
Abstract
Fatty acids have been recognized as regulators of immune function in addition to their known metabolic role. Long-chain fatty acids bind free fatty acid receptor (FFAR)-1/GPR40, which is expressed on bovine neutrophils, and increase responses such as granule release and gene expression. In this study, we investigated the molecular mechanisms governing the up-regulation of cyclooxygenase-2 (COX-2) and IL-8, as well as matrix metalloproteinase (MMP)-9 granule release in FFAR1/GPR40 agonist-stimulated neutrophils. Our results showed that natural (oleic and linoleic acid) and synthetic (GW9508) FFAR1/GPR40 agonists increased ERK1/2, p38 MAPK and Akt phosphorylation, and that the FFAR1/GPR40 antagonist GW1100 reduced these responses. We evaluated the levels of IκBα, a component of the classical activation pathway of the transcription factor NF-κB, and we observed IκBα reduction after stimulation with FFAR1/GPR40 agonists, an effect that was inhibited by GW1100 or the inhibitors UO126, SB203580 or LY294002. FFAR1/GPR40 agonists increased COX-2 and IL-8 expression, which was inhibited by GW1100 and an NF-κB inhibitor. Finally, the FFAR1/GPR40 agonist-induced MMP-9 granule release was reduced by GW1100 and UO126. In conclusion, FFAR1/GPR40 agonists differentially stimulate neutrophil functions; COX-2 and IL-8 are expressed after FFAR1/GPR40 activation via NF-κB, IκBα reduction is FFAR1/GPR40- and PI3K/MAPK-dependent, and MMP-9 granule release is FFAR1/GPR40- and ERK1/2-dependent.
Introduction
Neutrophils are the first line of defence of the host against invasive microorganisms and are one of the first cells to migrate from the bloodstream into injured or infected tissues. 1 In the tissue, neutrophils perform their defensive role by engulfing pathogens, producing reactive oxygen species (ROS), releasing degradative enzymes stored in cytoplasmic granules and synthesizing proteins, thus contributing to the inflammatory process and inducing potential tissue damage; therefore, their activation need to be closely regulated.2,3
Fatty acids have extensively been studied as key metabolic components; however, a role in the immune and inflammatory response has begun to be recognized.4,5 Long-chain fatty acids (LCFA) are increased in the plasma in cows around partum, a time where these animals are more susceptible to acquiring infectious diseases, and an association between LCFA and infectious disease incidence has been suggested.6,7 In bovine neutrophils, the unsaturated fatty acids oleic (OLA) and linoleic (LA) acid increase ROS production, matrix metalloproteinase (MMP)-9 release, and cyclooxygenase (COX)-2 and IL-8 mRNA levels.8–10
OLA and LA-induced ROS and MMP-9 release are mediated through free fatty acid receptor-1 (FFAR1/GPR40). FFAR1/GPR40 is a G protein-coupled receptor that has been identified in pancreatic cells,11–13 monkey neurons, 14 breast cancer cells,15,16 and bovine mammary epithelial cells and neutrophils.8,17 In pancreatic β cells and breast cancer cells, FFAR1/GPR40 is coupled to intracellular Gq and Gi proteins, respectively,15,18 whereas in bovine neutrophils, it is coupled to both Gq and Gi proteins because phospholipase C (PLC) inhibition reduced MMP-9 and ROS production induced by LCFA, and pertussis toxin only partially reduced OLA-induced intracellular calcium mobilization.8,10 In addition, we observed that FFAR1/GPR40 activation induces protein kinase C (PKC) activation in bovine neutrophils; 10 however, the downstream signalling pathways activated by FFAR1/GPR40 in bovine neutrophils remain unknown. Some reports have suggested the participation of FFAR1/GPR40 in MAPK and PI3K pathway activation; synthetic and endogenous ligands of FFAR1/GPR40 induce ERK1/2, JNK and p38 MAPK, and PI3K/Akt phosphorylation in β pancreatic cells, breast cancer cells, bovine mammary epithelial cells or murine neurons.13,15,17,19,20 The MAPK and PI3K pathways have been involved in various cellular processes important in inflammation, such as migration, respiratory burst, granule release and gene expression.21,22 Previous studies have shown that ERK1/2 and p38 MAPK are involved in the up-regulation of COX-2 expression induced by IgE in human neutrophils. 23 In addition, LA induces COX-2 expression through the activation of ERK1/2 in retinal epithelial cells. 24 Besides, LA increased IL-8 expression in intestinal smooth muscle in Crohn’s disease. 25 Although fatty acids (FFAR1/GPR40 agonists) are known to induce MAPK phosphorylation and gene expression, 9 a role of FFAR1/GPR40 in signalling in bovine neutrophils has not yet been demonstrated. Therefore, this study established the participation of FFAR1/GPR40 in the phosphorylation of intracellular signalling pathways such as ERK1/2, p38 MAPK, and PI3K/Akt, and their role in MMP-9 release. We also demonstrated a role for FFAR1/GPR40 in the signalling pathway of NF-κB, a transcription factor that controls COX-2 and IL-8 gene expression.
Materials and methods
Neutrophil isolation
Blood was collected by jugular venipuncture of five healthy Holstein heifers from a Universidad Austral de Chile herd, and samples were collected in ACD Blood Collection Tubes (Becton Dickinson, Franklin Lake, NJ, USA). All experiments were conducted in strict accordance with protocols approved by the ethical committee of the Universidad Austral de Chile (permit number: 216/2015). Neutrophils were isolated according to a previously described method. 26 Viability was determined by trypan blue exclusion assays and was at least 97% for all experiments. Neutrophil purity was at least 95%, as assessed by flow cytometry (BD FACSCanto II; Becton Dickinson) using a forward-scatter vs. side-scatter dot plot to determine the relative size and granularity of the cells. 27
Immunoblot
Five million bovine neutrophils were suspended in Hank’s balanced salt solution (HBSS) plus Ca2+ (HBSS + 0.9 mM Ca2+) and incubated with vehicle (0.1% DMSO) or 10 µM GW1100 (FFAR1/GPR40 antagonist) for 15 min at 37℃. Then, FFAR1/GPR40 agonists (10 µM GW9508, 100 µM LA or 300 µM OLA) were added and incubated for 5 min at 37℃ (for phosphorylation experiments). As a control, 100 nM platelet-activating factor (PAF) was used. To analyse the COX-2 levels, neutrophils were incubated with vehicle (0.1% DMSO), 10 µM GW1100 or 50 µM andrographolide (NF-κB inhibitor) for 15 min at 37℃, and then FFAR1/GPR40 agonists (10 µM GW9508, 100 µM LA or 300 µM OLA) were added and incubated for 3 h at 37℃. Total proteins were obtained, as previously described. 9 Total proteins (80 µg) were analysed by 12% SDS–PAGE and transferred onto a nitrocellulose membrane. Immunoblotting was performed according to a protocol as previously described. 9 Primary Abs against phospho-p38 MAPK, phospho-ERK1/2 and phospho-Akt (Cell Signaling, Beverly, MA, USA) or COX-2 (Cayman Technologies, Pickerington, OH, USA) were used according to the instructions provided by the manufacturer. A secondary anti-rabbit HRP-conjugated Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used, and the membranes were developed using an enhanced chemiluminescence system (Perkin-Elmer, Waltham, MA, USA). The primary Abs were stripped, 9 and each membrane was re-probed with an Ab recognizing total p38, ERK1/2, Akt or β-actin (Cell Signaling). Re-probed signal was detected as described above.
Determination of IκBα levels by flow cytometry
Neutrophils (1 × 106 cells) were incubated with vehicle (0.1% DMSO) or 10 µM GW1100 for 15 min, and then FFAR1/GPR40 agonists (10 µM GW9508, 100 µM LA or 300 µM OLA) were added and incubated for 30 min at 37℃. To analyse the effect of the MAPK and PI3K pathways on IκBα levels, the pharmacological inhibitors UO126 (10 µM, MEK1/2 inhibitor), SB203580 (10 µM, p38 MAPK inhibitor), LY294002 (10 µM, PI3K inhibitor) or vehicle (0.1% DMSO) were incubated for 15 min at 37℃. Next, FFAR1/GPR40 agonists were added and incubated for 30 min at 37℃. The neutrophils were fixed with 4% paraformaldehyde, washed with PBS, permeabilized with Perm/Wash Buffer I (BD™ Phosflow; Becton Dickinson) and incubated with an AlexaFluor 488-conjugated IκBα Mouse mAb (Cell Signaling) for 1 h. The cells were analysed using a FACSCanto II flow cytometer (Becton Dickinson) and FlowJo 7.6 software (TreeStar Inc., Ashland, OR, USA).
RT-qPCR
Neutrophils (1 × 107) were incubated with vehicle (0.1% DMSO), 10 µM GW1100 or 50 µM andrographolide (AP; NF-κB inhibitor) for 15 min at 37℃. Afterwards, FFAR1/GPR40 agonists (10 µM GW9508, 100 µM LA or 300 µM OLA) were added and incubated for 2 h at 37℃. Total RNA was isolated using a Total RNA Kit (E.Z.N.A; Promega, Madison, WI, USA). For the cDNA synthesis reaction, 90 ng total RNA was reverse transcribed using the Affinity Script QPCR cDNA Synthesis Kit (Agilent Technologies, Cedar Creek, TX, USA). Real-time PCR was performed using SYBR Green qPCR Master mix (Fermentas Life Sciences, Waltham, MA, USA) and primers specific for bovine COX-2, IL-8 and GAPDH. The primers used for the PCR reaction were as follows: COX-2 R 5′-ACCGTTTTGGTGAGGTGCGTAT-3′, COX-2 F 5′-AGCACCATTCTCCCTGAAACT-3′; IL-8 R 5′-TCATGGATCTTGCTTCTCAGC-3′, IL-8 F 5′-TGGGCCACACTGTGAAAAT-3′; GAPDH R 5′-CCCTCCACGATGCCAAAGT-3′, GAPDH F 5′-GGCGTGAACCACGAGAAGTATAA-3′. 9 The following conditions were used: 95℃ for 10 min and 40 cycles of 30 s at 95℃, 30 s at 55℃ and 30 s at 72℃. This was followed by three additional steps (dissociation curve): 95℃ for 1 min, 60℃ for 30 s and 95℃ for 30 s.
ELISA
Neutrophils (5 × 106) were incubated with vehicle (0.1% DMSO), 10 µM GW1100 or 50 µM AP for 15 min at 37℃. Afterwards, FFAR1/GPR40 agonists (10 µM GW9508, 100 µM LA or 300 µM OLA) were added and incubated for 5 h at 37℃. Then, the supernatants were obtained and stored at –80℃. The IL-8 levels in the supernatants were assayed using a commercially available IL-8 ELISA kit (R&D Systems, Inc., Minneapolis, MN, USA). The Ab pairs used in this kit have been previously shown to cross-react with bovine IL-8. 28 The absorbance was measured in a Varioskan microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) at 450 nm. The results were expressed as the IL-8 concentration (pg/ml).
Determination of MMP-9 activity by zymography
Neutrophils (1 × 106) were incubated with 10 µM UO126, 10 µM SB203580, 10 µM LY294002 or vehicle (0.1% DMSO) for 5 min at 37℃, and then with FFAR1/GPR40 agonists (10 µM GW9508, 100 µM LA or 300 µM OLA) for 5 min at 37℃. After incubation, the neutrophils were centrifuged at 600 g for 6 min, and equal amounts of supernatants were assayed for gelatinase activity by zymography, as described. 10 Briefly, 10 μl supernatant was loaded on 10% polyacrylamide gels (0.75-mm thick) containing 0.2% gelatine. The gels were run at 200 V for 1 h in a Bio-Rad Mini Protean II apparatus (Bio-Rad Laboratories, Richmond, CA, USA) and then soaked twice in 2.5% Triton X-100 in distilled water on a shaker at room temperature for 30 min. Then, the gels were soaked in reaction buffer consisting of 100 mM Tris (pH 7.5) and 10 mM CaCl2 at 37℃ overnight. The gels were stained in 0.5% Coomassie Brilliant Blue R-250 (Winkler, Santiago, Chile) in acetic acid:methanol:water (1:3:6). Evidence of enzymatic activity was determined by non-staining areas in which the gelatine was degraded. The gelatinolytic bands were compared with a recombinant MMP-9 standard (Sigma-Aldrich, St. Louis, MO, USA). 9 To measure the activity, the gels were digitalized, and the intensity of the bands was determined using ImageJ 1.35 s software (NIH, Bethesda, MD, USA).
Statistical analysis
All experimental protocols were performed in quintuplicate. The results are illustrated in bar graphs as the mean ± SEM. For statistical analysis, an ANOVA and Dunnett’s multiple comparison or t-tests were performed. All analyses were performed using Graph Pad Prism v6.01 software (GraphPad Inc., La Jolla, CA, USA) using a significance level of 5%.
Results
FFAR1/GPR40 agonists induce ERK1/2, p38 MAPK and Akt phosphorylation
To study the intracellular signalling pathways related to the FFAR1/GPR40 receptor, we used the endogenous agonists LA and OLA, and the synthetic agonist GW9508, and the phosphorylation of MAPK p38, ERK1/2 and Akt was evaluated. Previously, we demonstrated that the ERK1/2 and p38 phosphorylation induced by LA is a rapid process, reaching a maximum at 5 min of treatment.
9
Neutrophils were incubated with 10 µM GW9508, 100 µM LA and 300 µM OLA for 5 min, and total proteins were analysed by immunoblot. The LA and OLA concentrations used in the experiments are in the range of the concentration in healthy and ketotic cows;29,30 furthermore, these LA, OLA and GW9508 concentrations were previously used to induce neutrophil functions.
10
We observed a significant increase in the phosphorylation of ERK1/2, p38 MAPK and Akt (Figure 1). The pretreatment of neutrophils with the FFAR1/GPR40 antagonist GW1100 significantly reduced the phosphorylation stimulated by the agonists. As a control, we used neutrophils treated with GW1100 and then stimulated with PAF, a ligand of an unrelated fatty acid receptor, and no reduction in PAF-induced phosphorylation was observed.
The FFAR1/GPR40 receptor participates in ERK1/2, p38 MAPK and Akt phosphorylation induced by OLA, LA or GW9508. Neutrophils were treated with GW1100 (10 µM) for 15 min and then stimulated with GW9508 (10 µM), LA (100 µM) or OLA (300 µM) for 5 min. PAF (100 nM) was used as a control for the specificity of GW1100. Total proteins were obtained, and the phosphorylation of (a) ERK1/2, (b) p38 MAPK and (c) Akt was analysed by immunoblot. Densitometric analyses of five independent experiments are shown in bar graphs (mean ± SEM). *P < 0.05, **P < 0.01, ***P < 0.001.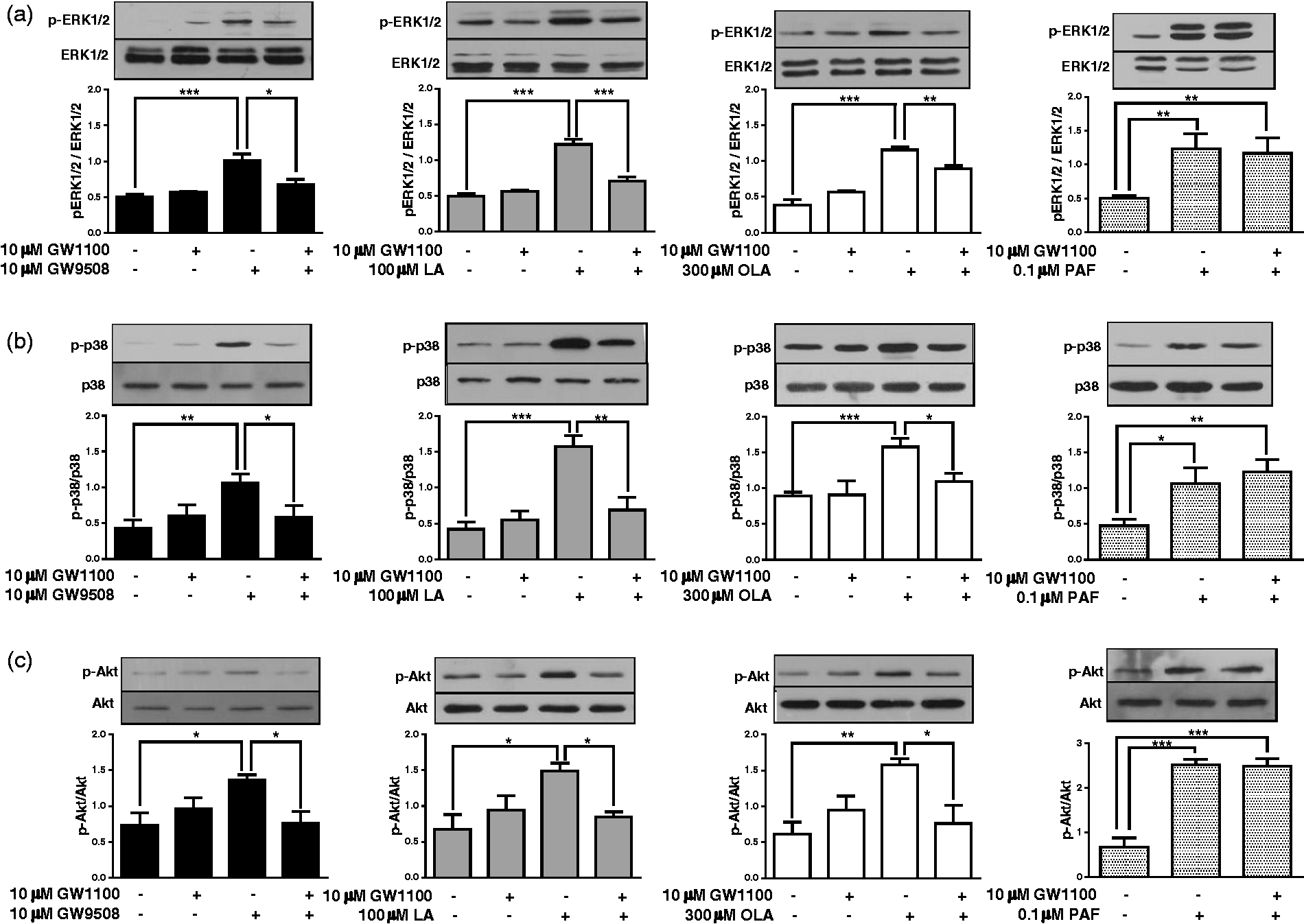
FFAR1/GPR40 stimulation reduces IκBα levels
The NF-κB transcription factor pathway is important for the up-regulation of pro-inflammatory genes.31–33 Therefore, we studied whether agonists of FFAR1/GPR40 participate in the activation of this pathway. The classical NF-κB pathway involves the protein IκBα, which is degraded when the pathway is activated.
34
Neutrophils were incubated with FFAR1/GPR40 agonists for 30 min, and levels of IκBα were analysed by flow cytometry. A significant reduction in IκBα levels was observed in neutrophils treated with GW9508, LA or OLA (Figure 2). The pretreatment of neutrophils with GW1100 and then with the agonists showed significantly higher levels of IκBα than the agonists, suggesting the participation of FFAR1/GPR40 in the activation of the classical NF-κB pathway.
Natural and synthetic FFAR1/GPR40 agonists reduce IκBα levels via FFAR1/GPR40. Neutrophils were treated with GW1100 (10 µM) for 15 min and then stimulated with GW9508 (10 µM) (a), LA (100 µM) (b) or OLA (300 µM) (c) for 30 min. Detection of IκBα was performed by flow cytometry, as described in the ‘Materials and methods’. Bar graphs represent the mean ± SEM of five independent experiments. MFI: mean fluorescence intensity. *P < 0.05, **P < 0.01, ***P < 0.001.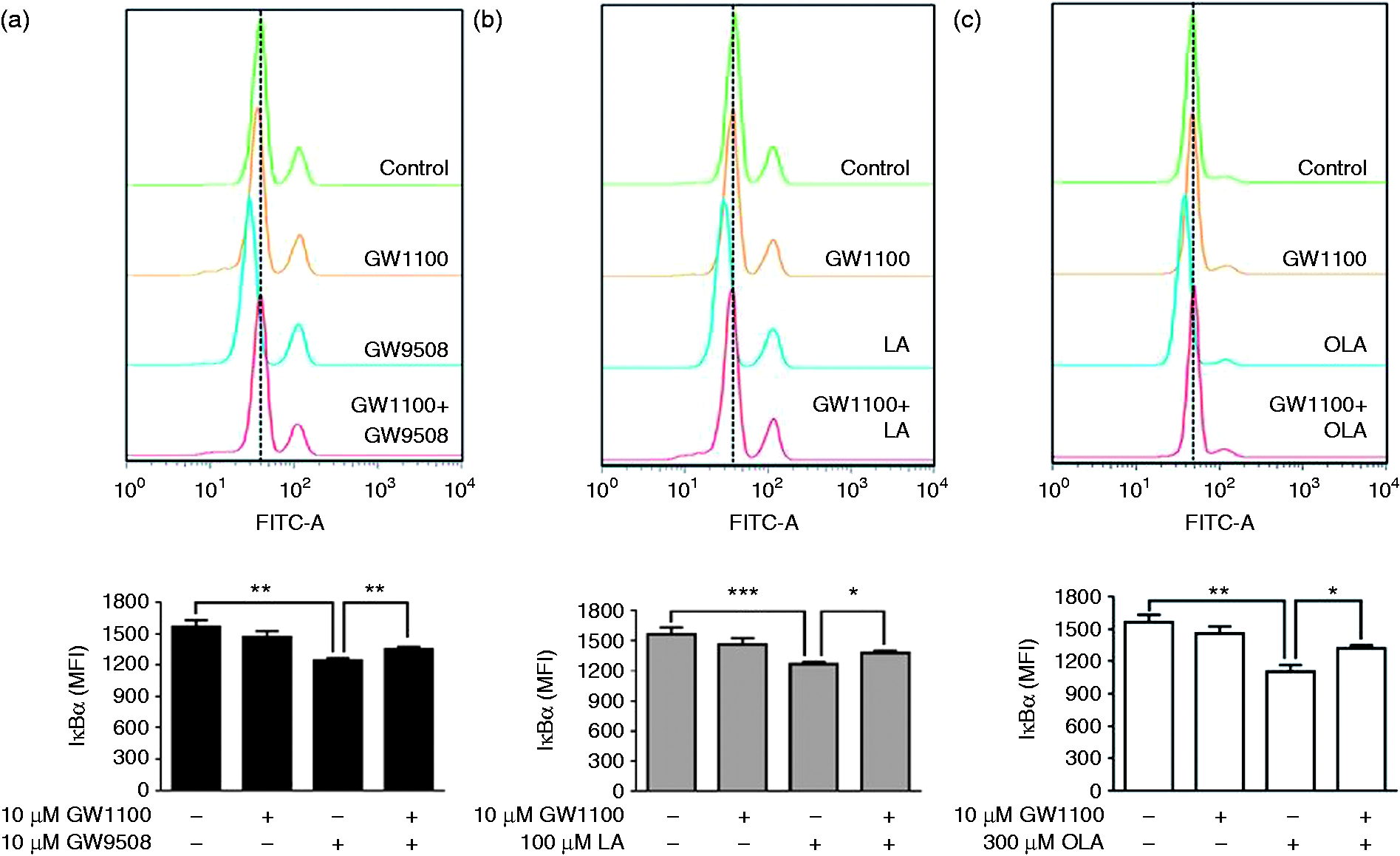
We next evaluated the role of the pathways activated by FFAR1/GPR40, MAPK and PI3K/Akt, on the IκBα levels. For this, we used pharmacological inhibitors of MEK1/2, p38 MAPK and PI3K, prior to stimulation with GW9508, LA or OLA, and the IκBα level was determined by flow cytometry. The inhibitors UO126 (MEK1/2 inhibitor), SB203580 (p38 MAPK inhibitor) and LY294002 (PI3K inhibitor) significantly reduced the effects of GW9508, LA and OLA (Figure 3), increasing the IκBα levels compared with the agonists alone.
ERK1/2, p38 MAPK and PI3K participate in the reduction of IκBα levels. Neutrophils were treated with (a) 10 µM UO126, (b) 10 µM SB203580 or (c) 10 µM LY294002 for 15 min and then stimulated with GW9508, LA or OLA for 30 min. Detection of IκBα was performed by flow cytometry, as described in the ‘Materials and methods’. Bar graphs represent the mean ± SEM of five independent experiments. MFI: mean fluorescence intensity. *P < 0.05, **P < 0.01, ***P < 0.001.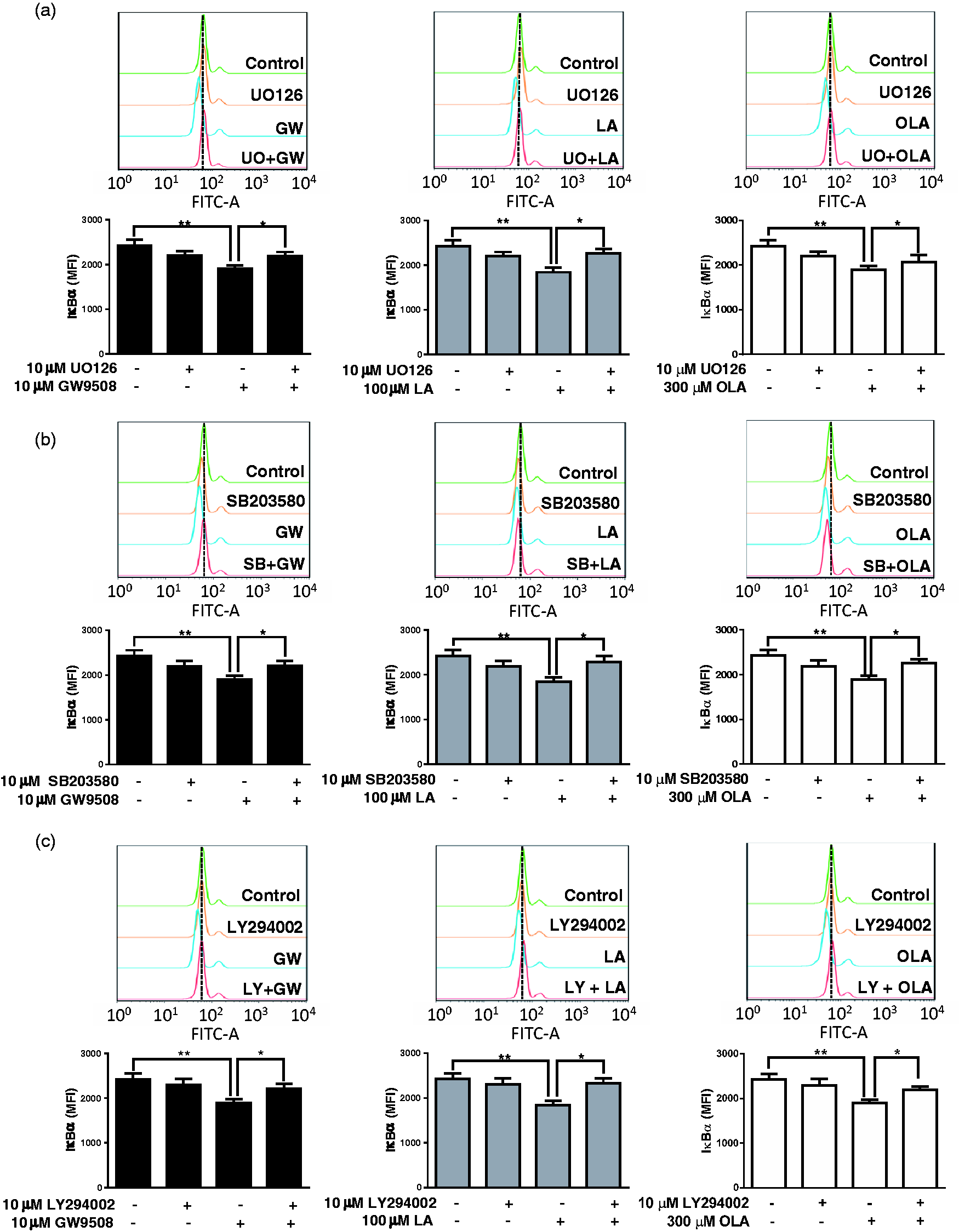
FFAR1/GPR40 stimulation increases the expression of the pro-inflammatory genes IL-8 and COX-2 via NF-κB
Previous data showed that in bovine neutrophils LA increases the expression of the pro-inflammatory genes IL-8 and COX-2,
9
two important mediators with a role in the inflammatory response and neutrophil chemotaxis. However, the participation of FFAR1/GPR40 in this response has not been demonstrated. Neutrophils were incubated with GW1100 for 15 min, and then GW9508, LA or OLA were added and incubated for 2 h (for mRNA analysis), 3 h (for immunoblot of COX-2) or 5 h (for IL-8 ELISA). COX-2 mRNA and protein were analysed by RT-qPCR or immunoblot, respectively, and IL-8 mRNA and protein were analysed by RT-qPCR or ELISA, respectively. IL-8 mRNA was significantly increased in neutrophils treated with GW9508, LA and OLA (Figure 4a); LA and OLA induced a greater increase in IL-8 mRNA than the synthetic agonist GW9508. Similarly, the IL-8 released into the culture medium was also increased in neutrophils stimulated with LA and OLA compared with those stimulated with GW9508 (Figure 4b). The pretreatment of neutrophils with GW1100 prior to agonist treatment significantly reduced the levels of IL-8 mRNA and protein.
Induction of COX-2 and IL-8 expression by FFAR1/GPR40 agonists. Neutrophils were incubated with GW1100 (10 µM) for 15 min prior to stimulation with GW9508 (10 µM), LA (100 µM) or OLA (300 µM) for 2 h (for RNA isolation), 3 h (for COX-2 immunoblot) or 5 h (for IL-8 ELISA). (a) IL-8 and (c) COX-2 mRNA expression was analysed by RT-qPCR using GAPDH as an internal control. The results are depicted as fold changes relative to the control (without GW1100 and agonists). (b) IL-8 production was analysed by ELISA. (d) COX-2 protein levels were analysed by immunoblot using β-actin for normalization, and densitometric analysis is shown in bar graphs. The mean ± SEM of five independent experiments is shown. *P < 0.05, **P < 0.01, ***P < 0.001.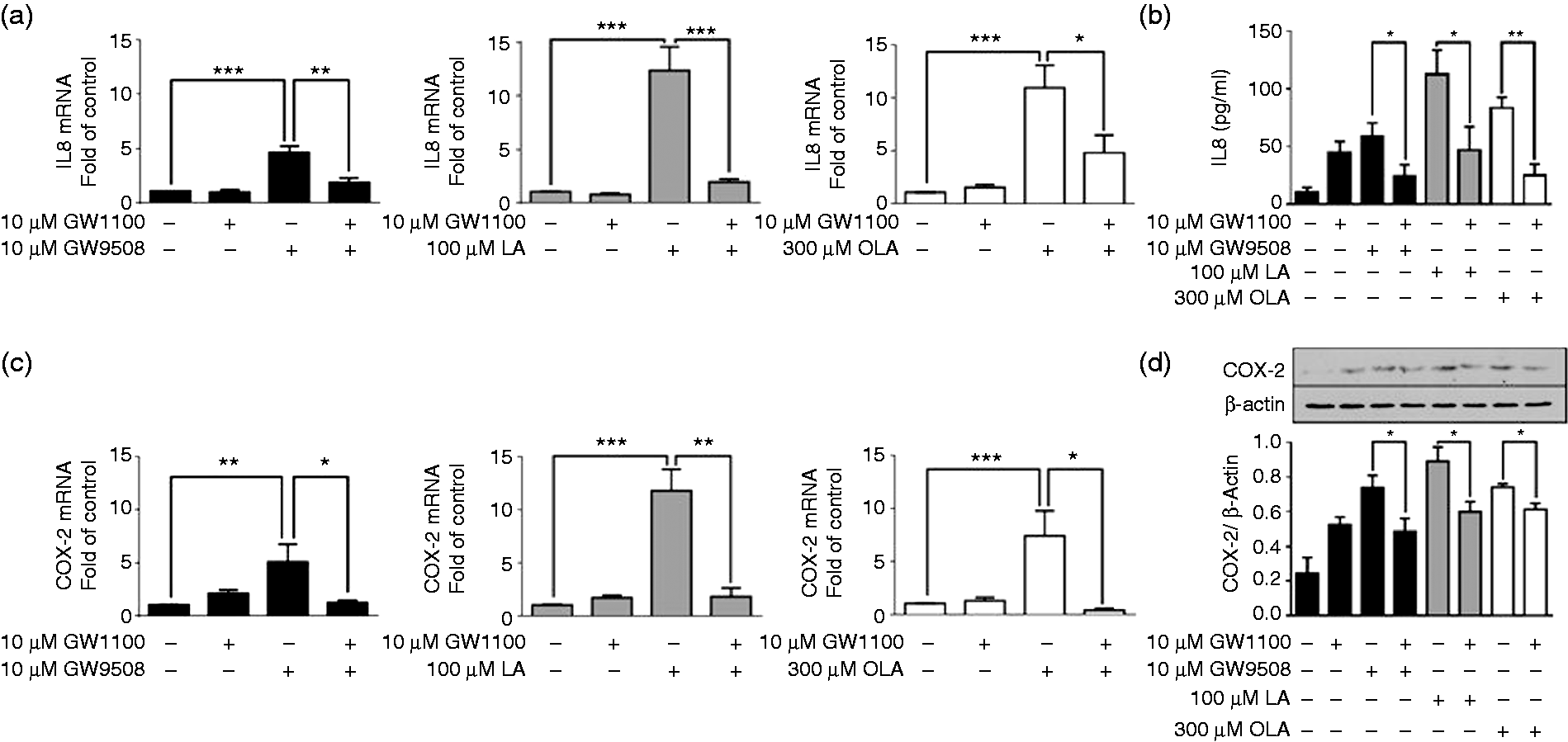
The analysis of COX-2 (Figure 4c, d) showed a significant increase in the mRNA and protein in neutrophils treated with all agonists, and the greatest increase was obtained with LA. Pretreatment with GW1100 reduced the mRNA to near basal levels, and the protein levels were also significantly reduced.
FFAR1/GPR40 activated the NF-κB pathway, and COX-2 and IL-8 possess sites in their promoters for NF-κB binding.35,36 Therefore, we assessed the participation of this pathway in IL-8 and COX-2 expression induced by FFAR1/GPR40 agonists. We pretreated neutrophils with 50 µM AP, an NF-κB inhibitor,37,38 prior to treatment with GW9508, LA or OLA. AP significantly reduced the IL-8 mRNA levels induced by GW9508 and OLA, while a non-significant reduction was observed in neutrophils stimulated with LA (Figure 5a). Similar results were observed in the analysis of the protein by ELISA (Figure 5b). COX-2 mRNA and protein were significantly reduced by AP when all agonists were evaluated (Figure 5c, d).
FFAR1/GPR40 agonist-stimulated COX-2 and IL-8 expression is dependent on the NF-κB pathway. Neutrophils were incubated with 50 µM AP for 15 min prior to stimulation with GW9508 (10 µM), LA (100 µM) or OLA (300 µM), as described in the ‘Materials and methods’. (a) IL-8 and (c) COX-2 mRNA expression was analysed by RT-qPCR using GAPDH as an internal control. The results are depicted as fold changes relative to the control (without AP and agonists). (c) IL-8 production was analysed by ELISA. (d) COX-2 protein levels were analysed by immunoblot using β-actin for normalization, and densitometric analysis is shown in bar graphs. The mean ± SEM of five independent experiments is shown. NS: not significant. *P < 0.05, **P < 0.01, ***P < 0.001.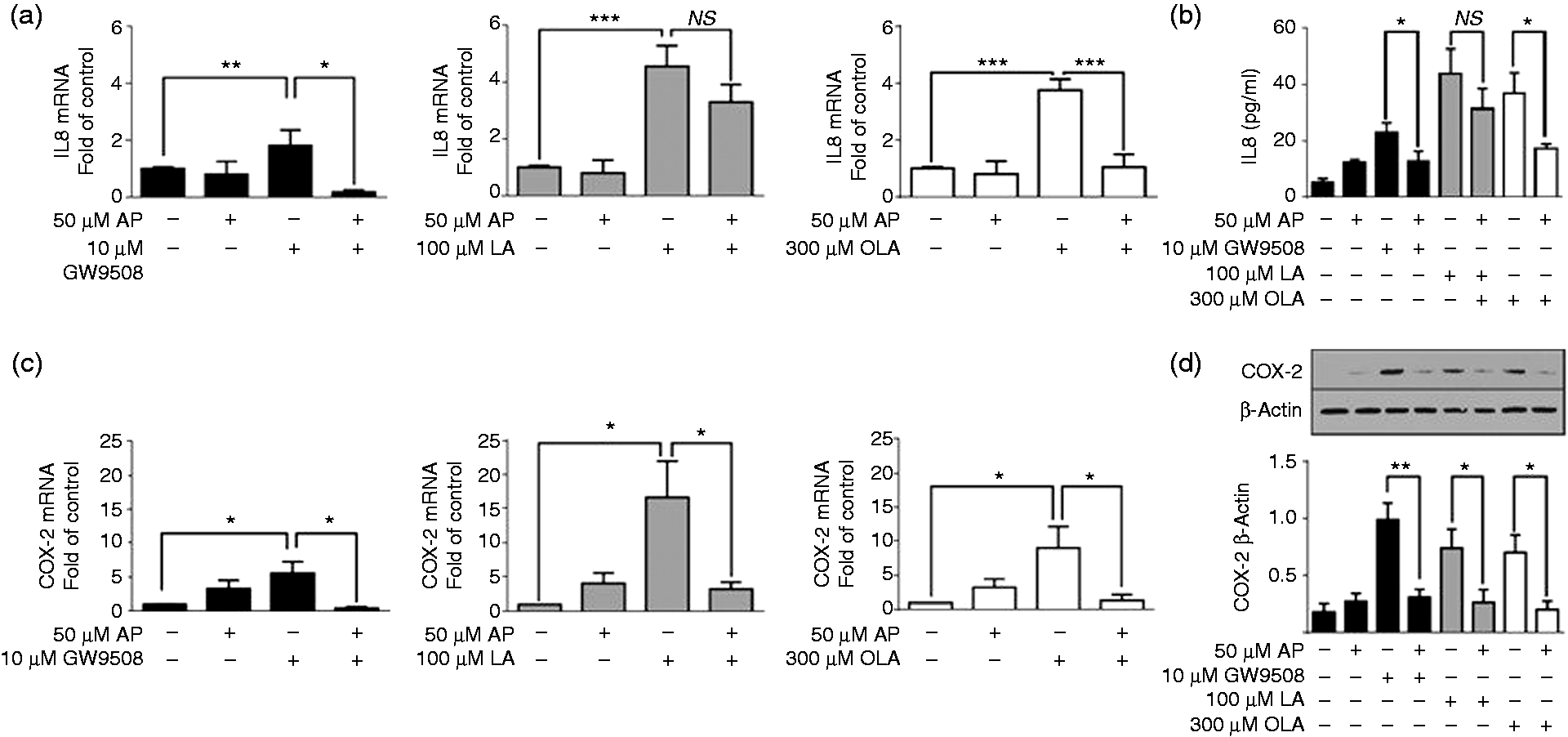
FFAR1/GPR40 stimulation increases MMP-9 granule release
Granule release is an important defensive mechanism that occurs very rapidly in neutrophils,8,39,40 and a role of MAPK and PI3K in the release of MMP-9 in human neutrophils induced by a kinin receptor agonist has been demonstrated.
41
We evaluated the participation of FFAR1/GPR40 in the release of MMP-9 into the supernatants of neutrophils incubated with GW1100 and then with FFAR1/GPR40 agonists for 5 min. Zymography analysis showed that GW9508, LA and OLA significantly increased the secretion of MMP-9, visualized as gelatinase activity. GW1100 significantly reduced the release of MMP-9, demonstrating the involvement of the receptor in this process (Figure 6a).
The ERK1/2 pathway controls MMP-9 granule release induced by FFAR1/GPR40 agonists. Neutrophils were treated with (a) 10 µM GW1100 or (b) 10 µM UO126, 10 µM SB203580 or 10 µM LY294002 for 15 min and then stimulated with GW9508, LA or OLA for 5 min. The MMP-9 activity released into the supernatants was analysed by zymography. Densitometric analysis is shown in the bar graphs. The mean ± SEM of five independent experiments is shown. AU: arbitrary units. NS: not significant. *P < 0.05, **P < 0.01, ***P < 0.001.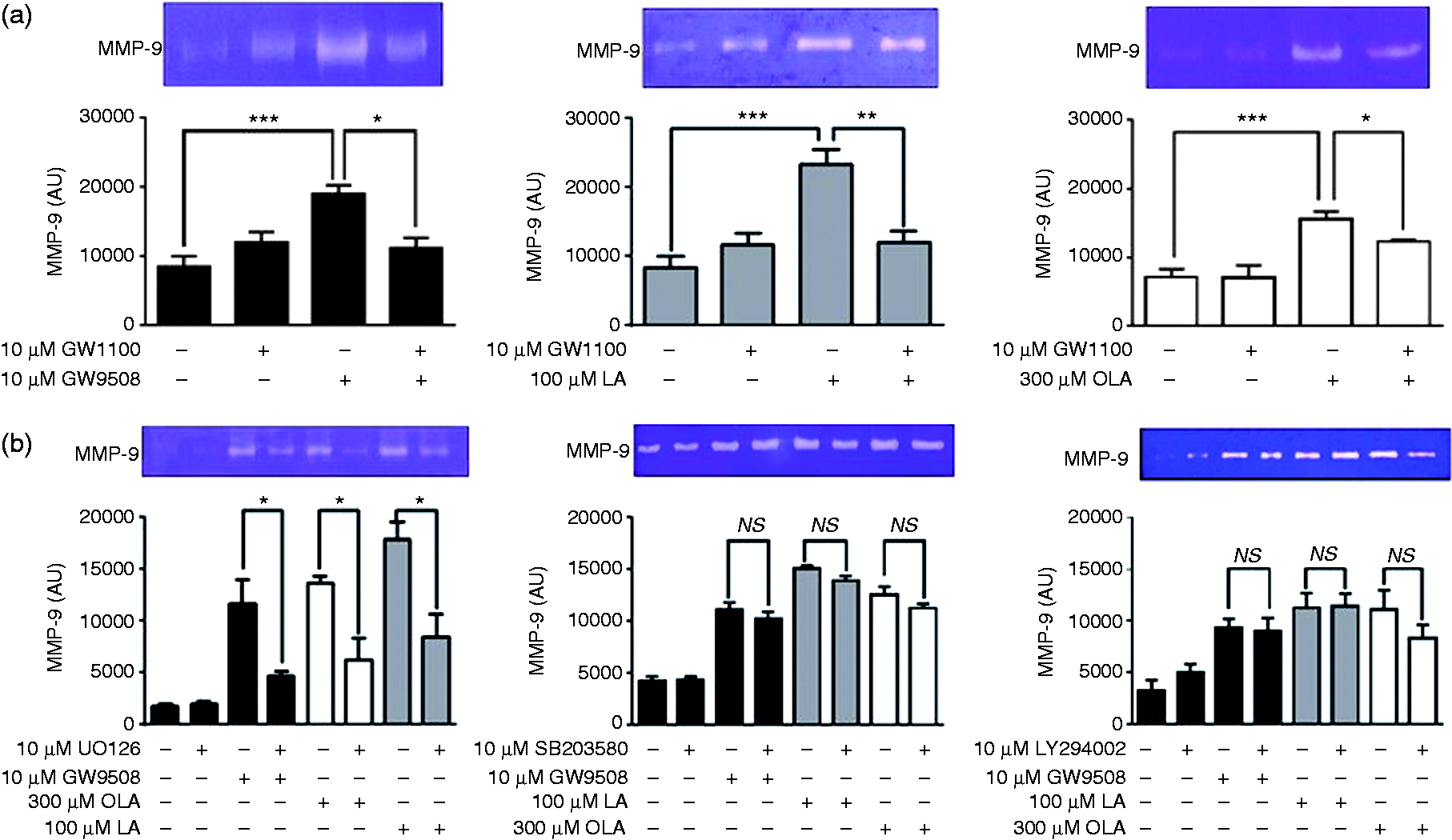
Because FFAR1/GPR40 activation stimulates the rapid phosphorylation of MAPK and Akt, we assessed the role of these protein kinases in MMP-9 release. Neutrophils were treated with UO126, SB203580 or LY294002, and then with FFAR1/GPR40 agonists, and the MMP-9 activity in the supernatants was evaluated by zymography. We observed that only UO126 significantly reduced the MMP-9 activity induced by GW9508, LA or OLA (Figure 6b), indicating the role of MEK1/2-ERK1/2 pathway in gelatinase granule release.
Discussion
Our results revealed the intracellular signalling pathways activated by FFAR1/GPR40 ligands in bovine neutrophils and their link to gene expression and MMP-9 granule release. We observed that natural and synthetic ligands of FFAR1/GPR40 induced the phosphorylation of ERK1/2, p38 MAPK and Akt via FFAR1/GPR40 in bovine neutrophils. Studies of FFAR1/GPR40 in pancreatic β cells (human, mouse or rat) have demonstrated that FFAR1/GPR40 activation stimulates PLC and intracellular calcium. 42 Human FFAR1, which shows 85% identity with the bovine receptor, 10 induced PI3K, ERK1/2 and p38 MAPK activation. 19 In bovine neutrophils, natural and synthetic ligands of FFAR1/GPR40 stimulated intracellular calcium mobilization and PLC and PKC activation, 10 pathways that have been associated with MAPK activation. The absence of external calcium and the store-operated calcium entry inhibitor 2-aminoethoxydiphenyl borate reduced the ERK1/2 phosphorylation induced by platelet-activating factor in bovine neutrophils. 43 The role of PKC in MAPK activation has been demonstrated using the PKC inhibitor GF109203X, which decreased kinin receptor agonist-induced ERK1/2 phosphorylation in human neutrophils. 41
We studied the activation of the NF-κB downstream of ERK1/2, p38 MAPK and PI3K/Akt because this transcription factor is important in the gene expression of pro-inflammatory mediators, such as COX-2 and IL-8. Our results showed for the first time that FFAR1/GPR40 ligands reduced IκBα levels via FFAR1/GPR40 in neutrophils, suggesting the activation of the classical NF-κB pathway, and the inhibitors UO126, SB203580 and LY294002 reduced this effect, indicating the participation of ERK1/2, p38 MAPK and PI3K. Previous reports have demonstrated the participation of MAPK and PI3K in the classical NF-κB pathway activation induced by different stimuli in neutrophils. A role for ERK1/2 has been shown in NF-κB activation in zymosan-stimulated mouse neutrophils because UO126 inhibited the phosphorylation of p65 NF-κB, a subunit of the transcription factor that is phosphorylated upon activation of the pathway. 44 The role of p38 MAPK in NF-κB activation was demonstrated through p38 MAPK inhibition, which reduced the NF-κB nuclear translocation induced by LPS or high levels of superoxide in human or mouse neutrophils, respectively.45,46 Recently, we demonstrated that the inhibition of the ERK1/2, p38 MAPK or PI3K/Akt pathways reduced the NF-κB nuclear translocation induced by fMLP in human neutrophils. 47
Fatty acid-induced NF-κB activation has scarcely been studied. A recent study showed that palmitic acid, a saturated fatty acid, increased NF-κB activity in microvascular endothelial cells, 48 and LA induced NF-κB transcriptional activation in retinal pigment epithelial cells. 49 In contrast, omega-3 polyunsaturated fatty acids (PUFAs) do not affect NF-κB in Caco-2 cells by themselves, however omega-3 PUFAs inhibited the IL-1β-induced degradation of IκBα. 50 In addition, another study suggested the participation of FFAR1/GPR40 in the NF-κB-mediated activation of human renal epithelial cells, showing that cisplatin-induced NF-κB activation was attenuated by pretreatment with GW9508. 51 These studies provide initial data about the effects of fatty acids on NF-κB, which could vary depending on the fatty acid used, the cell type and the presence or absence of a stimulus.
COX-2 and IL-8 genes have NF-κB binding sites in their promoter regions that regulate their expression.36,52 Our results showed that FFAR1/GPR40 agonists increased the gene and protein levels of COX-2 and IL-8. Moreover, we observed that the NF-κB inhibitor AP significantly reduced FFAR1/GPR40 agonist-induced COX-2 and IL-8 expression, suggesting the participation of NF-κB. In addition, the FFAR1/GPR40 antagonist reduced the COX-2 and IL-8 levels, suggesting the involvement of FFAR1/GPR40 in the expression of these pro-inflammatory mediators. Previous studies have demonstrated a role for fatty acids in COX-2 and IL-8 expression in other cells; LA and OLA significantly increased the COX-2 protein expression in chicken hepatocytes and human retinal pigment epithelium, but the participation of FFAR1/GPR40 was not studied.49,53 Our recent study in bovine endothelial cells showed that LA increased IL-8 mRNA, and GW1100 significantly reduced LA-induced IL-8. 54 From our analysis using the MAPK and PI3K inhibitors in FFAR1/GPR40 agonist-treated neutrophils (Supplementary Figure), we did not observe a reduction in COX-2 or IL-8 expression, although a trend towards a decrease in some experiments was observed, which indicates that the inhibition of a single pathway is not enough to reduce the levels of COX-2 or IL-8.
Finally, we studied whether the MAPK or PI3K pathways could play roles in MMP-9 release because MAPK and Akt phosphorylation are rapid events and MMP-9 release also occurs quickly,8,39,40 because active MMP-9 is stored in granules into neutrophils. 55 First, we demonstrated the participation of FFAR1/GPR40 in FFAR1/GPR40 agonist-induced MMP-9 release (Figure 6). Our experiments using MAPK and PI3K inhibitors demonstrated that only the ERK1/2 pathway is involved in MMP-9 release induced by FFAR1/GPR40 agonists. This result is consistent with previous studies in which the ERK1/2 pathway is required for active MMP-9 protein release in aldosterone-stimulated HL-60 cells; however, PI3K and p38 MAPK also played roles in MMP-9 release in that model. 56 In kinin receptor agonist-stimulated human neutrophils, MMP-9 release was dependent on ERK1/2 and p38 MAPK. 41 Thus, our results and those of others show that specific signalling pathways involved in MMP-9 granule release are dependent on receptor activation.
In conclusion, we demonstrated that COX-2 and IL-8 are expressed after FFAR1/GPR40 activation, and NF-κB controls this response. MMP-9 granule release is FFAR1/GPR40- and ERK1/2-dependent in bovine neutrophils. Overall, natural and synthetic FFAR1/GPR40 agonists activate the receptor and differentially activate intracellular signalling pathways to mediate the appropriate bovine neutrophil functions (Figure 7), which is specific of the bovine because FFAR1/GPR40 is not expressed in human neutrophils. These results suggest a link between metabolism and innate immunity, via FFAR1/GPR40 on bovine neutrophils activation, which could contribute to the risk developing infectious diseases at calving, by mechanisms that have been involved in tissue damage.
Summary illustration of the signalling pathways activated by the receptor FFAR1/GPR40 in bovine neutrophils. COX-2 and IL-8 expression are controlled by the NF-κB pathway, IκBα levels are controlled by the ERK1/2, p38 MAPK and PI3K pathways, whereas MMP-9 granule release is dependent on ERK1/2.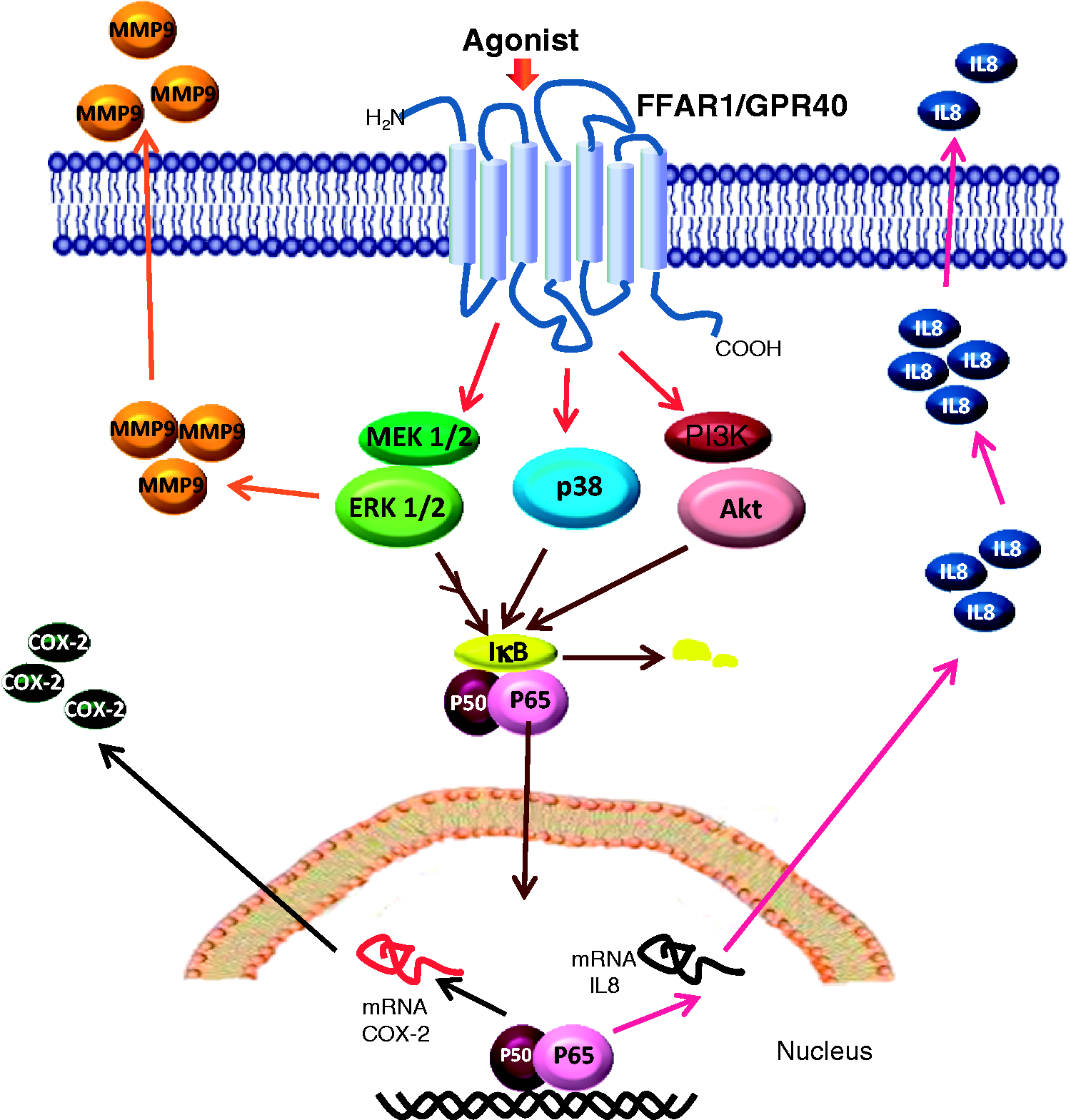
Supplemental Material
Supplemental material for Differential free fatty acid receptor-1 (FFAR1/GPR40) signalling is associated with gene expression or gelatinase granule release in bovine neutrophils
Supplemental Material for Differential free fatty acid receptor-1 (FFAR1/GPR40) signalling is associated with gene expression or gelatinase granule release in bovine neutrophils by Sandra J Mena, Carolina Manosalva, Maria D Carretta, Stefanie Teuber, Iván Olmo, Rafael A Burgos and Maria A Hidalgo in Innate Immunity
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Fondo Nacional de Desarrollo Científico y Tecnológico (Grant FONDECYT No. 1151047 and 11100413); Fondo de Fomento al Desarrollo Científico y Tecnológico (Grant FONDEF IDeA ID14I10050); Mecesup (Scholarship No. AUS0704 and AUS1203); Dirección de Investigación y Desarrollo (Grant DID-UACH No. D-201302); and the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT No. 21100559).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.