
Abstract
Dermatophytoses are chronic fungal infections, the main causative agent of which is Trichophyton rubrum (T. rubrum). Despite their high occurrence worldwide, the immunological mechanisms underlying these diseases remain largely unknown. Here, we uncovered the C-type lectin receptors, Dectin-1 and Dectin-2, as key elements in the immune response to T. rubrum infection in a model of deep dermatophytosis. In vitro, we observed that deficiency in Dectin-1 and Dectin-2 severely compromised cytokine production by dendritic cells. In vivo, mice lacking Dectin-1 and/or Dectin-2 showed an inadequate pro-inflammatory cytokine production in response to T. rubrum infection, impairing its resolution. Strikingly, neither adaptive immunity nor IL-17 response were required for fungal clearance, highlighting innate immunity as the main checkpoint in the pathogenesis of T. rubrum infection.
Introduction
Co-senior authors.
Even though T. rubrum infections are restrained to the cutaneous compartment, recently described mutations in CARD9 were associated to the development of deep dermatophytosis, a condition in which the infection overcomes the skin barrier and reaches internal tissues and organs.4,5 However, it is still unknown which CARD9-dependent receptors are specifically involved and compromised in these conditions.
In the immune response to fungi, C-type lectin receptors (CLRs) and the IL-17 response are mandatory topics. CLRs are considered important initiators of inflammation by driving the production of cytokines, recruiting and activating phagocytes, and also shaping the adaptive immunity, particularly, Th17-biased responses. 6
The IL-17 family cytokines are memorable for their abilities to recruit polymorphonuclear cells and induce production of antimicrobial peptides. 7 Although traditional T lymphocytes (Th17 cells) are their classical source, IL-17 can also be produced by innate, or ‘innate-like’ cells, as neutrophils and innate lymphoid cells,8,9 participating in the interface between innate and adaptive responses.
While the importance of these systems in the immunity to fungal pathogens such as Candida albicans and Pneumocystis carinii, Aspergillus fumigatus, Cryptococcus neoformans and Fonsecaea pedrosoi is well established,7,9–14 their roles against T. rubrum have not yet been characterized. Considering that CARD9 is a key molecule in the CLR signaling pathway, we hypothesized that Dectin-1 and Dectin-2 could also be involved in deep dermatophytosis.
Herein, we uncovered an essential role of these receptors in the immunity to T. rubrum in a murine model of deep dermatophytosis. In vitro, cytokine production by bone marrow-derived dendritic cells (BMDCs) was greatly dependent on both CLRs. In vivo, their deficiency compromised fungal clearance. Strikingly, neither the adaptive immunity nor the IL-17 response were necessary for fungal control.
Materials and methods
Mice
C57BL/6 J background Dectin-1-deficient (Δclec7a), 15 Dectin-2-deficient (Δclec4n), 10 IL-17A and IL-17F double-deficient (Δil-17) 16 and rag2-deficient (Δrag) mice were used in this study. Dectin-1 and Dectin-2 double-deficient mice (dko) were produced by crossing each singly deficient mice and Dectin-1/Dectin-2 deletion was confirmed by FACS (Supplementary Figure 1). Wild type (WT) C57BL/6 J mice were obtained from CLEA Japan (Tokyo, Japan). All mice were kept under specific pathogen-free conditions with a gamma ray-sterilized diet, acidified tap water (0.002 N HCl, pH 3.0), and autoclaved wood chip bed in environmentally controlled clean rooms at the Bio-medical Research Center or Medical Mycology Research Center, Chiba University, Japan. Animals were euthanized by cervical dislocation.
Ethics statement
Animal experiments were performed in strict accordance with the Act on Welfare and Management of Animals (Law No. 105 dated 19 October 1973, revised 30 May 2014), the guidelines under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology, Japan (Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions, dated 1 June 2006) and the ‘Regulation of Animal Experimentation at Chiba University’ (dated 25 April 2007, revised 1 October 2014). The protocols were approved by Chiba University Institutional Animal Care and Use Committee (CUIACUC authorization numbers D26-230 and D27-274).
Fungal preparation
T. rubrum (Castellani) Sabouraud ATCC 28188 (American Type Culture Collection) was employed throughout this study. Production of T. rubrum suspensions was performed as described. 17 Briefly, colonies from 14–21-d-old cultures grown on potato dextrose agar plates (Eiken Chemical, Tokyo, Japan) were incubated for 3 d in potato dextrose broth (BD, Franklin Lakes, NJ, USA) at 30℃ and 150 rpm. Suspensions were filtered in 70-µm cell strainers (BD), and recovered cells were washed with 0.1% Tween 80 in PBS and re-suspended in PBS.
BMDC
The generation of BMDCs was performed as previously described. 18 Bone marrow was collected from tibia and femur, and aliquots of 4–6 × 106 cells were cultured in 10 ml R10 medium supplemented with 20 ng/ml GM-CSF (Invitrogen, Carlsbad, CA, USA) at 37℃ and 5% CO2 for 3 d. On d 3, cells were supplemented with additional 10 ml medium. On d 6, 10 ml medium was collected and centrifuged, and pellet cells were re-suspended in 10 ml fresh R10 medium supplemented with 10 ng/ml GM-CSF (Invitrogen) and returned to the original plates. On d 8, this step was repeated. On d 10, non-adherent cells were collected for assay. Dendritic cell differentiation was confirmed by FACS analysis and BMDCs were defined as CD11b+ CD11c+ cells (Supplementary Figure 2).
In vitro assays
BMDCs (105 cells per well) were plated on 96-wells plates and incubated with T. rubrum suspensions (MOI 1:1) for 4, 6 and 8 h. At each time point, supernatants were collected for cytokine measurements.
Dectin-1 and Dectin-2 Fc chimeras
Soluble Dectin-1-human IgG Fc (Dec-1 Fc) and Dectin-2-human IgG Fc (Dec-2 Fc) chimeras were produced by cloning the extracellular domain of murine Dectin-1/Dectin-2 into a pFUSE vector containing a mutated Fc portion of human IgG2 (pFUSE-hIgG2-Fc2 vector; Invivogen, San Diego, CA, USA). The construct was transfected into HEK293T cells and the protein was purified from culture supernatants by affinity chromatography on Protein A Sepharose.
In binding assays, fungal cells, produced as described above, were re-suspended in TSA buffer [Tris Buffer Saline with 0.5% (m/v) BSA, 2 mM MgCl2 and 2 mM CaCl2] at a concentration of 107 cells/ml and were incubated with Fc chimera (1 µg/ml for Dectin-1 and 10 µg/ml for Dectin-2), or empty Fc as a control, for 1 h at 4℃. Binding was detected using PE anti-human IgG Fc Ab (Biolegend, San Diego, CA, USA) by FACS (FACS Verse; BD).
In vivo infection
For in vivo studies, we employed our previously murine model. 17 Briefly, mice were infected intraperitoneally with 107 fungal cells/animal and kept for up to 14 d, with food and water ad libitum. Animals were euthanized for spleen removal 7 and 14 d post-infection (dpi).
Organs were macerated in PBS and aliquots of final suspensions were plated on Sabouraud Dextrose Agar (BD). Plates were kept at 30℃ for 5–7 d and numbers of recovered colonies were determined. Results were then expressed as log of CFU/g of organ.
Organ suspensions were centrifuged at 2465 g for 10 min and supernatants were collected for cytokine measurements.
Cytokine measurements
Cytokines were quantified by BD Cytometric Bead Array, according to the manufacturer’s instructions. Data were collected in the flow cytometer FACS Verse (BD) and analyzed with FCAP Array (v.3.0.1; BD). The detection limits for the cytokines analyzed were as follows: IL-1β = 1.9 pg/ml; TNF-α = 2.8 pg/ml; IL-10 = 9.6 pg/ml; IL-6 = 1.4 pg/ml; IL-12p40/IL-23 = 13.6 pg/ml; IFN-γ = 0.5 pg/ml; IL-4 = 0.3 pg/m l; IL-17 A = 0.95 pg/ml; IL-17 F = 0.81 pg/ml.
Statistical analysis
Statistical analysis was performed in the software GraphPad Prism (version 6 for OSX; GraphPad Inc., La Jolla, CA, USA). Cytokine data from in vitro assays were analyzed by two-way ANOVA and Sidak’s multiple comparison test; fungal burden data were analyzed by paired t-test, and cytokine data from in vivo assays were analyzed by two-way ANOVA and Dunnett’s multiple comparisons test. Data are expressed as mean ± SEM and P < 0.05 was considered statistically significant.
Results
Dectin-1 and Dectin-2 recognize T. rubrum
Dectin-1 and Dectin-2 are known to recognize sugar-associated molecules in the surface of fungi, but some fungal strains can evade this recognition through agonist masking, as has already been described for C. albicans and A. fumigatus.19,20 In order to confirm that T. rubrum is, indeed, recognized by these CLRs, we constructed soluble Fc chimeras and investigated their binding through FACS analysis (Figure 1). As expected, we confirmed that both receptors directly bound to our strain and therefore they could be involved in the generation of an immune response against this pathogen.
Trichophyton rubrum is recognized by Dectin-1 and Dectin-2. Fungal suspensions were incubated with Dectin-1 Fc (1 µg/ml), Dectin-2 Fc (10 µg/ml) or empty Fc, and binding was stained with PE anti-human IgG and analyzed by FACS. Histograms are representative of three independent experiments.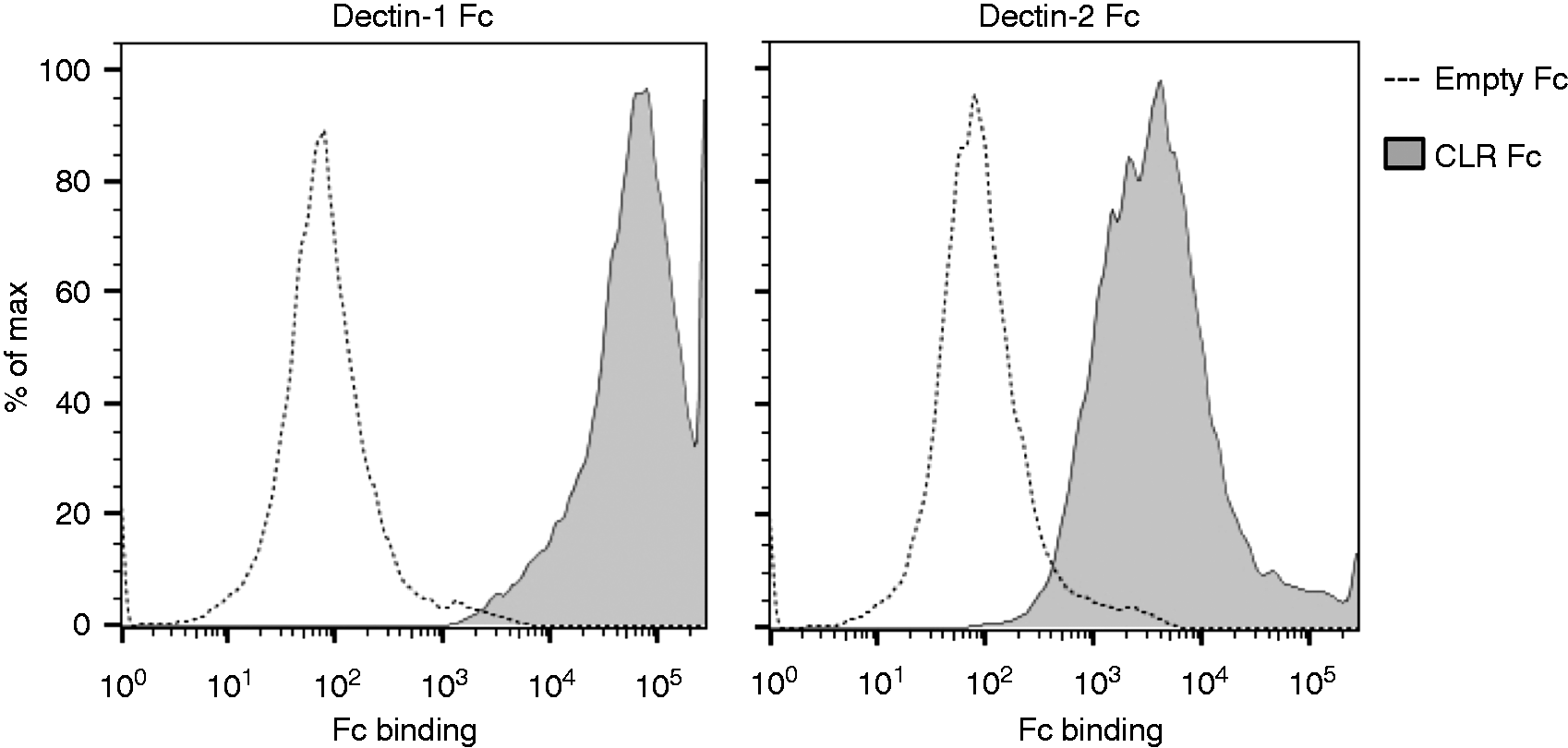
Dectin-1 and Dectin-2 are required for cytokine production
We then continued our investigation by analyzing the interaction between T. rubrum and BMDCs. When incubated with the dermatophyte, BMDCs readily produced IL-1β, TNF-α, IL-10 and IL-6 in a Dectin-1 - and Dectin-2-dependent fashion (Figure 2).
Dectin-1 and Dectin-2 regulate cytokine production by BMDCs in response to T. rubrum. BMDCs [wt, Δclec7a (Dectin-1 ko), Δclec4n (dectin-2 ko) and dko (Dectin-1/-2 ko)] were incubated with T. rubrum (MOI 1), and cytokines were quantified from supernatants after 4, 6 and 8 h of interaction. Data, expressed as mean ± SEM, are representative of three independent experiments (triplicate wells each). Two-way ANOVA and Sidak’s multiple comparison test (wt vs. knockouts): * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001; n.s.: not significant.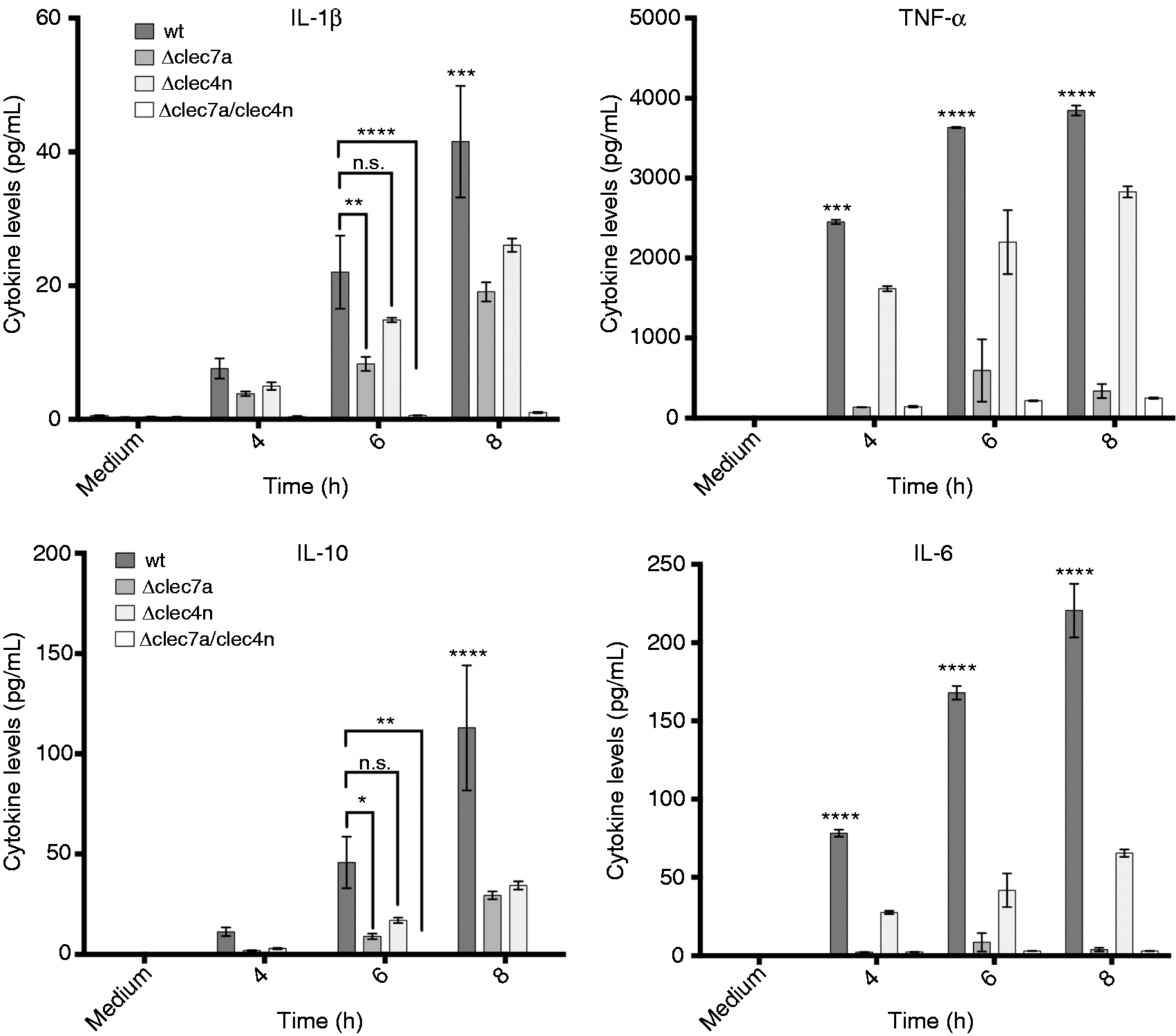
Although we observed a tendency toward reduction for IL-1β and IL-10 levels in Dectin-2-deficient BMDCs before 8 h, it should be highlighted that their cytokine profiles are very similar to TNF-α and IL-6 (where the influence of Dectin-2 is clearly detectable), suggesting that these findings may have a biological significance.
These results suggest that these CLRs are a main pathway for initiation of inflammation in response to T. rubrum.
Dectin-1 and Dectin-2 promote fungal clearance in vivo
Our in vitro data pointed to a potential role for these CLRs in the inflammatory response, prompting us to evaluate their contribution in vivo.
To confirm our hypothesis, we infected mice intraperitoneally with T. rubrum, allowing spleen dissemination, an approach previously employed for the establishment of invasive infection for fungi as Fonsecaea pedrosoi and Sporothrix schenckii.14,21 In this system, while wt animals readily controlled the pathogen after 14 d, Δclec7a, Δclec4n and dko counterparts were unable to do the same (Figure 3), implying a simultaneous requirement of both receptors for optimal response, as the single deficiency is enough to compromise the fungal clearance.
Deficiency in Dectin-1 and/or Dectin-2 impairs fungal clearance in vivo. Mice [wt, Δclec7a (Dectin-1 ko), Δclec4n (Dectin-2 ko) and dko (Dectin-1/-2 ko)] were infected i.p. with 107 fungal cells and fungal burden in spleen macerates were determined 7 and 14 dpi. Each point represents one animal, dark line represents mean (nwt = 15 animals; nΔclec7a = 15 animals; nΔclec4n = 14 animals; ndko = 10 animals). Paired t-test: **P < 0.01.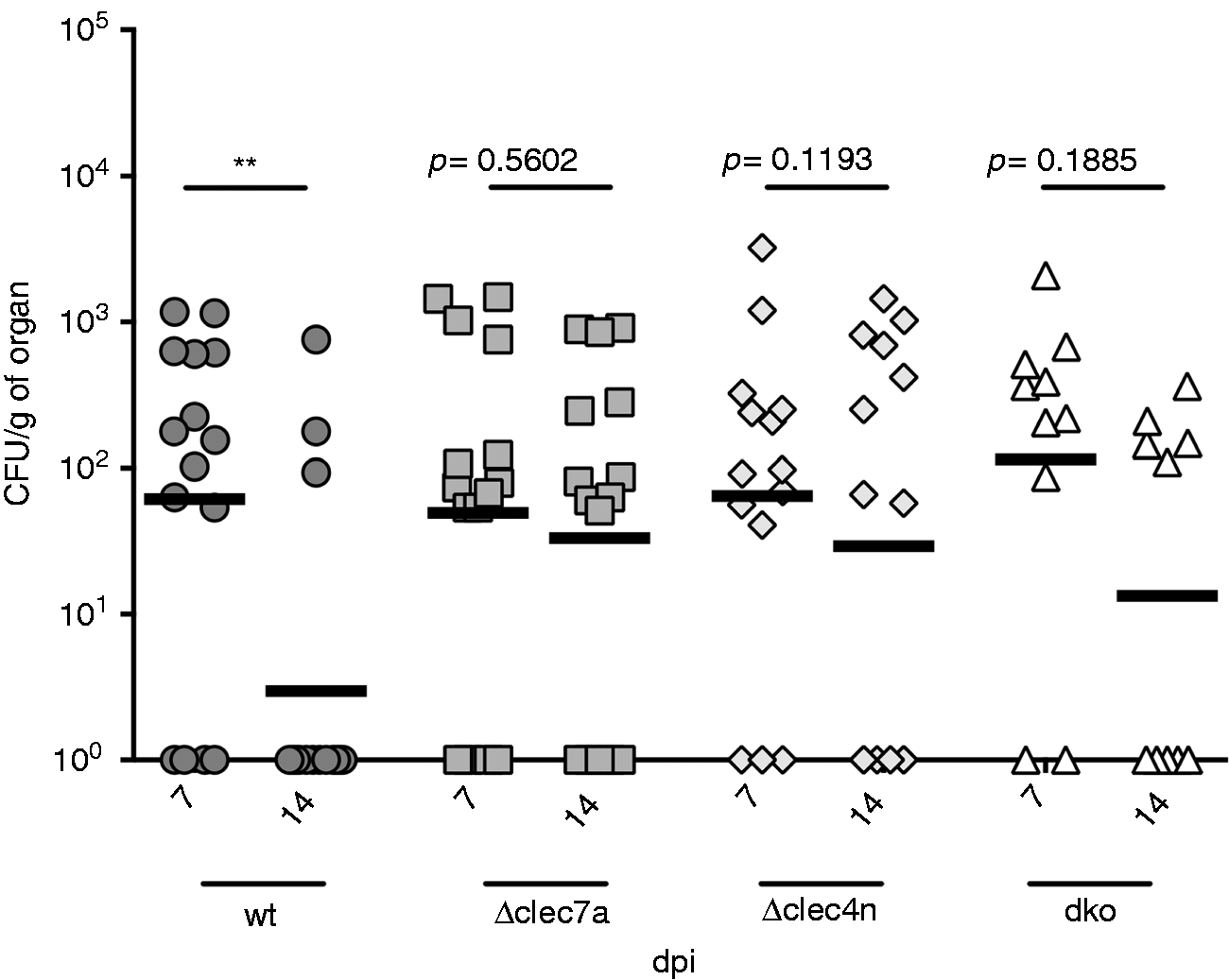
The profiling of the cytokines in wt mice indicated a high production of IL-1β and TNF-α, which was compromised by the lack of the CLRs (Figure 4a). IL-6 production also showed a tendency toward reduction in the knockouts, although it did not reach statistical significance. Curiously, IL-10 production was not compromised by the lack of Dectin-1 and Dectin-2, suggesting that the contribution of other immune pathways/receptors may be responsible for the cytokine pool in the in vivo settings or may, at least, compensate the CLR deficiency in these animals.
In vivo production of inflammatory cytokines is impaired by the lack of Dectin-1 and Dectin-2. Mice [wt, Δclec7a (Dectin-1 ko), Δclec4n (Dectin-2 ko) and dko (Dectin-1/-2 ko)] were infected i.p. with 107 fungal cells and cytokines in spleen macerates were quantified 7 dpi. (a) Levels of IL-1β, TNF-α, IL-10, IL-6 and IL-23. (b) Levels of IFN-γ, IL-4, IL-17 A and IL-17 F. Data expressed as mean ± SEM. Two-way ANOVA and Dunnett’s multiple comparisons test: *P < 0.05; ** P < 0.01; *** P < 0.001 (nwt = 15 animals; nΔclec7a = 15 animals, nΔclec4n = 14 animals; ndko = 10 animals).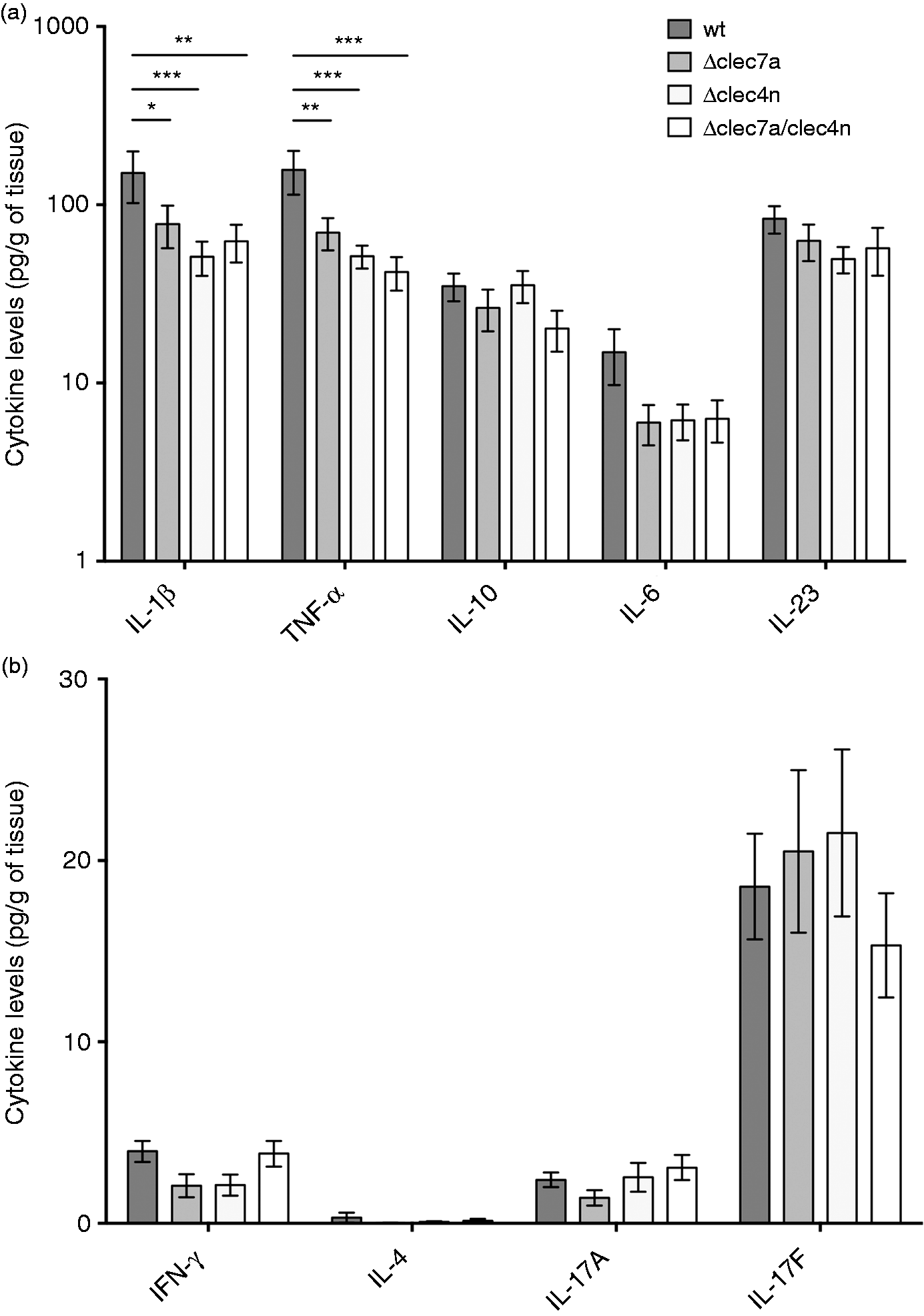
These results indicate that Dectin-1 and Dectin-2 promotes clearance of T. rubrum infection and triggers a robust inflammatory environment.
IL-17 response and adaptive immunity are dispensable
Consistent with our previous work, 17 IL-17 response also prevailed in our system, overcoming IFN-γ and IL-4 responses (Figure 4b). Yet, IL-17 F production was independent of Dectin-1 and Dectin-2. Considering that IL-23 levels, an important cytokine for IL-17 response generation, 22 was not affected by CLR deficiency in vivo (Figure 4a), it is conceivable that this response is not disturbed in our system.
Even though IL-17 response is considered an essential component in the immune response to fungal infections, 7 our data suggest that, against T. rubrum, IL-17 by itself may not be sufficient to promote fungal control.
In order to confirm this hypothesis, we next analyzed the infection outcome in mice lacking this cytokine family.
Strikingly, IL-17 was completely dispensable to promote fungal clearance, as IL-17 A and IL-17 F double-deficient (Δil17) mice were as efficient as their wt counterparts in controlling the infection (Figure 5a).
Neither IL-17 nor T/B lymphocytes are required to T. rubrum control. Mice [wt, Δil17 (IL-17 A/F ko), Δrag2 (Rag2 ko)] were infected i.p. with 107 fungal cells and spleens were harvested for analysis 7 dpi. (a) Fungal burden in spleen macerates 7 and 14 dpi. Each point represents one animal, dark line represents mean (n = 15 animals in each group). Paired t-test: **** P < 0.0001. (b) Cytokines quantified in spleen macerates 7 dpi. Data expressed as mean ± SEM. Two-way ANOVA and Dunnett’s multiple comparisons test: no significance found (n = 15 animals in each group).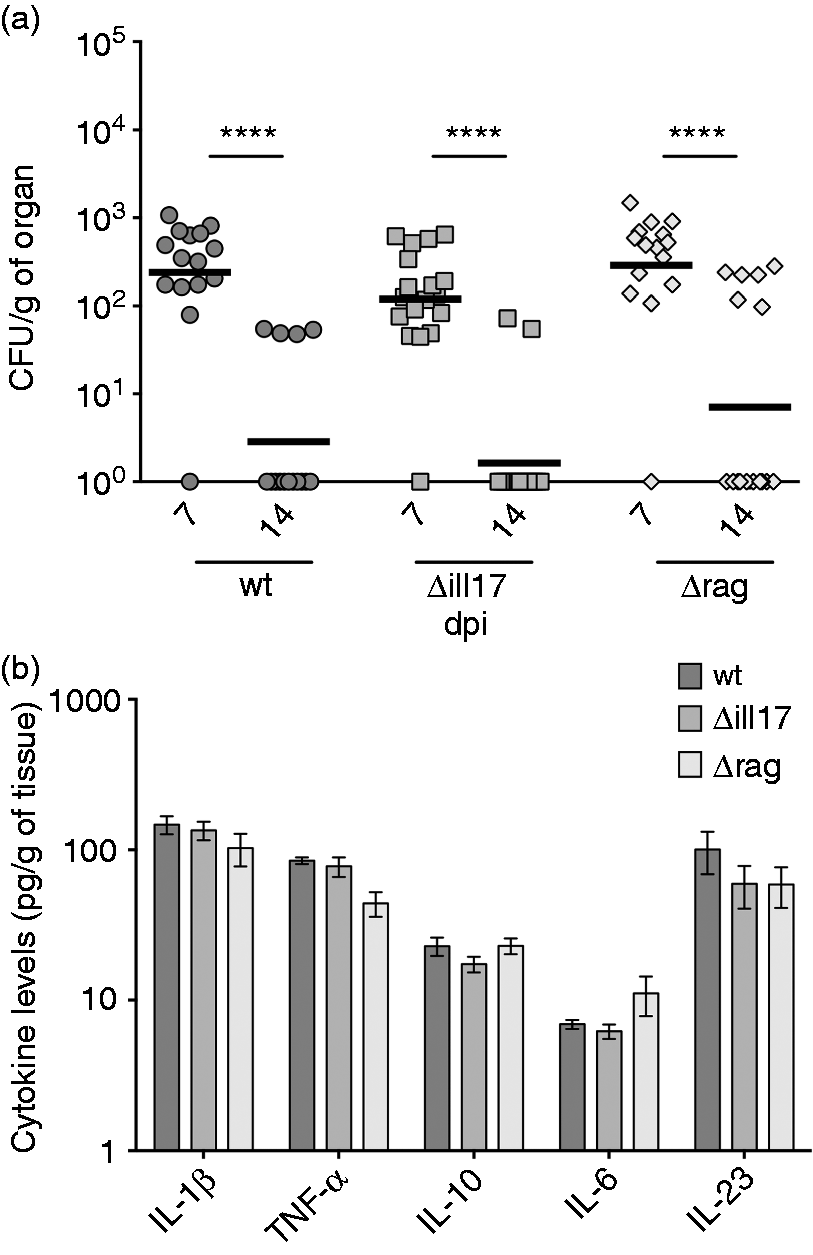
Interestingly, we also observed that mice deficient in the molecule rag2 (a protein involved in the V(D)J recombination step, essential for B and T cells development) were also competently able to clear the infection (Figure 5a). Therefore, our results suggest that adaptive immunity as whole was not essential in the infection resolution and that T. rubrum control may rely specifically on innate immune mechanisms. Indeed, as expected, no impairments in the production of innate inflammatory cytokines were observed, neither by the lack of IL-17 nor of T and B lymphocytes (Figure 5b). Therefore, we conclude that a robust inflammation, triggered by CLRs activation, is the main driving force promoting T. rubrum elimination.
Discussion
Knowledge gaps surrounding the immune recognition of T. rubrum are a barrier to understand these infections fully. Our data uncovered a remarkable role for Dectin-1 and Dectin-2 in the immunity to this pathogen.
Sato et al. reported that Dectin-1 and Dectin-2 could also recognize the ATCC 14001 strain of T. rubrum (we employed the ATCC 28188 strain) and another kind of dermatophyte, Microsporum audouinni, 23 suggesting that the mechanisms we uncovered may be applied to dermatophytosis by other etiological agents. Strain-specific factors are important variables as they were shown to affect the contribution of Dectin-1 in response to C. albicans. 15
The recently described mutations in CARD9, whose product is a key molecule in CLR signaling, 6 were associated with the development of deep dermatophytosis, a severe clinical form that disseminates to internal organs.4,5 Although these works suggest that multiple CARD9-dependent receptors could be involved, our findings have highlighted Dectin-1 and Dectin-2 as outstanding CLRs in immunity to T. rubrum. Their deficiency prevented a prompt resolution of the infection (Figure 3), owing to inadequate production of inflammatory cytokines (Figure 4). In support of our data, skin biopsies from patients with superficial dermatophytosis show higher expression of β-defensins, TLR2 and TLR4, but no changes in Dectin-2 levels, 24 hinting CLR unresponsiveness as a possible risk factor for these infections.
Although Dectin-1 and Dectin-2 recognize different pathogen-associated molecular patterns (PAMPs; the former recognizes β-glucans, the latter binds to Man), they both signal through a same pathway (Syk-dependent) 25 and can work in synergy to promote a robust activation of the signaling cascade. As we did not observe differences in cytokine production across our knockouts, we speculate that a receptor working alone cannot provide a signal with enough strength to reach the required threshold.
In vitro, the contribution of Dectin-2 seems to be less important than Dectin-1. As Dectin-2 ligands (Man-associated molecules) could be recognized by other PRRs (Man receptor, and DC-SIGN, TLR2/4), 26 the lack of the CLR could be compensated for and its contribution might be underappreciated. However, it should be acknowledged that this ‘reduced contribution’ is limited to the in vitro system. In the in vivo setting, Dectin-2 is as important as Dectin-1, reinforcing the cooperative nature of both receptors.
Supporting our hypothesis of an innate, CLR-driven inflammation as a main determinant to infection control, we observed that adaptive immunity was dispensable for T. rubrum clearance (Figure 5). Even though immunosuppression, for example with HIV infection/AIDS, is associated with a higher susceptibility to dermatophytosis,27,28 there is, in fact, no correlation, between the T-cell count and dermatophytosis incidence but to the viral load. 29 Therefore, immunosuppressed patients may become prone to dermatophyte infections due to concurrent (innate) immunological defects. Accordingly, HIV was shown to act directly on macrophages, compromising the signaling pathways of TLRs and Dectin-1. 30
Moreover, while lymphocytes from immunocompetent patients with dermatophytosis can proliferate in response to T. rubrum antigens and still show some fungicidal activity in vitro,31,32 patient-derived dendritic cells are also able to induce T-cell proliferation, indicating normal antigen presentation and co-stimulatory capabilities, 33 and suggesting that dermatophytosis is not necessarily associated with depressed or altered adaptive immunity.
Recently, Baltazar et al. showed IL-12 and IFN-γ responses are required to combat T. rubrum. 34 In our system, however, the IFN-γ response was negligible (Figure 4). Even though differences in modeling design must be considered, the system of Baltazar et al. also showed defective IL-1β and TNF-α production, blurring a direct contribution of IL-12 and IFN-γ in favor of our hypothesis of innate inflammation.
Finally, we specifically ruled out the contribution of the IL-17 response in our system. Even though IL-17 F was, indeed, preferentially induced in response to T. rubrum (Figure 4), its deficiency did not impair pathogen elimination (Figure 5).
However, it was shown that patients with adult T-cell leukemia/lymphoma are prone to superficial dermatophytosis, presenting reduced levels of circulating IL-17 and Th17 cells but increased production of IL-10 and TGF-β1. 35 However, considering that the observed correlation may not imply causation, while inborn errors in IL-17 genes and the receptor IL-17RA are well-known predisposing factors for Candida infection, 36 no similar parallel was described for dermatophytosis incidence.
Curiously, while in mucocutaneous candidiasis due to C. albicans, the IL-17 axis was critical for disease restriction, 37 in systemic infection by Candida tropicalis, IL-17 was dispensable for infection control. 38 Therefore, it is also tempting to speculate that a similar situation could occur in T. rubrum infection: in the invasive disease, as in our model, IL-17 is not as important as in the cutaneous context. Future studies focusing on skin models may shed more light on these immunological discrepancies among these different clinical forms.
As long as the IL-17 family is composed of multiple members, 39 it is also feasible that other IL-17 cytokines could play a more relevant role. Nonetheless, one of these understudied members, IL-17 C, was shown to be dispensable for C. albicans control in models of oropharyngeal, cutaneous and disseminated infection. 40
In conclusion, we defined Dectin-1 and Dectin-2 as key players in immunity to T. rubrum, driving inflammation and pathogen clearance, while adaptive immunity and IL-17 were not required (Figure 6).
Proposed model for the role of Dectin-1 and Dectin-2 in the response to T. rubrum. Trichophyton rubrum hyphae are recognized by Dectin-1 and Dectin-2 in dendritic cells, triggering production of inflammatory cytokines, mainly IL-1β and TNF-α. This inflammation is able to promote clearance of the pathogen in vivo, without the involvement of lymphocytes. Even though IL-17 is induced, it is not essential for infection resolution.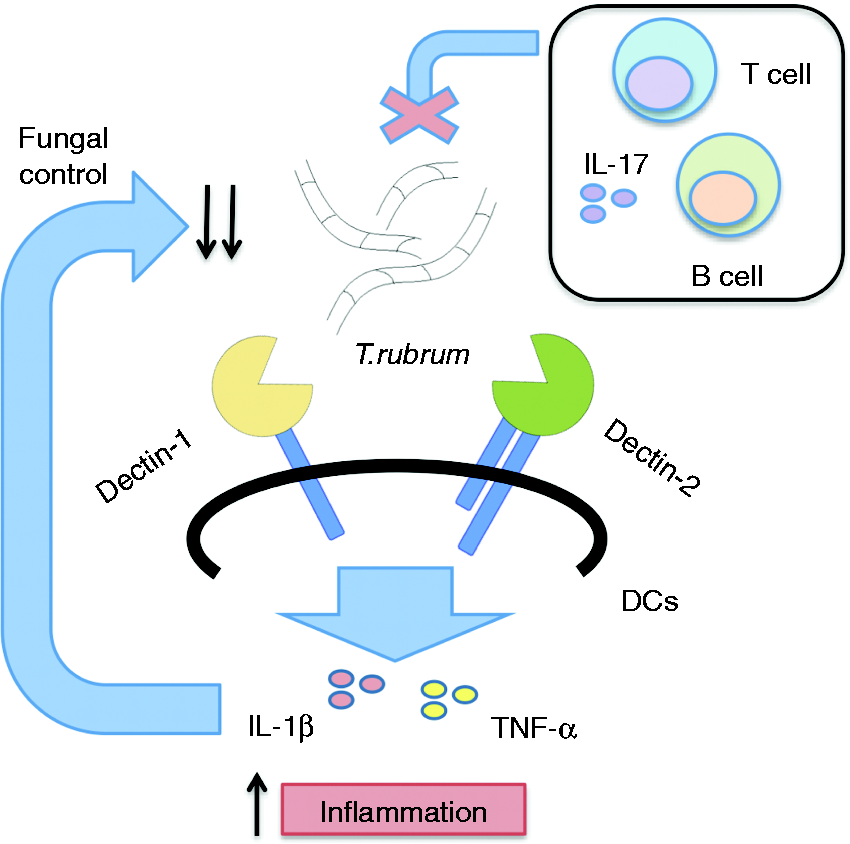
While deficiencies in other PRRs also impair the fungal control and promote the infection—as we and others have previously shown for mannose receptor and DC-SIGN, 41 the NLRP3 inflammasome 17 and DC-HIL 42 —here we stress the great and cooperative contribution of Dectin-1 and Dectin-2 against this pathogen. Collectively, these works indicate that the immunity to T. rubrum is a complex, rather than a straightforward, framework that relies on multiple inputs for appropriate control and where individual interferences may compromise its performance.
Notably, secreted mannans from T. rubrum are well known for their immunomodulatory properties, despite being highly antigenic in patients. 43 It is conceivable that mannan-producing strains would employ the sugar production as a strategy to avoid CLR recognition and establish a chronic infection. Further studies translating our findings to the human system may open the venue to development of improved therapeutic approaches.
Supplemental Material
Supplemental material for Dectin-1 and Dectin-2 promote control of the fungal pathogen Trichophyton rubrum independently of IL-17 and adaptive immunity in experimental deep dermatophytosis
Supplemental Material for Dectin-1 and Dectin-2 promote control of the fungal pathogen Trichophyton rubrum independently of IL-17 and adaptive immunity in experimental deep dermatophytosis by Fabio SY Yoshikawa, Rikio Yabe, Yoichiro Iwakura, Sandro R de Almeida and Shinobu Saijo in Innate Immunity
Footnotes
Acknowledgements
We thank M. Morimoto for technical assistance and T. Suzuki for animal care support.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: F. Y. was supported by fellowship from CAPES Foundation (Grant Number 99999.004279/2014-00). This work was supported by JAPS KAKENHI Grant Numbers 2629304, 16K15271 to S. S., 16K19147, 16K19147 to R.Y., and the Uehara Memorial Foundation Grant-in-Aid for Young Scientists (B) to R. Y.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.