
Abstract
Enterococcal surface protein (Esp) is encoded on a pathogenicity island in Enterococcus faecalis and E. faecium and is involved in biofilm formation and binding to epithelial cells. In this study, using Esp-expressing E. faecalis MMH594 and its isogenic Esp-deficient strain, as well as purified Esp, we show that Esp is sufficient for activation of NF-κB and the subsequent production of pro-inflammatory cytokines IL-1β and TNF-α in macrophages in vitro. In a mouse peritonitis model, we also show that mice infected with Esp-expressing E. faecalis showed comparatively higher levels of cytokines TNF-α, IL-1β and IL-6 in peritoneal fluid, and IL-6 in serum. Moreover, neutrophil infiltration and tissue damage in the liver was higher in the mice infected with the Esp-expressing strain compared with mice infected with the Esp-deficient mutant. These results add Esp to the growing list of enterococcal virulence factors that can modulate inflammation during infection and has implications for enterococcal pathogenesis.
Introduction
Enterococci are part of the commensal flora of the gastrointestinal tract of most metazoans, where they occur in numbers of approximately 104–108 organisms per gram of feces. In addition to the well-recognized intrinsic and acquired antimicrobial resistance within this genus, these organisms are also endowed with virulence traits that make them an opportunistic pathogen. 1 Over the last couple of decades enterococci have thus emerged as a leading cause of antibiotic-resistant infections and now rank among the most common nosocomial pathogens infecting the bloodstream, surgical sites and urinary tract. 2 Despite the general notion that these organisms possess low virulence, enterococci are frequently the causative organism identified with catheter-associated urinary tract infections (UTIs) and infective endocarditis. Although the independent capability of this organism to cause sepsis has been questioned earlier,3–6 recent reports have shown that Enterococcus faecalis infection can lead to severe sepsis or septic shock in immunocompromised patients, burn patients and thermally injured mice, by modulation of the host systemic inflammatory response.7–9
Lipoteichoic acid (LTA) has been reported earlier as an enterococcal PAMP recognized by host TLR2 to induce a signaling cascade that culminates in the activation of transcription factors and induction of pro-inflammatory cytokine expression.10,11 More recent studies have, however, shown that LTA is not a TLR2 stimulant, and such perceived activation is a result of small amounts of LTA-associated lipoproteins in these preparations that bind to TLR2.12,13 Other studies have suggested earlier that the host can employ TLR2-independent pathways to stimulate inflammatory cascades during Gram-positive bacterial infection, 14 implying that in addition to lipoproteins, there could be other enterococcal virulence factors participating in stimulating the inflammatory response. Enterococcal aggregation substance and binding substance have been implicated in the stimulation of an inflammatory response in vitro, 15 and, more recently, enterococcal leucine-rich repeat-containing protein (ElrA) has been shown to stimulate host inflammation and contribute to E. faecalis virulence. 16
Enterococcal surface protein (Esp) was identified initially as a surface protein in a highly virulent, gentamicin-resistant E. faecalis isolate from a bacteremia. 17 The esp gene is found to be enriched in infection-derived E. faecalis isolates and an esp homolog is present in E. faecium isolates. 18 Epidemiological studies have determined that esp is generally associated with and enriched among infection-derived isolates compared with commensal isolates and carried on a large pathogenicity island.1,19,20
We, and others,21,22 found a significant correlation between the presence of esp and the ability of enterococci to form biofilms on abiotic surfaces. Esp, as a cell surface exposed protein of E. faecalis and E. faecium can interact directly with host cells during infection, and is therefore a good candidate to challenge the host immune system. Previous studies in a murine UTI model have found that higher levels of TNF-α, IL-6 and MCP-1 were present in homogenates of kidneys from mice infected with an Esp-positive strain of E. faecium compared with levels for mice infected with an isogenic Esp-deficient mutant. 23 By investigating the role of Esp in triggering the host innate immune response in this study, we demonstrate that Esp can induce pro-inflammatory cytokine expression via activation of the NF-κB pathway in vitro. Consistent with this, we also show that mice infected intraperitoneally with an Esp-positive E. faecalis showed comparatively higher levels of pro-inflammatory cytokines in peritoneal fluid and more neutrophil infiltration in the liver compared with mice infected with an isogenic Esp-deficient mutant strain.
Materials and methods
Bacterial strains and eukaryotic cells
Esp-positive E. faecalis MMH594 and its isogenic Esp-deficient mutant (MMH594b) have been described previously. 24 These strains were grown in THB containing 1% Glc supplemented with the appropriate antibiotics for 16 h and bacterial numbers counted by serial dilution and plating. Bacteria were pelleted by centrifugation and washed with PBS before use. RAW264.7 macrophages were cultivated initially in DMEM plus 10% FBS. Bone marrow-derived macrophages (BMDMs) were isolated and cultured according to a method previously described. 25
Purification of Esp
Expression and purification of the N-terminal region of Esp has been previously described. 17 Endotoxin removal from the purified protein was conducted by using a ToxinEraser TM Endotoxin Removal Kit (GenScript, Piscataway, NJ, USA) and the final concentration of endotoxin was estimated using the Endosafe-PTS system (Charles River, Charleston, SC, USA) to be less than 0.1 EU/ml. TLR2–/– and MyD88–/– BMDM were incubated with 50 µg/ml Esp for 5 h before harvesting for RT-PCR.
In vitro infection assays and siRNA
RAW264.7 macrophages were cultivated initially in DMEM plus 10% FBS to confluence in tissue culture dishes. The cells were seeded in six-well or 24-well plates and then infected with E. faecalis wild type or Esp-deficient strain for 1 h or 5 h at an MOI of 10. Infection with E. faecalis, assay for bacterial viability and Western blot were done as described earlier. 26 For transfection with siRNA, the RAW264.7 cells were transiently transfected with control siRNA or p65 siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) according to the manufacturer’s protocols.
RNA extraction and RT-PCR
For RT-PCR analysis, RNA was extracted with an RNeasy Mini kit (Qiagen, Valencia, CA, USA). cDNA generated from randomly primed RNA using iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA) was used as template to measure the levels of TNF-α and IL-1β by PCR.
ELISA
Cytokine levels in the supernatant of RAW264.7 cells, serum and peritoneal lavage were measured by ELISA (R&D Systems, Minneapolis, MN, USA).
Immunofluorescence
For immunofluorescence staining, the treated RAW264.7 macrophages were fixed with 4% paraformaldehyde for 15 min before permeabilizing with 0.2% Triton X-100. After blocking with 2% BSA, the slides were incubated with rabbit anti-p65 (Cell Signaling Technologies, Beverly, MA, USA) diluted 1:200, followed by secondary Ab coupled with fluorochrome. The images were collected by using an Olympus IX73 microscope.
In vivo peritonitis model
In vivo peritonitis studies were conducted under an animal protocol approved by the University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee, as described earlier. 27 Bacteria were cultivated overnight at 37℃ in THB containing 1% Glc supplemented with appropriate antibiotics. BALB/c mice were infected with E. faecalis MMH594 and its isogenic Esp-deficient strain at 2.0 × 108 CFU per mice via i.p. injection as described earlier. 27 Control animals were treated in a manner identical to the experimental groups in all respects except for bacterial infection. Groups of four mice per strain were euthanized at 6 h or 48 h by isoflurane overdose followed by cervical dislocation. The liver and kidney tissues were washed with physiological saline to remove residual blood, and then fixed in 4% formalin and embedded in paraffin for histological examination by hematoxylin and eosin (H&E) staining as described previously. 27
Neutrophils in the liver or bacteria in the kidney were detected by immunohistochemistry as described. 27 For flow cytometry analysis, the cells collected from peritoneal cavity were incubated with Fc Block (anti-CD16/CD32) and then stained for neutrophil and macrophage markers using FITC anti-mouse Ly-6G/Ly-6C (Gr-1) and Alexa Fluor 647 anti-mouse F4/80 Abs (BioLegend, San Diego, CA, USA).
Isolation and analysis of peritoneal macrophages
At 6 h post-infection, mice were euthanized by isoflurane overdose followed by cervical dislocation. Five ml sterile PBS was injected into the peritoneal cavity of the mice to recover peritoneal exudate cells (PEC). The suspension was centrifuged for 5 min at 500 g at 4℃, and PEC resuspended in DMEM containing 10% FBS, vancomycin (16 µg/ml) and gentamicin (150 µg/ml). About 106 cells were added per well of a 24-well tissue culture plate. After 1 h, the cells in the plate were washed twice to remove non-adherent cells, and the adherent macrophages were collected for cytokine analysis by RT-PCR.
Statistical analysis
Experimental values are represented as the mean of triplicates ± SD from at least three independent experiments. Student’s t-test was used to determine statistical significance. A value of P < 0.05 was considered statistically significant.
Results
Esp stimulates TNF-α and IL-1β through NF-κB activation
To investigate the role of Esp in expression of pro-inflammatory cytokines during E. faecalis infection, direct comparison of MMH594 wild type (Esp-WT) and Esp-knockout (Esp-KO) strains showed an appreciable increase in mRNA (Figure 1a) and corresponding protein levels (Figure 1b) of pro-inflammatory cytokines TNF-α and IL-1β, but not IL-6 (data not shown) in Esp-WT infected cells compared with levels in cells infected with the Esp-KO strain. Comparison of phagocytosis of Esp-WT and Esp-KO strains by macrophages revealed similar levels, thus ruling out differences in phagocytosis levels to be the cause of any differences in pro-inflammatory cytokine expression levels (Figure S1A) To analyze NF-ĸB activation in cells infected with Esp-WT and Esp-KO strains, the degradation of IĸB-α and nuclear translocation of NF-kB p65 were assayed by Western blot and immunofluorescence, respectively. The total IĸB-α protein levels in Esp-WT infected cells was lower than that in Esp-KO strain infected cells (Figure 1c). Quantitative estimation of NF-κB p65 translocation into the nucleus of cells by immunofluorescence showed that significantly higher levels of NF-κB p65 translocated into the nucleus of cells infected with the Esp-WT strain compared with Esp-KO strain (Figure 1d).
Comparison of the pro-inflammatory cytokines TNF-α and IL-1β, and NF-κB activation in RAW264.7 cells infected by Esp-WT and Esp-KO strains. RAW264.7 cells were stimulated with Esp wild type (Esp-WT) or Esp knockout (Esp-KO) strain. (a) Intracellular RNAs were isolated, and the amounts of TNF-α and IL-1β mRNA were determined by qRT-PCR. The results are expressed as the levels of TNF-α and IL-1β normalized to actin from three independent experiments. (b) The amounts of TNF-α and IL-1β protein in the supernatants at 5 h post-infection were determined by ELISA. (c) The cells after 5 h infection were subjected to Western blot with an anti-IκB Ab, IκB levels were normalized by actin densitometry, and fold relative to untreated samples were determined. A representative blot and graph depicting mean and SD of three independent experiments are shown. (d) The cells after 5 h post-infection were stained with an anti-p65 Ab and the amount of p65 translocation was quantified by counting 100 cells over three independent experiments and the percentage of cells with p65 in the nucleus was determined. *P < 0.05.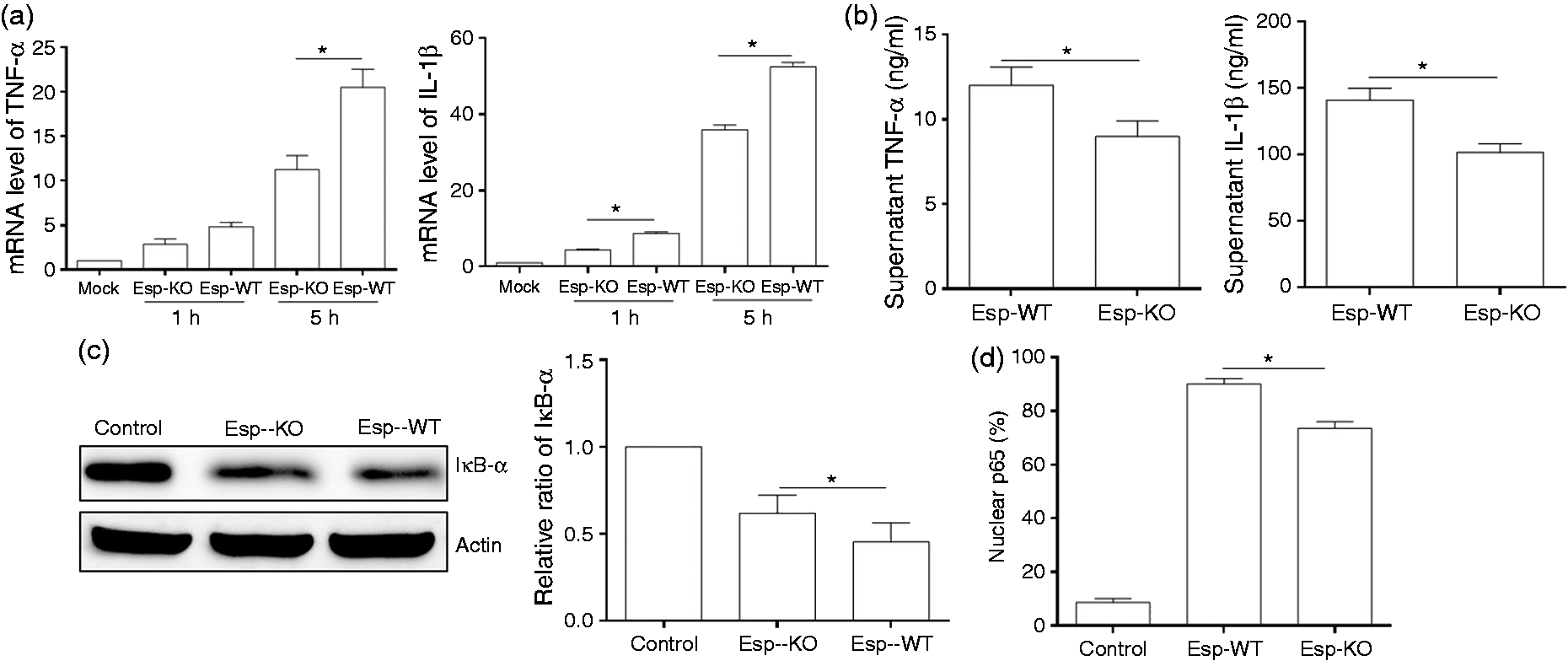
To investigate whether Esp could directly induce cytokine expression, treatment of RAW264.7 macrophages with recombinant Esp (rEsp) for 1 h or 5 h followed by quantitative RT-PCR analysis revealed the induction of TNF-α and IL-1β (Figure 2a). The protein level of induced IL-1β was confirmed to be consistent with mRNA expression data by Western blot (data not shown). To investigate whether Esp stimulates TNF-α and IL-1β expression via the NF-κB signaling pathway, we analyzed the activation of NF-κB during rEsp stimulation. NF-κB p65 translocation into the nucleus was assayed by immunofluorescence staining, which showed the nuclear translocation of NF-κB p65 in Esp-treated cells (Figure 2b). We then transfected RAW264.7 cells with siRNAs specific for NF-κB p65. At 24 h post-transfection, Western blot analysis confirmed the effectiveness of p65 knockdown by specific siRNA (data not shown). Next we examined whether knockdown of p65 by siRNAs would affect TNF-α and IL-1β expression, and found that p65 siRNAs significantly inhibited TNF-α and IL-1β production induced by rEsp (Figure 2c). Furthermore, it was demonstrated that the production of inflammatory cytokines induced by rESP was abolished in MyD88–/– BMDM, the expression of TNF-α and IL-1β was still significantly increased in TLR2–/– BMDM (Figure 2d). Consistent with this, we found that the Esp-WT strain could stimulate higher levels of TNF-α in primary TLR2–/– BMDM cells compared to the Esp-KO strain (Figure S1B). Thus, these results suggested that induction of inflammatory cytokines by Esp is mediated by MyD88 and through the activation of the NF-κB signaling pathway.
Induction of pro-inflammatory cytokines TNF-α and IL-1β by Esp mediated via MyD88 and through NF-κB signaling pathway. (a) RAW264.7 cells were treated with 10 µg/ml of Esp for 1 h or 5 h, and the cells were collected for extraction of mRNA to analyze the expression of TNF-α and IL-1β by qRT-PCR. (b) Following Esp stimulation for 5 h, RAW264.7 cells were stained with an anti-p65 Ab for immunofluorescence. (c) RAW264.7 cells transfected with p65-specific siRNAs (+) or with a non-specific siRNA (-), after 24 h, the cells were then stimulated with Esp for 5 h. Intracellular RNAs were isolated, and the amounts of TNF-α and IL-1β mRNA were determined by qRT-PCR. Error bars show SD of the means. (d) BMDM isolated from TLR2–/– or MyD88–/– mice were treated with Esp for 5 h, and the cells were collected for extraction of mRNA to analyze the expression of TNF-α and IL-1β by qRT-PCR. *P < 0.05.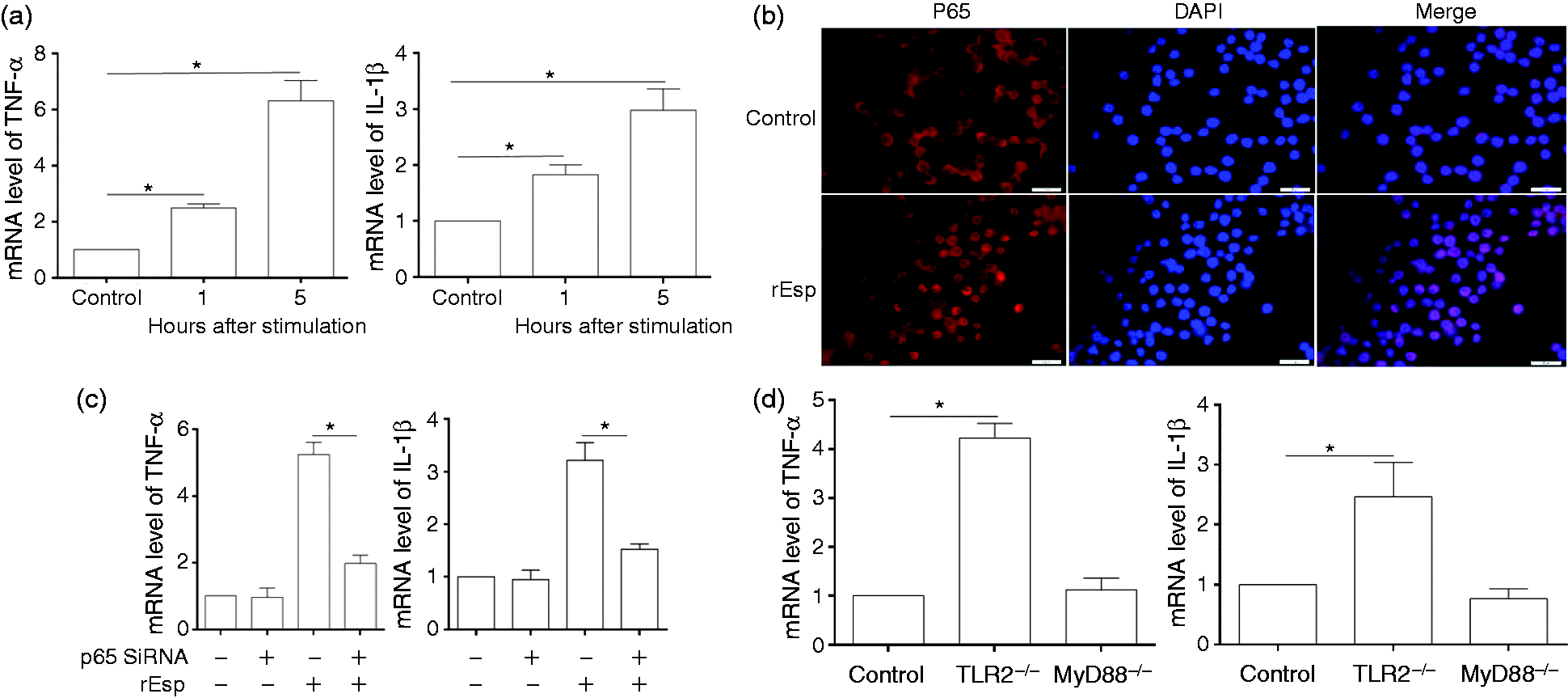
Esp enhances pro-inflammatory cytokine levels in the peritoneal cavity
Our peritonitis model, designed to investigate the role of Esp in inflammation during infection in vivo, revealed that at 6 h post-infection, IL-1β, TNF-α and IL-6 in the peritoneal cavity were significantly increased compared with that in uninfected mice, and the level of these cytokines in the peritoneal fluid of Esp-WT-infected mice were significantly higher than that in Esp-KO-infected mice (Figure 3a–c). Levels of these measured cytokines in peritoneal fluid decreased almost to the levels in uninfected group at 48 h post infection (data not shown). In contrast, while serum levels of IL-1β and TNF-α in the infected mice were not significantly increased at 6 h post-infection (data not shown), the IL-6 response at 6 h post-infection was more pronounced in the serum of Esp-WT-infected mice compared with that in Esp-KO-infected mice (Figure 3d). In these mice, a rapid increase in the number of peritoneal cells was observed 6 h after E. faecalis peritoneal infection. Flow cytometry identified the different cell types as F4/80+ macrophages, Gr-1+ neutrophils and found that there was no statistical significance in the number of macrophages and neutrophils in the peritoneal cavity between Esp-WT and Esp-KO-infected mice (Figure S2A, B). The increase in the total number of peritoneal cells at 6 h post-infection was mainly caused by a strong influx of neutrophils (Figure S2A), while the number of macrophages was slightly decreased (Figure S2B). This is consistent with a previous study, which showed similar influx of neutrophils that were essential for rapid clearance of E. faecium in mice.
28
Although macrophages may not be the dominant immune cells to control E. faecalis at the early stage of infection, by injecting GFP-expressing E. faecalis into the peritoneal cavity of mice we observed that macrophages could efficiently phagocytize E. faecalis in the peritoneal cavity, with about 70% of macrophages containing internalized bacteria. Furthermore, we show that macrophages isolated from the peritoneal cavity of Esp-WT infected mice produced more abundant IL-1β (Figure 3e) and TNF-α (Figure 3f) compared with that from Esp-KO infected mice, suggesting that Esp enhanced pro-inflammatory cytokine levels in peritoneal cavity was at least partially attributable to macrophages.
The role of Esp during E. faecalis infection in vivo. The level of (a) IL-1β, (b) TNF-α and (c) IL-6 in peritoneal fluid, and (d) IL-6 in blood following intraperitoneal infection with E. faecalis. Peritoneal lavage fluid and blood were collected from mice infected with E. faecalis Esp wild type (Esp-WT) and Esp knockout (Esp-KO) strains at 6 h post-infection. The levels of IL-1β, TNF-α and IL-6 was measured by ELISA. The peritoneal exudate cells were isolated from uninfected (Control) and infected mice at 6 h post-infection, and seeded in 24-well plates for 1 h to let macrophages adhere. Non-adherent cells were washed away and the attached macrophages were collected for analysis of (e) IL-1β and (f) TNF-α expression by RT-PCR. Error bars show SDs. *P < 0.05.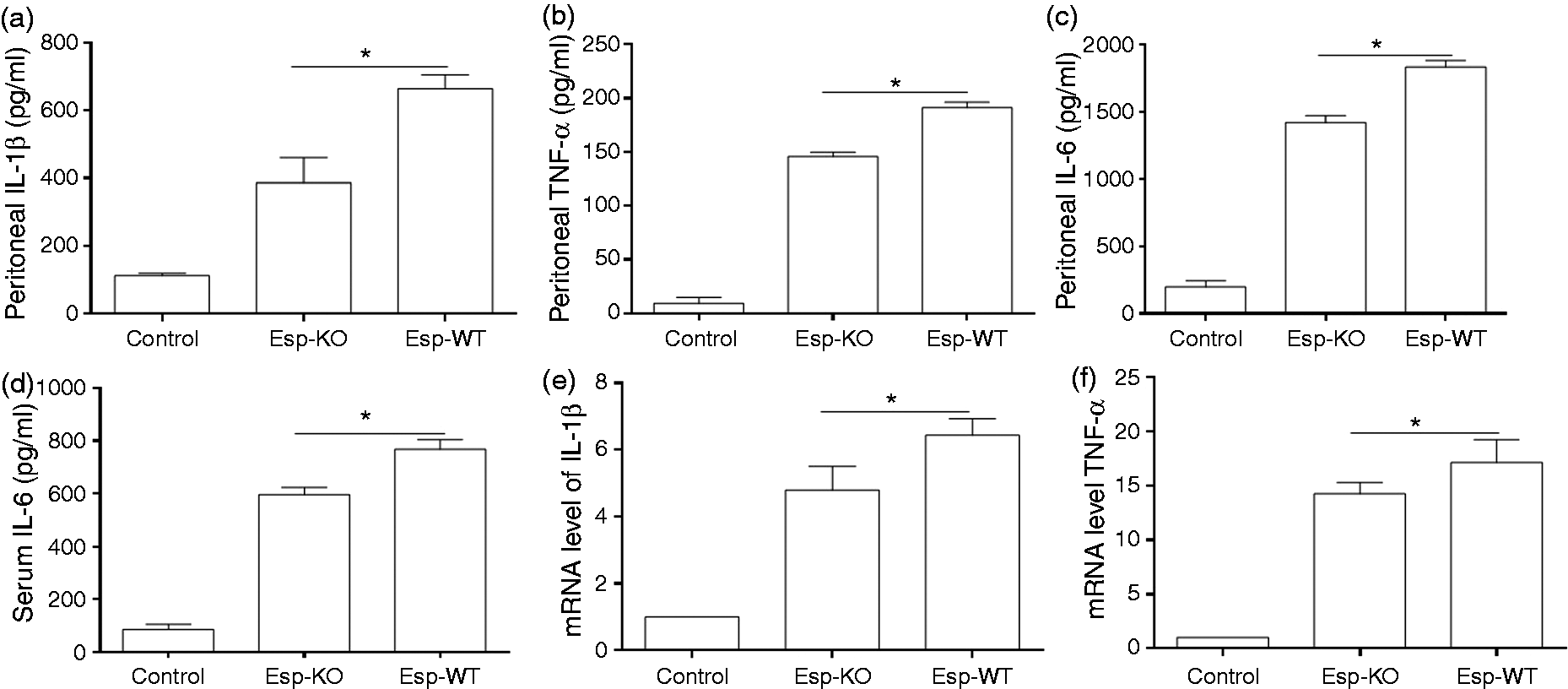
Histological analysis of liver and kidney of E. faecalis infected mice
Histological examination of infected tissue revealed that the pathological changes in the liver, including cellular degeneration and necrosis, were more serious in Esp-WT-infected mice than in Esp-KO-infected mice at 6 h post-infection (Figure 4a; Figure S3A). More neutrophils were detected in cellular infiltrates from the liver of Esp-WT-infected mice than that in Esp-KO infected mice at 6 h post-infection (Figure 4a; Figure S3B). No significant differences in bacterial numbers were observed in livers of mice infected with either Esp-WT or Esp-KO (Figure S3C), suggesting that the differences observed in pathology at 6 h post-infection were not due to differences in the numbers of colonizing bacteria but likely caused by inflammation. While there was not a striking difference in the severity of lesions in the kidney at 48 h post-infection, microscopic observation of kidney sections revealed numerous bacterial foci, in both Esp-WT- and Esp-KO-infected mice (Figure 4b). The bacterial foci formed by E. faecalis within the kidney, confirmed by immunohistochemistry with polyclonal Abs to E. faecalis, showed that they were fewer in number in Esp-WT infected mice than those observed in mice infected with the Esp-KO strain (Figure 4c).
Histology and neutrophil infiltration in the liver and bacteria in the kidney. Mice were intraperitoneally infected with E. faecalis Esp wild type (Esp-WT) and Esp knockout (Esp-KO) strains. (a) The samples of livers were harvested at 6 h post infection and sections stained with rabbit anti-neutrophil elastase to observe neutrophil infiltration (arrow), also, cellular degeneration and necrosis (arrowhead) can be observed. (b) The kidney sections at 48 h post-infection were stained with H&E. (c) Immunohistochemical detection of bacteria in the kidney of mice infected with Esp-WT or Esp-KO strain using anti-E. faecalis serum.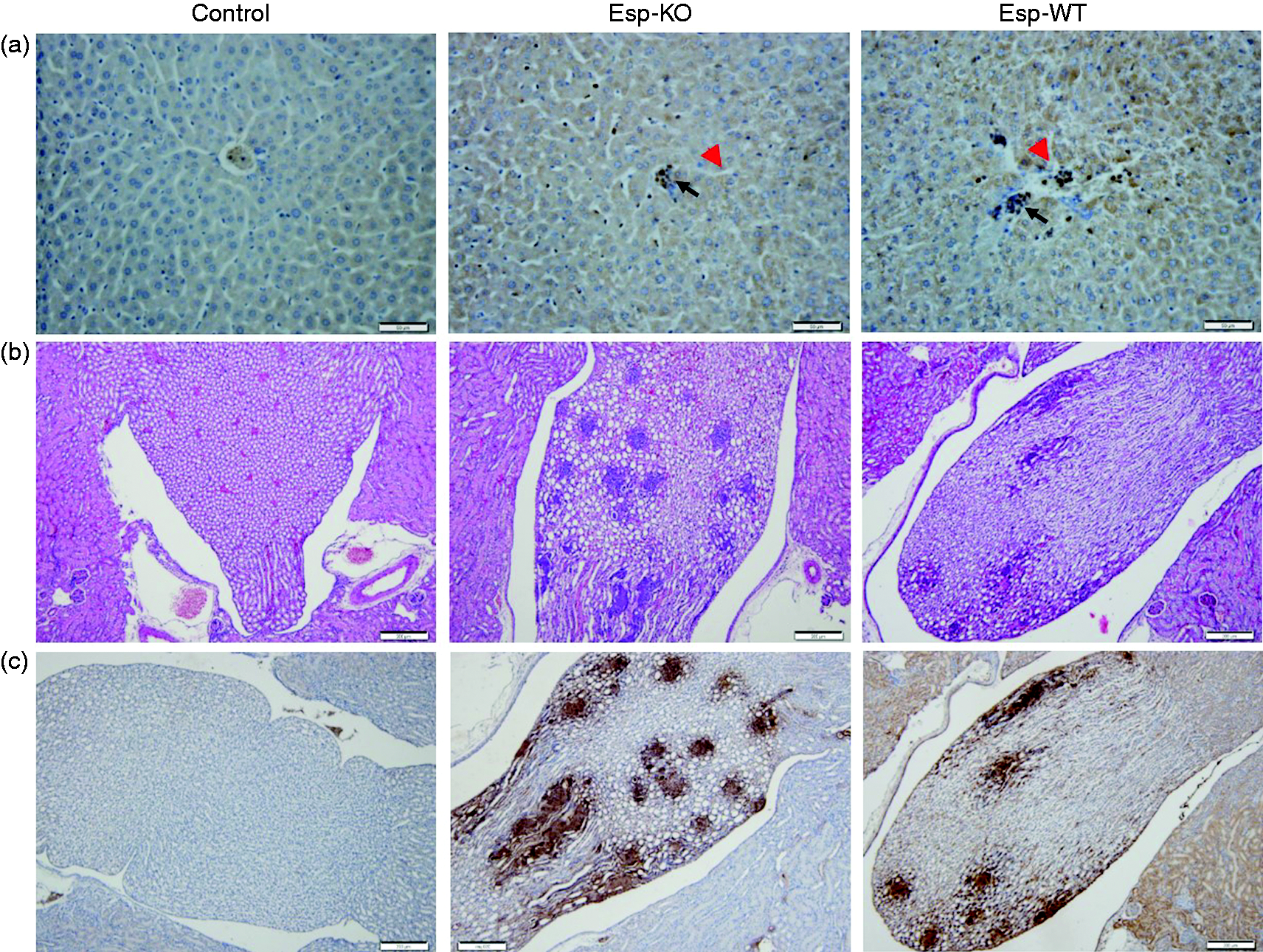
Discussion
Cytokines are well known for their wide-ranging effects on eukaryotic cell function and also are recognized as important mediators of tissue pathology during infection by microorganisms. In this study, we have shown that the Esp protein, which is encoded within the pathogenicity island of many E. faecalis and E. faecium isolates, can participate in the activation of the NF-κB signaling pathway and subsequent pro-inflammatory cytokine expression. Further, we have shown that rEsp can promote TNF-α and IL-1β expression in a NF-κB-dependent manner. Our in vivo infection experiments revealed a higher level of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6, and more severe pathology in the liver of the mice infected with the Esp-WT strain.
Our results from the cell culture experiments and the in vivo peritonitis experiments confirm the previously observed discrepancy between pro-inflammatory cytokine induction profiles seen during enterococcal infection in vitro and in vivo. 29 Peritoneal levels of TNF-α, IL-1β and IL-6 were significantly elevated at 6 h post-infection in the peritoneal cavity of mice infected with the Esp-WT strain compared with those infected with the Esp-KO strain. In a similar study using a wild type E. faecalis strain OG1RF and its isogenic ElrA-deficient mutant, a role for ElrA in E. faecalis-mediated IL-6 induction was established. 16 In this study, although other time points were not evaluated, the level of IL-6 at 20 h post-infection was at least twofold lower in mice infected with an ElrA-deficient mutant compared with the parent strain, OG1RF. Interestingly, no differences were observed in the expression of a number of other cytokines, including TNF-α and IL-1β. These observations let the authors to conclude that ElrA positively influenced IL-6 expression. Although the cytokine expression data represent snapshots at different time points, the differences seen in the levels of TNF-α and IL-1β between this earlier study and ours could be attributed to the different strain backgrounds. While wild type strain OG1RF is ElrA+, Esp–, the MMH594 strain used in our study is ElrA+, Esp+. Coupled with the in vitro results (Figure 1), we conclude that Esp expression has a direct bearing on the expression of TNF-α and IL-1β.
While we have shown that Esp is sufficient to directly activate NF-κB and promote inflammatory cytokine expression, the exact mechanism of how Esp activates NF-κB is yet to be established. Analysis of Esp structure shows it to be similar to many bacterial surface protein adhesins that are involved in binding to host ligands. Leendertse et al. have shown that Esp expression in E. faecium could enhance the in vitro binding of bacteria to bladder and kidney epithelial cells. 23 We propose that the mediation of E. faecalis attachment to host cells by Esp may also play an important role in activation of host cell signaling pathway(s) that may induce pro-inflammatory cytokines during infection. Along these lines, recent in vitro work has shown the E. faecalis infection of gastric cancer cells up-regulates the expression of many NF-κB regulated pro-inflammatory cytokines and chemokines. 30
It is also interesting to consider how Esp is localized at the bacterial cell surface by virtue of a slight variation (YPxTG) in the signature carboxy terminal anchoring motif (LPxTG) that allows anchoring of Gram-positive surface proteins to the cell wall in a sortase-dependent fashion. 31 Recently emerging evidence has suggested that Protein A from Staphylococcus aureus, also anchored at the cell surface by the action of sortase A, may be released into the extracellular medium by a process that is not yet fully understood and thus contribute to immune evasion. 32 In this regard it must be pointed out that earlier studies looking at environmental cues that promote E. faecalis gene expression found that esp gene expression increased when grown in serum (10-fold) and urine (4.9-fold) compared with growth in 2xYT medium. 33 Although the in vivo expression and release of enterococcal Esp into the extracellular medium has not been thoroughly investigated, it raises some important possibilities for enterococci to promote pro-inflammatory cytokine induction through released Esp. Therefore, the enhanced expression of Esp in vivo may have a more profound influence on inflammatory mediators than in vitro.
An earlier study employing a mouse model of UTI showed that E. faecalis strains had a tropism for the kidneys and persisted in this organ for at least 2 wk in preference to the bladder. 14 Histological examination further revealed an inflammatory response in the kidneys but not in the bladder. Additionally, this study also concluded that the host employed TLR2-independent pathways to stimulate inflammatory cascades during Gram-positive infection, although specific virulence determinants were not evaluated. In our present study, we found that Esp could stimulate high levels of pro-inflammatory cytokines in primary TLR2–/– BMDM cells, suggesting that Esp may be at least one factor in E. faecalis that influences inflammation during infection in a TLR2-independent manner. In the peritonitis model, the injected enterococci in peritoneal cavity could not be eliminated immediately by innate immune cells such as neutrophils and macrophages, and some bacteria will enter the blood and spread to other organs through systemic circulation. 27 In the present study it was observed that at 48 h post-peritoneal infection a lot of bacterial foci appeared within the glomeruli of infected mice kidneys, confirming the tropism of E. faecalis for the kidneys, which was described by Kau et al. 14 The observation of the lower bacterial numbers in the kidneys of mice infected with the Esp-WT strain in our model, compared with that in mice infected with the Esp-KO strain, may be a result of more efficient clearance of Esp-WT bacteria due to the enhanced systemic inflammatory immune response. These observations are consistent with the earlier report from the UTI model, which showed that inflammation contributes to the control of host infection, and induction of inflammation in the absence of an implanted catheter promoted bacteria clearance. 34
It is also intriguing to consider inflammation in the broader context of the dual roles that enterococci seem to play in the host—that of a commensal organism in the gut, as well as of an opportunistic pathogen. These contrasting roles suggest an interplay between the bacterium and the host that is environment-specific. For instance, enterococci in the gut may have more of an immunomodulatory role orchestrated to suppress inflammation in general, or particularly during tissue injury to the intestinal epithelium. This is supported by the use of enterococci as a probiotic.35–37 Other reports have also shown that E. faecalis as first colonizers of healthy infant guts can attenuate pro-inflammatory cytokine expression through MAPK signaling pathways in human intestinal epithelial cells and in a mouse model of DSS-induced inflammation. 38 However, pro-inflammatory responses induced by E. faecalis infection in host immune and other cell types may promote bacterial dissemination and host pathology. In this regard, in an IL-10 knockout model of colitis and cancer, long-term colonization with E. faecalis is known to promote carcinogenesis through a bystander effect involving macrophage-expressed TNF-α and COX-2. 39 Inflammation is obviously a double-edged sword and must be carefully resolved during the host–pathogen interaction and further studies would be required to carefully tease out the role of inflammation in enterococcal pathogenesis.
In summary, we have shown in this work that enterococcal surface protein Esp can induce the expression of pro-inflammatory cytokines through NF-κB activation. Further studies in this regard should aid in understanding the complexities of the enterococcal–host relationship.
Footnotes
Acknowledgements
We thank Drs. Michelle Callegan and Phil Coburn for providing the TLR2- and MyD88-deficient mice for isolation of BMDM. This work was funded, in part, by Public Health Services Grant AI 059673 from the National Institutes of Health and a President's Associates Presidential Professorship awarded by the University of Oklahoma to N.S.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflicts of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.