as in the present study) many samples are pooled for each array (i.e. each array itself samples the mean value) and if multiple conditions are assessed. We used two arrays per time point and experimental condition (NTHi versus saline injection). We also compared mice inoculated with NTHi for each time point against un-injected (0 h) controls, to generate sets of genes that change significantly over time following NTHi injection. Bonferroni multiple testing correction (αBonf < 0.05) was applied to identify only those genes with the most robust changes. Genes for which expression was statistically regulated greater than 2 × for at least two time points were considered to be significantly regulated.
ME mucosa tissue culture
For tissue culture, at 2 d after inoculation of rat MEs with NTHi, the animals were sacrificed and the ME mucosae were extracted and divided into roughly 1 mm2 explants. The explants were individually placed into the wells of 24-well culture plates containing 190 µl of culture media/well. A mixture of DMEM and Ham’s F12 medium was supplemented with FCS (5%), hydrocortisone (0.4 µg/ml), isoproterenol (10–6M), penicillin (100 IU/ml), and streptomycin (100 µg/ml). On d 2 of culture, all wells with healthy, attached explants were randomly divided into groups of 8–10 explants each. The RelA nuclear translocation inhibitor caffeic acid phenethyl ester (CAPE) was reconstituted in DMSO and added to one of three groups at 10, 50, and 250 µM in 400 µl of culture medium. A fourth group served as a negative control (DMSO alone added to the medium). Every other day, medium from each well was removed and 400 µl of fresh culture medium was added with the appropriate concentration of CAPE. Using the same procedure, explants were cultured with the pNIK inhibitor 1,3(2H,4H)-isoquinolinedione at 50, 250, and 1000 µM. The explants were kept in culture for 10 d. For each individual concentration of inhibitors both of classical and alternative NF-κB pathway, explants were photographed daily and the area of explants outgrowth was calculated using SPOT software program and analyzed out to 10 d in culture. This allowed us to assess absolute and relative area of the explants as a measure of tissue growth, regardless of the initial size of each explant. Only explants that maintained a healthy appearance and remained firmly attached to the well surface throughout the entire duration of culture were subjected to data analysis. Tissue growth data were analyzed by analysis of variance (ANOVA). Bonferroni multiple testing correction (αBonf < 0.05) was applied to identify individual group differences.
Results
ME mucosal hyperplasia
The proliferative response of the murine ME mucosa following bacterial infection has been extensively documented.20,21,34,35 The uninfected mucosa is a monolayer of simple squamous epithelium with a minimal stroma, between 5 and 10 µM in thickness. Within 12 h of NTHi inoculation, thickness increases to 15–20 µM, due primarily to sub-epithelial edema. By 24 h, proliferation has initiated and mucosal thickness increases to 25–35 µM. By 48 h, edema has resolved and proliferation peaks, with mucosal thickness reaching a maximum of 60–70 µM. At this point, the mucosa has developed into a pseudo-columnar epithelium containing secretory and ciliated cells, underlain by a highly organized stroma. The mucosa then gradually recovers as infection is resolved, returning to its normal thickness by 5 to 7d (Figure 2(a)).
Changes in expression of genes related to NF-κB signaling during the course of an episode of acute OM in the mouse. (a) Regulation of NF-κB subunit genes. (b), (c) Upstream signaling genes that lead to the activation of NF-κB from a variety of receptors. (d) IKK genes. (e) IκB genes. Panel (a) includes a representation of average mucosal thickness observed in several prior studies of NTHi-induced acute OM in the mouse.20–22 Of the genes illustrated in Figure 2 and Supplementary Table 1, only TAK1, NEMO, and BCL3 were also significantly regulated after sham injection of saline into the ME.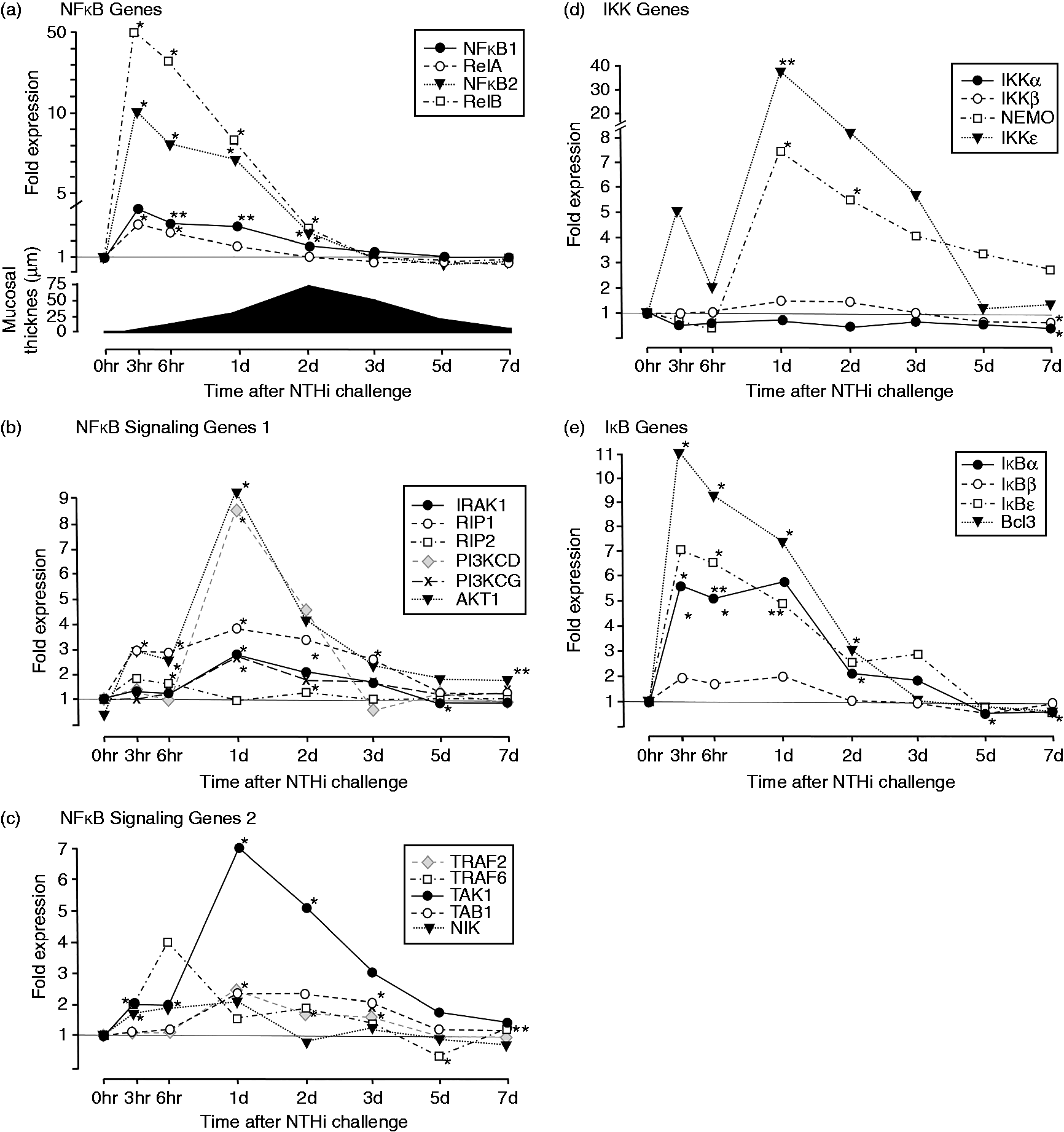
Expression of genes involved in canonical NF-κB activation
The expression of genes encoding NF-κB subunits (Figure 2(a) and Supplementary Table 1) exhibited up-regulation at 3h after infection, with a gradual decline thereafter, reaching pre-infection levels by around d 3 after ME inoculation. Genes involved in upstream signaling for the canonical NF-κB pathways were also regulated (Figure 2(b), (c) and Supplementary Table 1). Genes encoding RIP1, AKT1 and to a lesser extent RIP2 showed modest up-regulation in the first hours after infection, with RIP1 remaining elevated through d 3. Expression of IRAK1, the class 1 PI3K catalytic subunit PIK3CG, and especially PIK3CD, was enhanced at 24 h, accompanied by a dramatic further rise for AKT1. TRAF6 and TAK1 showed modest early increases, with TAK expression then peaking dramatically on d 1. TRAF2 and TAB2 increased expression modestly at 24 h. Expression of most signaling genes generally declined thereafter. The genes encoding PIK3CA, PIK3CB, the several class I PIK3R regulatory subunits, TRAF5, AKT2 and AKT3 were un-regulated during OM.
Of the IKK genes (Figure 2(d) and Supplementary Table 1), those encoding both NEMO (IKKγ) and IKKɛ showed dramatic up-regulation 1d after infection, which gradually declined thereafter. IKKα and IKKβ were largely unregulated, although each showed a slight but significant down-regulation at d 7.
Expression of the IκB genes (Figure 2(e), Supplementary Table 1) and BCL3 mirrored those of the NF-κB subunits themselves, showing strong up-regulation 3h after ME inoculation, with a gradual decline thereafter, reaching pre-infection levels by d 3.
Of the genes illustrated in Figure 2 and Supplementary Table 1, only TAK1, NEMO, and BCL3 were also significantly regulated after sham injection of saline into the ME.
Expression of genes involved in non-canonical NF-κB activation
The genes encoding the non-canonical NF-κB subunits RelB and NF-κB2 (Figure 2(a) and Supplementary Table 1) were rapidly regulated, with kinetics similar to that for RelA and NF-κB1, but the up-regulation was much stronger for the non-canonical subunits. Of the upstream signaling genes, NIK (Figure 2(c)) was modestly up-regulated from 3 to 24 h after inoculation, while TRAF3 was un-regulated.
Effect of canonical NF-κB pathway inhibition on the growth of previously infected ME mucosal explants
We have previously shown
29
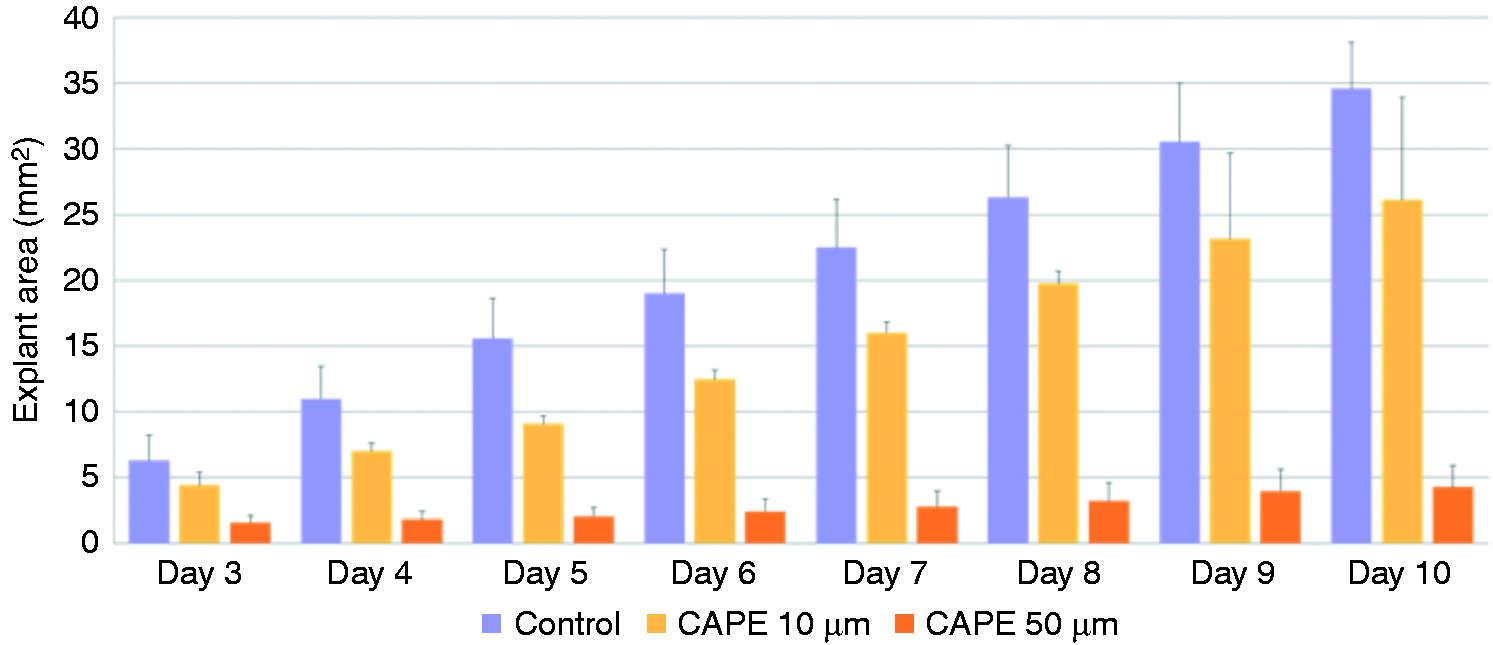
that ME mucosa harvested from animals infected 48 h previously with NTHi exhibited in vitro growth that is two to three times greater than that of mucosal samples harvested from uninfected mice. This hyperplastic growth is observed for at least 10 d of culture. The RelA nuclear translocation inhibitor CAPE was applied to explants of hyperplastic ME mucosa harvested from previously infected MEs. CAPE showed a significant inhibitory effect on mucosal outgrowth, in a dose-dependent manner. Figure 3 presents representative examples of explant outgrowth. Figure 3(a) and (b) illustrate explants of mucosa not treated with CAPE, on the 3rd and 8thd of culture, respectively. Figures 3(c) and (d) illustrate explants treated with 50 µM CAPE on the same days of culture. The infection-induced growth response is strongly suppressed by inhibitors of the canonical NF-κB pathway (ANOVA, P < 0.001). Figure 4 shows the average area of hyperplastic explants cultured with the two lowest concentrations of CAPE for 10 d, and documents a significant, dose-dependent, inhibitory effect on mucosal outgrowth over that observed initially. The effects of the inhibitors were obvious even after three days in culture. (Explants treated with 250 µM CAPE showed no increase in area throughout the duration of culture.)
Representative photomicrographs illustrating ME mucosal explants maintained in media ((a), (b)) and explants treated with 50 µM of the canonical NF-κB activation inhibitor CAPE ((c), (d)). The NTHi-induced hyperplastic growth response was suppressed by the inhibitor. Average explant area (mm2) of bacterially induced ME mucosa cultured with two concentrations of CAPE for 10 d. Dose-dependent inhibition was observed. (Explants treated with the highest dosage, 250 µM, showed no outgrowth through the entire duration of culture.) ANOVA identified a significant (P < 0.001) effect of inhibitor treatment.
Effect of non-canonical NF-κB pathway inhibition on the hyperplastic response of previously infected ME mucosal explants
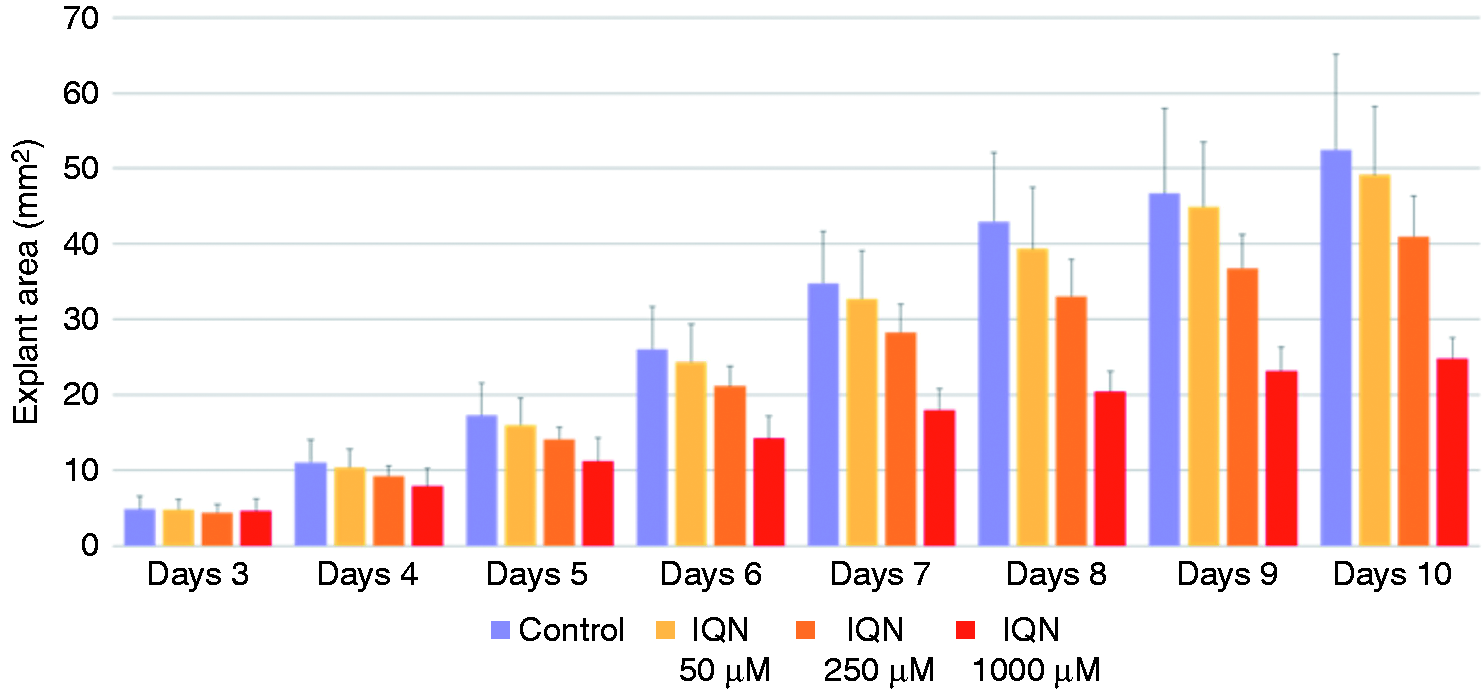
We also observed significant growth inhibition (ANOVA, P < 0.01) when explants of hyperplastic ME mucosa were treated with 1,3(2H,4H)-isoquinolinedione, compared with control explants, throughout 10 d of culture. Figure 5 shows representative examples of untreated explants ((a), (b)) or explants treated with 250 µM 1,3(2H,4H)-isoquinolinedione ((c), (d)) after 3 and 8d in culture. Figure 6 shows the average area of mucosal explant exposed to three concentrations of 1,3(2H,4H)-isoquinolinedione for 10 d. While no effect was observed after 3d in culture, a dose-dependent inhibition of mucosal growth was increasingly apparent at longer culture intervals. However, inhibition was not as great as that seen with CAPE.
Representative photomicrographs of ME explant outgrowth for untreated mucosa ((a), (b)) and after treatment with 250 µM of the NIK inhibitor 1,3(2H,4H)-isoquinolinedione (IQN) ((c), (d)). NTHi-induced hyperplastic growth response was partially suppressed by this inhibitor of alternative NF-κB activation. Average area (mm2) of bacterially induced ME mucosal explants cultured with different concentrations of 1,3(2H,4H)-isoquinolinedione (IQN) for 10 d. Significant inhibition of growth (ANOVA, P < 0.01) was observed, in a dose-dependent manner.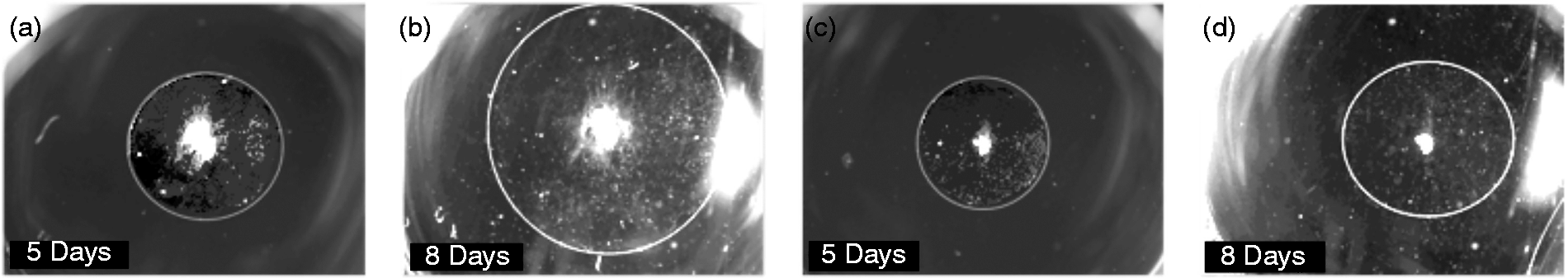
Discussion and conclusions
ME mucosal hyperplasia is a major characteristic of OM. To assess the role of NF-κB in this pathophysiologic response, we evaluated the expression of genes involved in the canonical and non-canonical pathways of NF-κB activation following ME infection with NTHi. We also determined the effects of inhibiting each pathway on the growth of ME mucosal explants in which growth has been accelerated by previous exposure to NTHi in vivo.
The regulation of many of the genes involved in NF-κB signaling and activation, as well as those encoding the NF-κB subunits themselves, provides strong evidence that NF-κB plays a major role during OM. Give the importance of innate immunity in normal recovery from OM,20,21,23–25 and the fact that canonical NF-κB activation is a major downstream target of many innate immune receptors, 36 our gene regulation results are perhaps not surprising. The dramatic regulation and kinetics of genes encoding the growth and survival factors PIK3CD and AKT1 also suggests that NF-κB plays a strong role in tissue hyperplasia. The significant regulation of genes that support non-canonical NF-κB activation was also expected, given the documented sensitization of cognate immunity that has been observed after Ag application to the ME. 37 However, their kinetics was not well related to the initiation of mucosal hyperplasia.
The high level of expression of inhibitory IKKɛ, concurrent with expression of NEMO (IKKγ), which is positively associated with NF-κB activation, presumably reflects the importance of controlling the downstream effects of NF-κB by negative regulation. This could include reducing the expression of NF-κB -induced pro-inflammatory mediators, to limit bystander tissue damage and prevent excessive ME mucosal hyperplasia.
The kinetics of NF-κB-related gene expression is also informative. Many NF-κB upstream signaling genes exhibit peak expression at 1 d after ME infection, at the time of initiation of mucosal hyperplasia. This was especially true of PIK3CD and AKT1, further implicating canonical NF-κB activation in mucosal hyperplasia. In contrast, IκB and NF-κB subunit genes were up-regulated earlier, perhaps reflecting the need for rapid innate immune response to bacteria, which would need to begin as soon as possible after infection.
Compared to the many NF-κB-related genes regulated after NTHi inoculation, only three were significantly affected by sham injection of saline alone into the ME. These genes are presumably responding to either surgical injury or the presence of fluid in the ME, but are not sufficient to induce hyperplasia, since sham injections do not induce ME mucosal growth. 28 While the additional genes regulated by NTHi potentially participate in mucosal hyperplasia, NF-κB also mediates many other responses to infection. 11 Our inhibitor studies provide more direct evidence of a function role for NF-κB in mucosal growth.
As expected, inhibition of canonical NF-κB signaling by CAPE reduced the growth of explants from infected MEs. We have previously shown that NTHI infection accelerates the growth of ME mucosal explants two- to threefold when compared to mucosal explants from uninfected MEs. This growth reflects the proliferation of mucosal epithelial cells, since the cells are positive both for epithelial cytokeratins and the cell proliferation marker proliferating cell nuclear Ag (PCNA). 29 The growth transformation appears to be permanent in that explants from infected ears continue to grow at a higher rate for at least 10 d, suggesting that signals that terminate growth in vivo are missing from our culture model. Classical activation is well known to enhance cell survival and proliferation in other tissues. 14 In particular, NF-κB acts downstream from many growth factor receptors, including several epidermal growth factor (EGF) receptors responsive to heparin-binding (HB)-EGF, which we have previously shown to be involved in ME mucosal growth during OM. 38
Unexpected was our finding that inhibition of the non-canonical NF-κB pathways also produced significant, dose-dependent suppression of bacterially stimulated growth of ME mucosal explants. As noted above, this pathway is generally associated with lymphoid tissue development, T-cell differentiation, and angiogenesis. 16 Since it seems highly unlikely that immune cells are responsible for mucosal growth in acute OM or in culture, our data support a direct role in mucosal hyperplasia. NIK and p100 mutations have been implicated in a number of cancers,39,40 supporting a role for the non-canonical pathway in tissue growth and survival.
Of course, the limitations of our study need to be acknowledged and considered when interpreting our data. NTHi is a human pathogen, and the response of animal models may not replicate that which occurs in patients. Similarly, our tissue culture model of ME mucosal growth, as in all in vitro systems, does not mimic in vivo responses. The permanent enhancement of mucosal explant growth induced by prior NTHi infection and the continuing involvement of NF-κB signaling, both of which resolve with OM recovery in vivo, are unique to the culture system. Finally, it should be noted that NTHi is only one of the three major pathogens associated with OM. Only further study would determine whether the results obtained with this bacterial species would be similar after infection with S. pneumonia or M. catarrhalis.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the NIH/National Institute on Deafness and Other Communication Disorders (NIDCD) (grant numbers DC000129 and DC012595), and by a grant from the VA (grant number BX001205).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.