
Abstract
The Gram-positive bacterium Enterococcus faecalis can cause life-threatening infections and is resistant to several commonly used antibiotics. The type II fatty acid pathway in bacteria is discussed as a potential target for antimicrobial therapy. However, it was shown that inhibition or deletion of its enzymes can be rescued in Gram-positive bacteria by supplementation with fatty acids. Here we show that by deletion of the fabN gene, which is essential for unsaturated fatty acid (UFA) synthesis in E. faecalis, growth is impaired but can be rescued by supplementation with oleic acid or human serum. Nonetheless, we demonstrate alterations of the UFA profile after supplementation with oleic acid in the ΔfabN mutant using a specific glycolipid. In addition, we demonstrate that cytokine release in vitro is almost abolished after stimulation of mouse macrophages by the mutant in comparison to the wild type. The results indicate that fabN is not a suitable target for antimicrobials as UFA auxotrophy can be overcome. However, deletion of fabN resulted in a decreased inflammatory response indicating that fabN and resulting UFA synthesis are relevant for virulence.
Introduction
The type II fatty acid synthesis (FASII) pathway is discussed as potential target for antimicrobial development,1,2 as there are significant structural differences to the type I fatty acid synthesis pathway in mammals. 2 The antimicrobials platencin and platensimycin inhibit the fatty acid acyl carrier protein synthase II and the fatty acid acyl carrier protein synthase III of the FASII pathway, and have been tested in preclinical trials against methicillin-resistant Staphylococcus aureus strains. 3 Platensimycin is also discussed as a potential anti-tuberculosis drug. 3 The selective antistaphylococcal enoyl-acyl carrier protein (ACP) reductase (FabI) inhibitor AFN-1252 is even already under investigation in human trials. 4 The fabN gene from Enterococcus faecalis encodes for a 3-hydroxyacyl-[acyl-carrier-protein]-dehydratase (3-hydroxyacyl-ACP-dehydratase) (EC: 4.2.1.59) that is also an enzyme in the FASII pathway. The FabN enzyme was first described by Wang and Cronan, 5 and defined as a bifunctional dehydratase/isomerase. It catalyzes the first step in the synthesis of unsaturated fatty acids by dehydration of β-hydroxydecanoyl–ACP and subsequent isomerization to cis-3-decenoyl–ACP, 6 which is a substrate for further unsaturated fatty acid (UFA) elongation. 7 In Streptococcus pneumoniae and Streptococcus mutans, the functional closely related FabM is an isomerase, essential for UFA synthesis in these pathogens. 8 Growth was impaired when fabM was deleted but was restored by the addition of UFAs to the media or by functional replacement of the deleted fabM by fabN from E. faecalis. 8 This strategy was also applied for the fabA deletion mutant in the Gram-negative bacterium Escherichia coli. 5 FabA is the essential enzyme for UFA synthesis in E. coli and catalyzes the isomerase/dehydratase reaction to cis-3-decenoyl–ACP.5,9 Upon replacement of fabA with fabN from E. faecalis, only small amounts of UFAs were produced, owing to substrate competition with the enoyl reductase FabI. Altogether, these findings demonstrate the versatility of FabN for the catalysis of unsaturated fatty acid synthesis in different bacterial species. We hypothesized that by deleting fabN, growth of E. faecalis would be impaired, owing to the described potential essentiality of the enzyme for UFA synthesis. Additionally, we wanted to find out whether the host inflammatory response was altered in the mutant. We have shown before that E. faecalis 12030 is a strong biofilm producer,10,11 and that glycolipids are involved in this process.11,12 In addition, we have detected a strong inflammatory reaction in vitro and in vivo, when the glycosyltransferase BgsA, transferring the second Glc moiety yielding diglycosyl-diacylglycerol (DGlcDAG) was deleted and the glycolipid monoglycosyl-diacylglycerol (MGlcDAG), containing unsaturated fatty acids, accumulated.11,13 Therefore, we wanted to investigate whether there is a connection between microbial UFAs and the immune response.
Materials and methods
Bacterial strains, plasmids and culture conditions
The pMAD Gram-positive, temperature-sensitive mutagenesis shuttle vector has been described previously. 14 Enterococcus faecalis 12030 strain was grown in tryptic soy broth (TSB; Carl Roth, Karlsruhe, Germany) medium or on TSA plates (Carl Roth) at 37℃ for 18 h. 15 For growth of E. faecalis 12030ΔfabN TSB media or TSA plates were supplemented with 0.1 mM oleic acid (Sigma Aldrich, St. Louis, MO, USA). When required, medium was supplemented with 50 µg/ml erythromycin (Carl Roth). Escherichia coli XL-1-blue (Invitrogen, Carlsbad, CA, USA), containing pMAD, was grown in lysogeny broth supplemented with 100 µg/ml ampicillin (Carl Roth) at 30℃ with agitation (200 rpm) for 48 h. For blue/white selection, agar plates were supplemented with 80 µg/ml X-gal (Applichem, Chicago, IL, USA).
Construction of ΔfabN
A non-polar deletion of a portion of fabN (ef 0284 in E. faecalis 12030), encoding for the enzyme (R)-3-hydroxymyristoyl–ACP dehydratase, was created using the method described previously.
16
Genomic DNA was isolated from E. faecalis 12030 via the MasterPure Gram Positive DNA Purification Kit (Illumina, Madison, WI, USA). Primer pair 0284_P1_fw_BamHI (CTCACCA
Growth kinetics
Overnight cultures of E. faecalis 12030 and the E. faecalis 12030ΔfabN mutant were adjusted to an OD600 of 0.1 in TSB medium without or with supplementation with 0.1 mM oleic acid (TSBO). For determination of growth by the mutant in human serum, medium was prepared out of 50% TSB and 50% human serum (filtered sterile). As before, 0.1 mM oleic acid was added to half of the samples. Again, the starting OD600 was adjusted with overnight cultures, grown for 18 h at 37℃, to 0.1. The cultures supplemented with human serum or only oleic acid were grown at same conditions at 37℃ at 200 rpm. OD600 was measured hourly over a time span of 6 h until stationary phase was reached.
Membrane lipid extraction
Crude lipid extraction was done following the method of Bligh and Dyer, 19 with a few modifications. Enterococcus faecalis 12030 or E. faecalis 12030ΔfabN were incubated for 18 h at 37℃ without agitation in TSB or TSBO, respectively. The cultures were spun down (1800 g, 20 min, 4℃) and the cell pellet was re-suspended in 20 ml NaAc-buffer [0.1% sodium acetate (w/v); pH 4.7] and pelleted again. The cell pellet was re-diluted in 10 ml NaAc buffer and glass beads with a diameter of 0.1 mm (Carl Roth) were added 1:1 to the suspension. With a BeadBeater (Glenn Mills, Clifton, NJ, USA) or, alternatively, by vortexing, cells were disrupted. After the glass beads had settled or had been spun down (42,000 g, 20 min, 4℃) the cell suspension was transferred to a new tube and centrifuged (7000 g, 20 min, 4℃). The supernatant was discarded and NaAc buffer, methanol and chloroform were added in ratios of 0.8:2:1 to the cell pellet and incubated for lipid extraction for 2 h at room temperature (RT; 22℃) while stirring. The suspension was centrifuged in polytetrafluoroethylene tubes (ThermoFisher Scientific, Waltham, MA, USA) (1800–7000 g, 15 min, 4℃). The supernatants, containing the extracted lipids, were combined and the cell pellet was re-diluted using the same ratio as before, stirred for an additional hour and separated again by centrifugation. The supernatants were combined and chloroform and NaAc buffer were added in volumes of 1:1:1 to the supernatant, mixed briefly and the organic and aqueous phase were separated again by centrifugation for 15 min. The lower organic phase containing the lipids was combined and separated again by centrifugation until only the organic solvent, containing the lipids, remained. By evaporation, using a rotary vacuum evaporator, the chloroform was separated from the extract. It was important that the temperature did not exceed 40–50℃, to decrease oxidation of double bonds. Subsequently, the lipid extract was dried under a fluent stream of nitrogen. Until use, lipids were stored in chloroform or under nitrogen atmosphere at −20℃.
RAW 264.7 mouse macrophage stimulation
For determination of cytokine formation, RAW264.7 mouse macrophages (a generous gift from the laboratory of Philip Bufler, Children’s Hospital, Munich, Germany) were seeded at a density of 1 × 105 cells/ml in 24-well dishes in DMEM (high Glc, GlutaMAX™, supplemented with 10% FCS and 100 U/ml Pen/Strep). The adherent cells were washed once with PBS and stimulated with sterile filtered (0.22 µm) supernatants of overnight cultures from mutant and wild type, adjusted to a protein concentration of 300 µg/ml by photometric determination. Cultures were incubated for 16 h at 37℃ in a 5% humidified CO2 environment. Stimulation assays were performed at cell passages 12–15. The TLR2 agonist Pam2CSK4 (R&D Systems, Minneapolis, MN, USA) was used as positive control. Cytokines were measured by ELISA using commercially available kits (eBioscience, San Diego, CA, USA). Statistical significance for two-way comparisons was determined by an unpaired t-test as indicated.
Visualization of lipids
Lipid extracts, diluted in chloroform, were applied onto a thin-layer chromatography (TLC) plate (0.1 mM Silica gel 60 F254nm; Merck, Darmstadt, Germany), which was developed in a TLC development chamber in CHCl3/MeOH/H2O [65:25:4; (v/v/v)] running buffer. To visualize total lipid extract, the plate was stained with molybdenum [5% H2SO4 (97%) (v/v); 0.1% Ce(SO4) × 4 H2O (w/v); 5% (NH4)6Mo7O24 × H2O (w/v)], 20 air-dried and developed at 150℃ for 5 min. For visualization of glycolipids, the plate was stained with Molisch’s reagent [82% MeOH (v/v), 10% H2SO4 (97%) (v/v); 3.2% α-naphthol C10H8O (w/v)] and developed at 150℃ for 5 min.
Purification of MGlcDAG via silica gel column chromatography
A glass column (NS 14/23 porosity: 0; Carl Roth) was filled with 7–10 ml silica gel (0.04–0.063 mm silica gel 60; Merck). The lipid extract, solved in chloroform, was added slowly onto the dry, even surface of the silica gel. By increasing the polarity of the eluent via decreasing the CHCl3/MeOH (v/v) ratio (100:0; 97:3; 95:5; 92:8; 90:10; 50:50) fractionation of the lipids occurred. The fractions were each eluted with a total volume of 100 ml. The eluents were removed again by rotary evaporation. Via TLC the combined fractions were analyzed for purity.
Determining the fatty acid composition of MGlcDAG
The fatty acid composition of MGlcDAG of the wild type and the ΔFabN mutant was determined after methanolysis (2 M HCl/MeOH, 85℃, 2 h), acetylation (85℃, 10 min) and detection by GC/MS. GC/MS analyses of all samples were performed on an Agilent Technologies 7890A gas chromatograph equipped with a dimethylpolysiloxane column [HP Ultra 1, 12 m × 0.2 mm × 0.33 µm film thickness and 5975C series MSD detector with electron impact ionization (EI) mode under autotune condition at 70 eV (Agilent, Santa Clara, CA, USA)]. The temperature program was 70℃ for 1.5 min, then 60℃ min−1 to 110℃ and 5℃ min−1 to 320℃ for 10 min. A reference probe (Bacterial Acid Methyl Ester Mix; Sigma Aldrich) with known lipid composition and elution profile was measured under the same conditions.
Preparation of picolinyl esters
For the exact determination of the position of the double bond in 18:1 fatty acid, fatty acids of MGlcDAG from the wild type and ΔFabN mutant were derivatized to 3-pyridylcarbinol (‘picolinyl’) esters. 21 The fatty acids were released (1 M NaOH–MeOH, 1 h, 85℃), recovered in CHCl3, treated with trifluoroacetic anhydride (1 h, 50℃) and subsequently with 20% (w/v) 3-pyridinemethanol solution in tetrahydrofuran (1 h, 50℃) and injected in GC/MS as described above.
Biofilm formation assay
Biofilm formation was measured as described previously. 22 Briefly, TSB media supplemented with 1% Glc (w/v) and 0.1 mM oleic acid (TSBGO) was inoculated and incubated at 37℃ for 18 h. Polystyrene tissue culture plates were filled with 198 µl TSBGO media and 2 µl of the overnight culture. The plate was incubated 18 h at 37℃. After the incubation period OD630 was measured. Supernatants were discarded and wells were washed twice with 200 µl PBS. The plate was dried for 1.5 h at 50–60℃ and subsequently 100 µl crystal violet solution (Sigma Aldrich) was added and incubated for 2 min at RT. The solution was discarded, the plate rinsed thoroughly with tap water and dried again at 50–60℃ for 15 min. OD630 was measured and normalized to growth with the biofilm index [biofilm index = OD biofilm × (0.5)/(OD growth)].
Results
Deletion of EF0284
The described fabN (ef 0284) gene from E. faecalis V583 shares 99.08% nucleotide identity to its homologue gene in E. faecalis 12030. 5 We hypothesized that by deleting fabN, E. faecalis 12030 would not be able to synthesize UFAs including vaccenic acid (18:1 Δ 11 ). To characterize the role of fabN, we created a non-polar deletion mutant using targeted mutagenesis. 16 Deletion mutants could only be successfully isolated by supplementation of TSB media with 0.1 mM oleic acid during the second cross-over event to overcome UFA auxotrophy. Without supplementation the fabN mutant had a pronounced growth defect and formed small colonies on tryptic soy agar plates approximately two-thirds smaller than the corresponding wild type.
Deletion of fabN results in UFA auxotrophy
Growth of the mutant in un-supplemented TSB resulted in a maximal (±SEM) growth to an OD600 of 0.52 ± 0.01, whereas maximum growth for the wild type was three times higher with an OD600 of 1.73 ± 0.03 (Figure 1). With addition of 0.1 mM oleic acid the growth defect was partly overcome. Maximal growth for ΔfabN doubled to an OD600 of 1.13 ± 0.04. However, growth of the wild type was not altered by supplementation of oleic acid (OD600 1.73 ± 0.01 vs. 1.73 ± 0.03 in the un-supplemented culture) (Figure 1). This shows that fabN is essential for growth, when no exogenous UFAs are available. As was also shown previously,
23
E. faecalis is capable of using exogenous UFAs.
Growth of wild type (wt) and E. faecalis 12030ΔfabN strain in TSB or TSBO, respectively. Values are represented as averages ± SEM (n = 3). Growth curve measurements were performed independently in triplicate.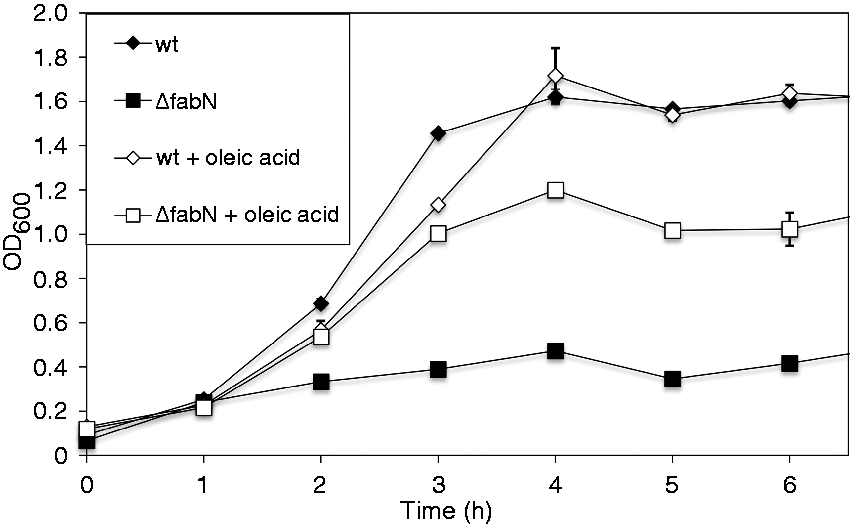
UFA auxotrophy can be overcome by growth in human serum
UFA auxotrophy in deletion mutants of Streptococcus agalactiae or by inhibition of the FASII pathway of Gram-positive pathogens was overcome under in vivo conditions by adding human serum to the growth media. Human serum contains usually about 1.88 mM oleic acid, which is the third most abundant fatty acid.
24
Therefore, we tested if growth of the fabN deletion mutant could also be restored by adding human serum (Figure 2). Growth of the ΔfabN mutant increased to similar levels (1.24 ± 0.02 with and 1.33 ± 0.11 without oleic acid) as the wild type (1.30 ± 0.11 with and 1.20 ± 0.01 without oleic acid). Altogether, these results show that the effect of fabN deletion and subsequent UFA auxotrophy can be overcome under in vivo conditions.
Growth of wild type (wt) and E. faecalis 12030ΔfabN strain in 50% (v/v) human serum in TSB or TSBO, respectively. Values are represented as averages ± SEM (n = 3). Growth curve measurements were performed independently in triplicate.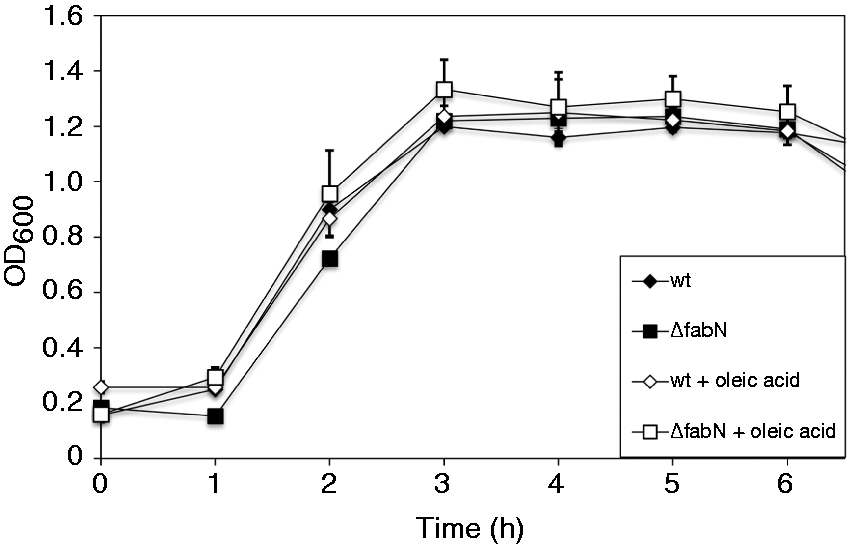
Deletion of fabN almost completely abolishes induction of TNF-α in RAW 264.7 mouse macrophages
To evaluate the biological effects of the deletion and the potentially changed lipid membrane composition, we stimulated mouse macrophages with supernatant from the wild type and mutant adjusted to same protein concentrations of 300 µg/ml (Figure 3). As in our previous work we observed that E. faecalis 12030 sheds cell membrane lipids into the media (personal observation),
25
we argued that changed lipid content in the supernatant would resemble changed membrane lipid composition. After 16 h, we measured the release of the cytokine TNF-α in the cell culture supernatant. TNF-α production in RAW264.7 cells was almost completely abrogated after stimulation with the mutant and, overall, sevenfold lower than the wild type strain. Therefore, our data suggest that impairment of UFA synthesis reduces the capability of culture supernatants to induce TNF-α production by macrophages. This indicates that microbial unsaturated acids potentially play a role in the induction of an inflammatory response.
Induction of TNF-α in RAW 264.7 mouse macrophages by E. faecalis 12030 wild type (wt) and ΔfabN mutant. Macrophages were stimulated with cell-free, sterile E. faecalis supernatants adjusted to a protein concentration of 300 µg/ml. After 16 h of incubation, supernatant from macrophage culture was analyzed for TNF-α by ELISA. Pam2CSK4 was used as positive control. Data represent mean ± SEM of triplicates. ***P < 0.001 E. faecalis 12030 wt vs. E. faecalis 12030 ΔfabN with t-test for unpaired comparisons. The assay was performed in duplicate independently.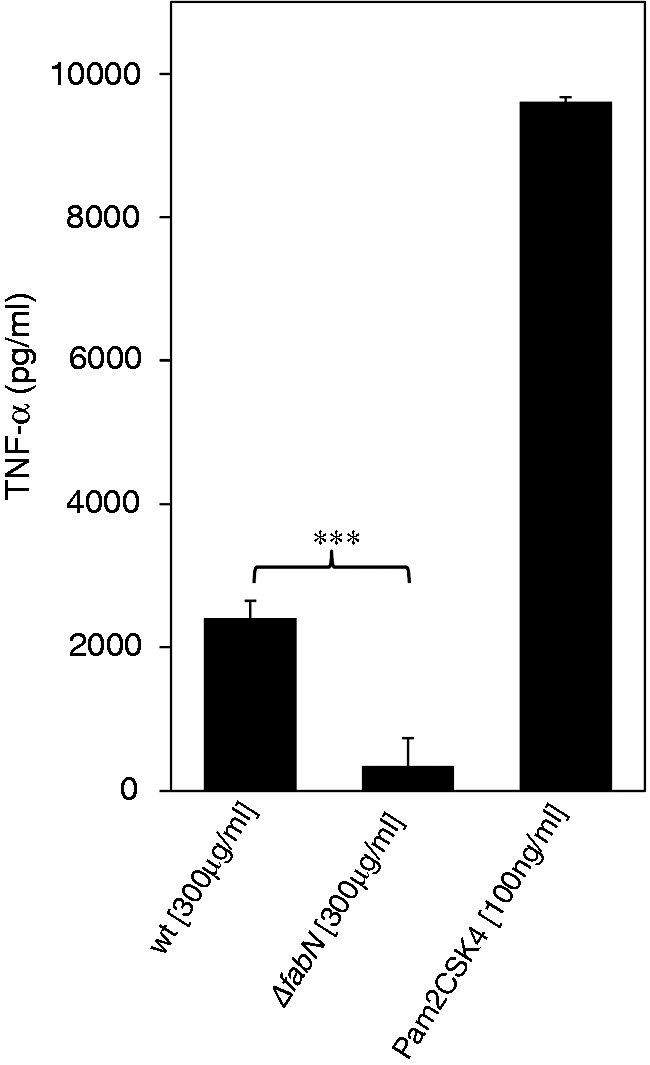
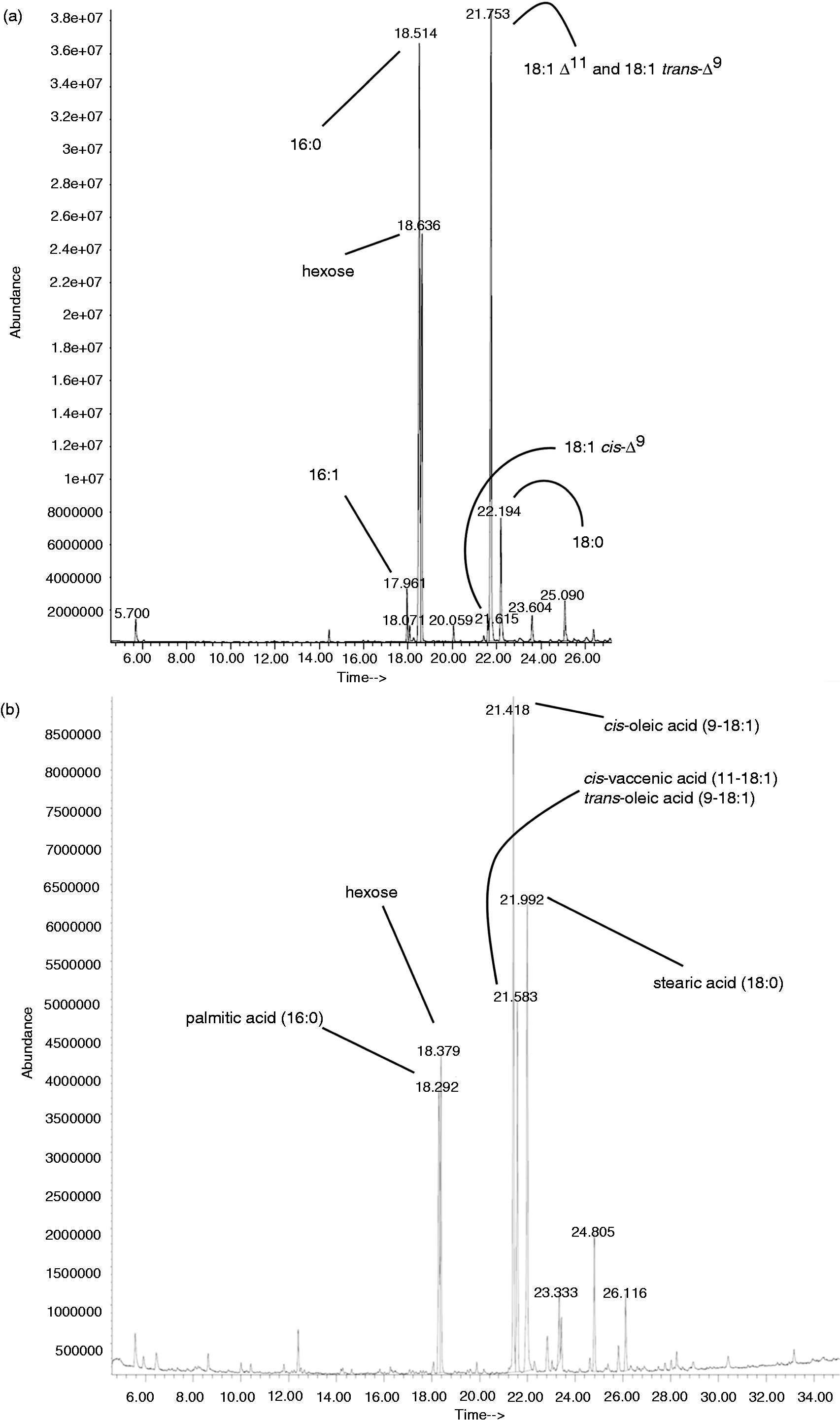
Fatty acid composition of MGlcDAG
In order to prove to what extent the mutation in the fabN gene changed the chemical composition of the fatty acids using the example of a single glycolipid, total lipids of the wild type and the ΔfabN mutant were extracted and fractionated as described. The α-naphthol stain (Figure 4B, D) shows that a glycolipid with an Rf ∼ 0.75 was eluted with 95:5 (Figure 4A, B, lane 3) or 92:8 CHCl3/MeOH (Figure 4C, D, lane 4) for the wild type or ΔfabN mutant, respectively, and was identified as MGlcDAG compared with the lipid profile of E. faecalis 12030ΔbsgA strain overproducing MGlcDAG (Figure 4A, B, lane 1).
11
The composition of the fatty acids of MGlcDAG from the wild type, as determined by GC/MS with the use of authentic standard of bacterial acid methyl ester (Sigma Aldrich), revealed the presence of glycerol, hexose, 16:1, 16:0, 18:1 and 18:0 (Figure 5A). This analysis, however, did not allow us to differentiate completely between 18:1 Δ
9
and 18:1 Δ
11
, as methyl esters of these fatty acids partially elute with the same retention time (21.753 min). Thus, picolinyl ester derivatives were prepared (data not shown) and it was shown that in the glycolipid from wild type, mainly Δ
11
of 18:1 was present, with only traces of 18:1 Δ
9
. The exact position (sn-1 or sn-2) of the different fatty acids on the glycerol backbone remains to be elucidated. The methanolysis of MGlcDAG from ΔfabN showed the presence of glycerol, hexose, 16:0, 18:1 Δ
9
, 18:1 Δ
11
and 18:0 (Figure 5B). The picolinyl derivates (data not shown) revealed the presence of both forms of 18:1, with the ratio 2:1 18:1 Δ
9
and 18:1 Δ
11
, respectively. Taken together the analyses showed that by deletion of fabN the composition of unsaturated fatty acid structures in E. faecalis changed (detection of supplemented 18:1 Δ
9
); however, the mutant still possessed a decreased amount of 18:1 Δ
11
.
TLC of fractionated lipid extracts of E. faecalis 12030 wild type (A, B). From silica gel column eluted fractions with CHCl3 (100) (lane 2), CHCl3/MeOH (95:5) (lane 3), CHCl3/MeOH (90:10) (lane 4), CHCl3/MeOH (85:15) (lane 5), CHCl3/MeOH (80:20) (lane 6) and CHCl3/MeOH (50:50) (lane 7). Twenty µg crude lipid extract of E. faecalis 12030ΔbgsA accumulating MGlcDAG (lane 1) was loaded as ladder. Twenty µg DGlcDAG (lane 8) as positive control. Fractions are concentrated, applied to a TLC plate, developed in CHCl3/MeOH/H2O [65:25:4 (v/v/v)] and visualized with (A) molybdenum or (B) α-naphthol. Arrows are indicating the glycolipid bands and the cardiolipin contamination. MGlcDAG; Rf ∼ 0.74 (a), DGlcDAG; Rf ∼ 0.49 (b) and cardiolipin (verified by MS) (c). TLC of fractionated lipid extracts of E. faecalis 12030ΔfabN (C, D). Fractions eluted with CHCl3 (100) (lane 1), CHCl3/MeOH (97:3) (lane 2), CHCl3/MeOH (95:5) (lane 3), CHCl3/MeOH (92:8) (lane 4), CHCl3/MeOH (90:10) (lane 5) and CHCl3/MeOH (50:50) (lane 6). Fractions were concentrated, applied to a TLC plate, developed in CHCl3/MeOH/H2O [65:25:4 (v/v/v)] and visualized with molybdenum (C) or α-naphthol (D). Arrows are indicating the MGlcDAG spots. GC/MS chromatogram of fatty acid methyl ester of MGlcDAG purified from wild type (a) and methyl esters MGlcDAG purified from the E. faecalis 12030ΔfabN mutant (b).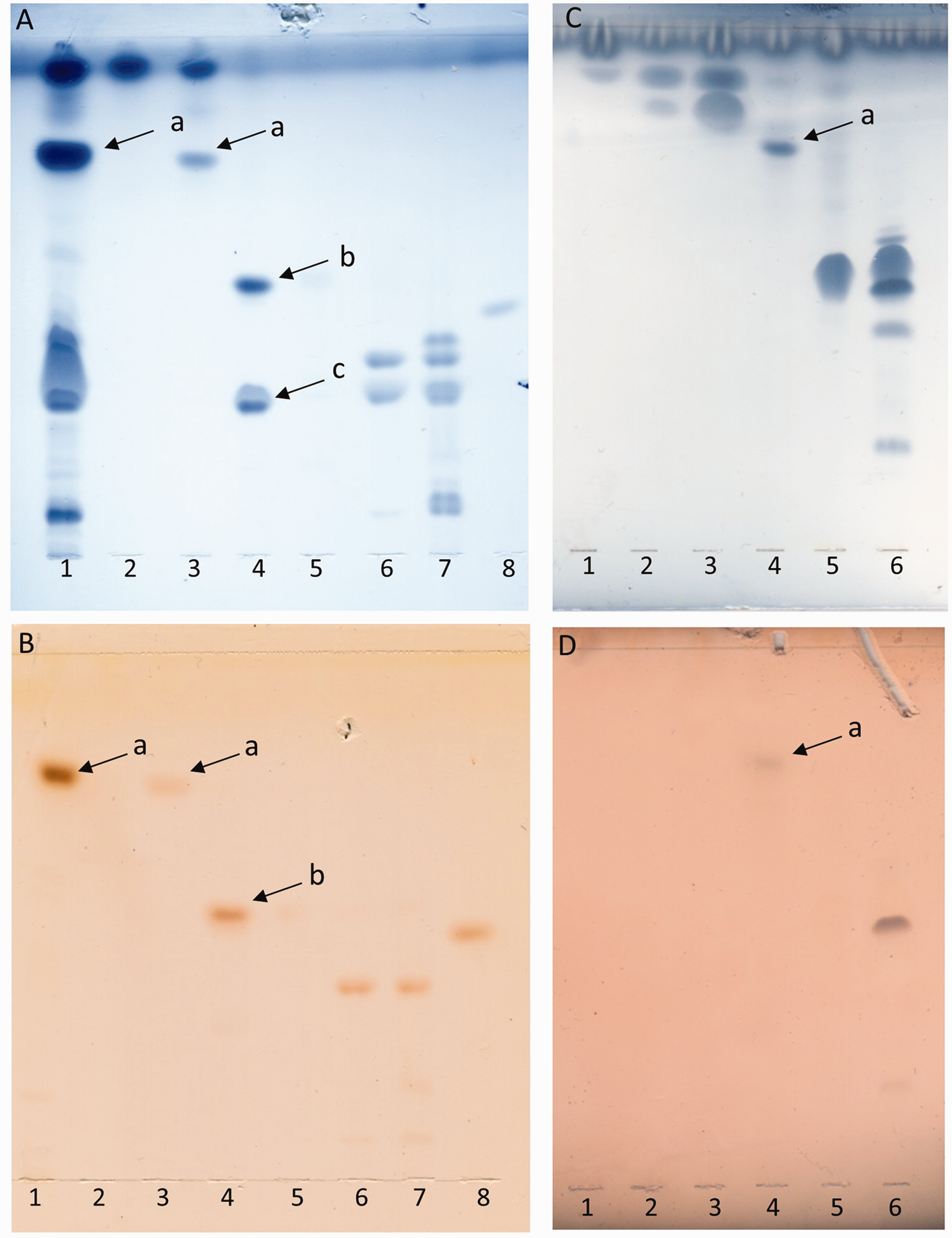
Biofilm formation of ΔfabN
Enterococcus faecalis mutant 12030ΔfabN did not show significant differences compared with the wild type in regard to biofilm formation on plastic surfaces (Figure 6).
Biofilm formation on plastic surfaces by E. faecalis 12030 wild type (wt) and ΔfabN grown in TSBGO for 18 h at 37℃. Biofilm index was calculated correlating growth to biofilm [biofilm index = OD biofilm × (0.5)/(OD growth)]. Data represent mean ± SEM (n = 6). ns: not significant; E. faecalis 12030 wild type vs. E. faecalis 12030ΔfabN with t-test for unpaired comparisons. The assay was performed in duplicate independently.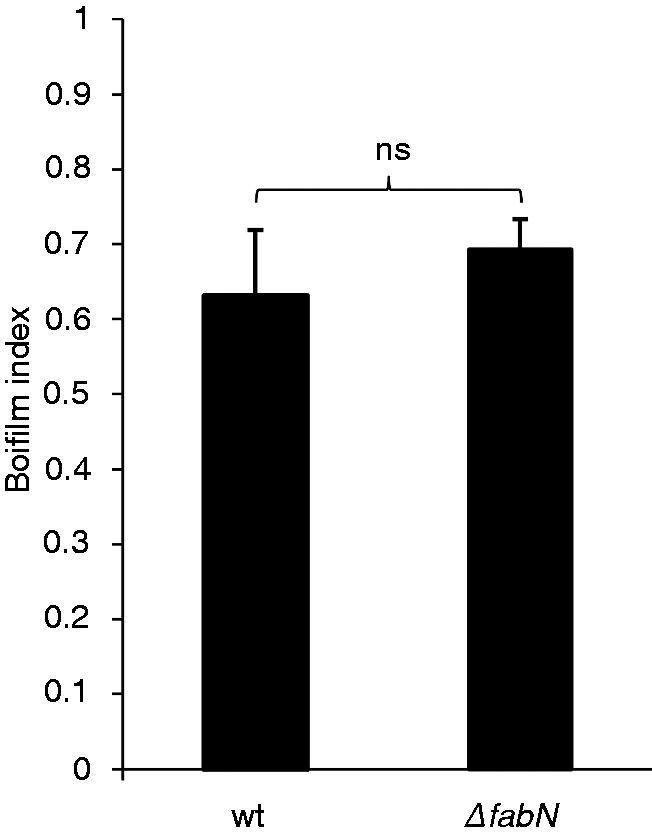
Discussion
Here we analyzed how deletion of the enzyme FabN, catalyzing the first step in unsaturated fatty acid synthesis in E. faecalis, affects growth kinetics, the inflammatory response of mouse macrophages and composition of fatty acids, using the glycolipid MGlcDAG as example. The deletion of fabN resulted in UFA auxotrophy, which was partly overcome by supplementation of the culture medium with the exogenous unsaturated fatty acid oleic acid or human serum. This phenotype was also previously reported for S. mutans, S. pneumoniae and S. agalactiae fabM-null mutants.8,26 It has been previously reported that UFA auxotrophy caused by deletion or inhibition through antimicrobials of different enzymes in the FASII pathway of Gram-positive pathogens could be overcome by supplementation with unsaturated fatty acids. 26 Our findings corroborate previous studies by Zhu et al., 27 who suggested that enzymes of the FASII pathway in E. faecalis are not a suitable target for antimicrobials. They reported that UFA auxotrophy in the enoyl-ACP reductase (FabI) deletion mutant was rescued by supplementation with oleic acid. 27 Similar observations were made in S. pneumoniae, in which exogenous fatty acids could replace de novo synthesized fatty acids. 28 Nevertheless, we could demonstrate that deletion of fabN resulted in a reduced inflammatory response by macrophages. Although growth cannot be inhibited by antimicrobials targeting the enzyme, they might possibly reduce the inflammatory potency of the pathogen. Targeting bacterial virulence (e.g. with Abs neutralizing toxins) reduces the pressure for drug-resistant mutations and protects the host microbiota against detrimental changes. 29 However, there are only a few antimicrobials that inhibit fabN homologs, which, to our knowledge, have not yet been tested in enterococci.1,30,31 Integration of UFAs into immunogenic compounds have an influence on their inflammatory potency. It has been shown recently that incorporation of exogenous unsaturated fatty acids into lipoproteins of S. aureus was connected to an enhanced inflammatory response by human monocytes and HEK–TLR2 cells. 32 Lipoproteins are the most pro-inflammatory compounds in the Gram-positive cell wall.33,34 Additionally, in some glycolipids the importance of the double bond of the fatty acids for recognition by iNKT cells and subsequent initiation of an inflammatory response was reported.35,36 Therefore, an altered unsaturated fatty acid profile in the ΔfabN mutant likely affects its inflammatory characteristics. We hypothesized that by deletion of fabN the structure of the glycolipid MGlcDAG would change from vaccenic acid to the supplemented oleic acid. We found recently that an E. faecalis 12030 deletion mutant, accumulating the glycolipid MGlcDAG, showed a strongly increased inflammatory phenotype due to an up-regulated lipoprotein content, 13 making the glycolipid an interesting target for analysis of a potential altered lipid composition. We found, in fact, a changed lipid profile for the glycolipid synthesized by the ΔfabN mutant, with oleic acid and palmitic acid being the most prominent fatty acids, but there was still vaccenic acid present. Analysis of the fatty acid composition of fabM mutants in S. pneumoniae and S. mutans showed that vaccenic acid is not detected when oleic acid is supplemented. 8 Taken together, these results suggest that in E. faecalis, FabN is not the only enzyme that is capable of synthesizing 18:1 Δ 11 . Another enzyme, designated as FabZ (ef2878) with 58.7% identical residues to FabN, 5 was identified in E. faecalis V583. Wang and Cronan showed that replacement with fabN by the functional homolog dehydratase/isomerase in the E. coli ΔfabA mutant restored unsaturated fatty acid synthesis. However, FabZ from E. faecalis could not restore synthesis of unsaturated fatty acids in the fabA deletion mutant, but traces of unsaturated fatty acids could be detected. Therefore, FabZ seems to be capable of synthesizing unsaturated fatty acids but with a rather low activity. The active site residues of FabA 8 are mostly conserved in FabN and FabZ. Domain swapping of FabN and FabZ 6 showed that β-sheets direct the form of the α-helix forming the substrate-binding tunnel. β-Sheets in FabN are differently positioned than in FabZ and are placing the substrate in the suitable position for isomerization. 6 Based on these findings it is conceivable that FabZ is able, with a rather low activity and affinity, to synthesize traces of unsaturated fatty acids.
In conclusion, we could demonstrate that the deletion of fabN in E. faecalis results in UFA auxotrophy, which can be complemented by supplementation either with oleic acid or human serum. Deletion of fabN results in a changed lipid profile in the glycolipid MGlcDAG, although the UFA vaccenic acid was still detected, suggesting another enzyme is capable of synthesizing vaccenic acid with lower activity than FabN. Cytokine release by stimulation of mouse macrophages with the mutant was almost completely abrogated. Hence, although UFA auxotrophy can be overcome and FabN is not a target for antimicrobials killing E. faecalis, inhibition of FabN could result in a less pathogenic phenotype and be an objective for anti-virulence strategies, which are a promising alternative to treat bacterial infections. 29 Further in vivo studies are required to explore the suitability of FabN and orthologue enzymes as targets for anti-virulence strategies.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.