
Abstract
TLRs and NLRs participate in the immune system recognition of Helicobacter pylori. However, little is known about the mechanisms leading to inflammasome activation by H. pylori and if NLRs in neutrophils are involved in the process. We studied how NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome components are involved in IL-1β maturation in human neutrophils in response to the infection and if they are dependent on T4SS (type IV secretion system) and TLRs. Human neutrophils were cultured and infected with the 26695 or the VirD4− H. pylori strains; the IL-1β concentration was analyzed by ELISA, and we also evaluated the activation of TLRs 2 and 4. The infection of neutrophils with both strains of H. pylori induced production of IL-1β and expression of the NLRP3 inflammasome components such as apoptosis-associated speck-like protein with CARD domain and NLRP3 protein. The infection also increased the activity of caspase-1, which is required for the maturation of IL-1β. Our study shows, for the first time, that H. pylori infection induces the expression and activation of components of NLRP3 inflammasomes in human neutrophils and that the activation is independent of a functional T4SS and TLR2 and TLR4.
Introduction
Helicobacter pylori plays an important role in the development of peptic ulcers, gastric adenocarcinoma and gastric mucosa-associated lymphoid tissue lymphoma.1–3 Infection with H. pylori strains that carry the Cag (cytotoxin-associated gene A) pathogenicity island is associated with more severe clinical outcomes. 4 The Cag pathogenicity island encoding the type IV secretion system (T4SS) is a macromolecular assembly used by bacteria to inject material into other cells. The T4SS usually comprises a set of 12 proteins (VirB1–11 and VirD4), of which VirD4 (HP0524) acts as a ‘coupling’ protein that recruits substrates to the T4SS apparatus. The H. pylori VirD4 acts as an adapter protein for the transfer of CagA protein and possibly other unknown proteins into the host cells.5,6
During H. pylori infection the innate immune response is activated and initiated by PAMPs, including LPS and flagellin. These PAMPs bind to PRRs, such as TLRs and NLRs. The activation of these receptors promotes pro-inflammatory cytokine gene expression and secretion by cells of the immune system, such as neutrophils. 7 The first pro-inflammatory cytokine produced is IL-1β, synthesized as an inactive precursor (pro-IL-1β) that requires cleavage by caspase-1. The activation of IL-1β, an important mediator of inflammation, is mediated, in part, by the inflammasomes.
Inflammasomes are molecular platforms assembled through hetero-oligomerization of an NLR, an ASC adaptor protein (apoptosis-associated speck-like protein with CARD domain), and an effector protein such as pro-caspase-1 or pro-caspase-5. The activation by caspases is followed by downstream maturation and secretion of the pro-inflammatory cytokines IL-1β and IL-18.8–11 The NLRP3 inflammasome is activated in response to a number of physical and chemical stimuli, including endogenous stress signals, such as extracellular ATP or uric acid crystals; exogenous compounds, such as aluminum salts and titanium dioxide; or pathogenic pore-forming toxins.12–15 The NLRP3 inflammasome is particularly important in the response to several bacterial pathogens, such as Staphylococcus aureus, Listeria monocytogenes, Legionella pneumophila and other bacteria. The NLRP3 inflammasome activates two effector mechanisms, the production of IL-1β and IL-18, and induction of pyroptosis and pyronecrosis, which have been shown relevant for protection against infection. 16
Kim et al. and Semper et al. confirmed that H. pylori induces IL-1β secretion in innate immune cells by means of the NLRP3 inflammasome and requires the Cag pathogenicity island,17,18 and we reported previously that H. pylori infection induced the release of IL-1β by human neutrophils. 19 However, little is known about the mechanisms leading to inflammasome activation by H. pylori and if the NLRs in neutrophils are involved in the process. In this work, we study whether NLRP3 inflammasome components are involved in IL-1β maturation in human neutrophils in response to H. pylori infection and if they are dependent on T4SS and TLRs.
Materials and methods
Ethics statement
The study was approved by the ethics committee of the Hospital de Pediatría, Centro Médico Nacional SXXI, Instituto Mexicano del Seguro Social, México City. The peripheral blood of healthy donors was obtained from the blood bank at the same institution. The bags of blood used in the study were intended for disposal, owing to an excessive filling, and therefore, informed consent was not necessary.
Isolation of human neutrophils
We selected blood from three donors that were H. pylori seronegative by rapid serological test for the detection of serum IgG Abs against H. pylori (DIAGNOMEX S.A. DE C.V., CA, USA) and further confirmed with a previously validated ELISA. 20 The neutrophils were isolated as described previously. 21 Briefly, blood was applied to a LymphoPrep density gradient (Nycomed, Oslo, Norway) and centrifuged at 800 g for 30 min at room temperature (24 ± 2℃). We discarded the mononuclear cells, obtained the cellular pellet and lysed the erythrocytes to obtain the granulocytes. After the lysis of the erythrocytes, the granulocytes were washed, re-suspended and cultured in six-well microplates at 5 × 106 cells/ml in RPMI-1640 medium (Life Technologies, Gaithersburg, MD, USA) supplemented with 2% FBS (Gibco BRL, USA), 100 IU/ml penicillin and 100 µg/ml streptomycin at 37℃. Cell viability was determined by the Trypan blue dye exclusion test, and neutrophil purity was determined by flow cytometry (FACScan; BD Biosciences, San Jose, CA, USA) (see Supplementary Figure 1).
H. pylori culture
The H. pylori 26695 ATCC strain and mutant VirD4– made in strain G27 as previously described (kindly supplied by N. Salama, FHCRC, Seattle, WA, USA) 22 were cultured on 5% defibrinated sheep blood agar base plates containing vancomycin, trimethoprim, nalidixic acid and amphotericin at 37℃ under microaerophilic conditions (9% CO2 atmosphere in incubator) for 24 h. At this time, we performed Gram staining and did urease and oxidase tests of each growth to confirm identity and purity of the strains. Next, the bacteria were harvested and washed with sterile saline solution, and the suspension was adjusted to 109 bacteria/ml.
Infection of neutrophil with H. pylori
Human neutrophils were cultured in RPMI media supplemented with 2% FBS, 100 IU/ml of penicillin and 100 µg/ml of streptomycin at 37℃ under microaerophilic conditions. After 18 h, neutrophils were infected with the 26695 strain or the VirD4− mutant at a corresponding MOI of 100 for 1, 3, 6 or 24 h. 23
Quantification of IL-1β
Cell-free culture supernatants were harvested after the H. pylori infection and the IL-1β concentration was measured by ELISA (Opt-EIATM; BD Pharmingen, San Diego, CA, USA), according to the manufacturer’s instructions; the sensitivity of this ELISA was of 3.9 pg/ml. We used RPMI-1640 medium alone as the negative control. In all tests, a standard recombinant cytokine preparation was used to estimate the cytokine concentrations in the samples. Each sample was run in duplicate, and the results are the average of at least three independent assays run on different days.
Blocking TLR2 and TLR4 activation
For Ab blocking experiments, neutrophils were first pretreated with anti-human IgG Ab (250 µg/ml) (Aventis Boehering GmbH, Marburgo, Germany) for 1 h at 37℃, in order to saturate Fc receptors and to avoid unspecific attachment of anti-TLRs mAbs. Next, neutrophils were treated with anti-TLR2 (clone TL2.1) or anti-TLR4 (clone HTA125) mAbs (IMGENEX BIOCARTA US, San Diego, CA, USA) at 5.0 and 10 µg/ml for 1 h at 37℃, followed by a 24 h of H. pylori challenge. As additional Ab control, neutrophils were treated with anti-human IgG Abs and challenged with H. pylori as described above. As negative controls, neutrophils were incubated with medium alone, with anti-human IgG, with anti-TLR2 or with anti-TLR4 mAbs but without H. pylori challenge.19 After each experiment, cell-free supernatants were collected and tested for IL-β. For the experiment, each sample was run in duplicate and the results are the average of three independent assays run on different days. We used lipoteichoic acid (10 µg) from Enterococcus faecalis, Pam3Cys (1 µg) and LPS from Escherichia coli 0111: B4 (100 ng) as positive controls; all received a pre-test to establish the optimal concentration of activation (Figure 1C; Supplementary Figure 2).
H. pylori induces the production of IL-1β independent of a functional T4SS and of TLR2. (a) Production of IL-1β by human neutrophils was determined after infection for 1, 3, 6 or 24 h with H. pylori strains 26695 and G27 mutant VirD4−. IL-1β concentration was measured by ELISA in cell-free culture supernatants. All assays were performed in duplicate, and the means ± SEMs of three independent experiments are shown. *P < 0.05 (two-way repeated-measures ANOVA and Student–Newman–Keuls post hoc test). (b) Viability of neutrophils after 1, 3, 6 and 24 h of H. pylori infection, as determined by CytoTox 96 test. The means ± SEMs are shown. *P < 0.05 (ANOVA and Dunnett’s post hoc test). (c) No inhibition of IL-1β production by anti-TLR2 Abs. Neutrophils were pretreated in presence or absence of human IgG, anti-TLR2 Abs followed by stimulation with lipoteichoic acid (10 µg) from E. faecalis, Pam3Cys (1 µg) or H. pylori (MOI 100), and the IL-1β production was determined by ELISA. The production of IL-1β induced by H. pylori was considered as 100%, and the corresponding inhibition by anti-TLR2 was calculated referring to this value. Data represent the mean ± SD of two independent experiments. *P < 0.05 with respect to control (100%).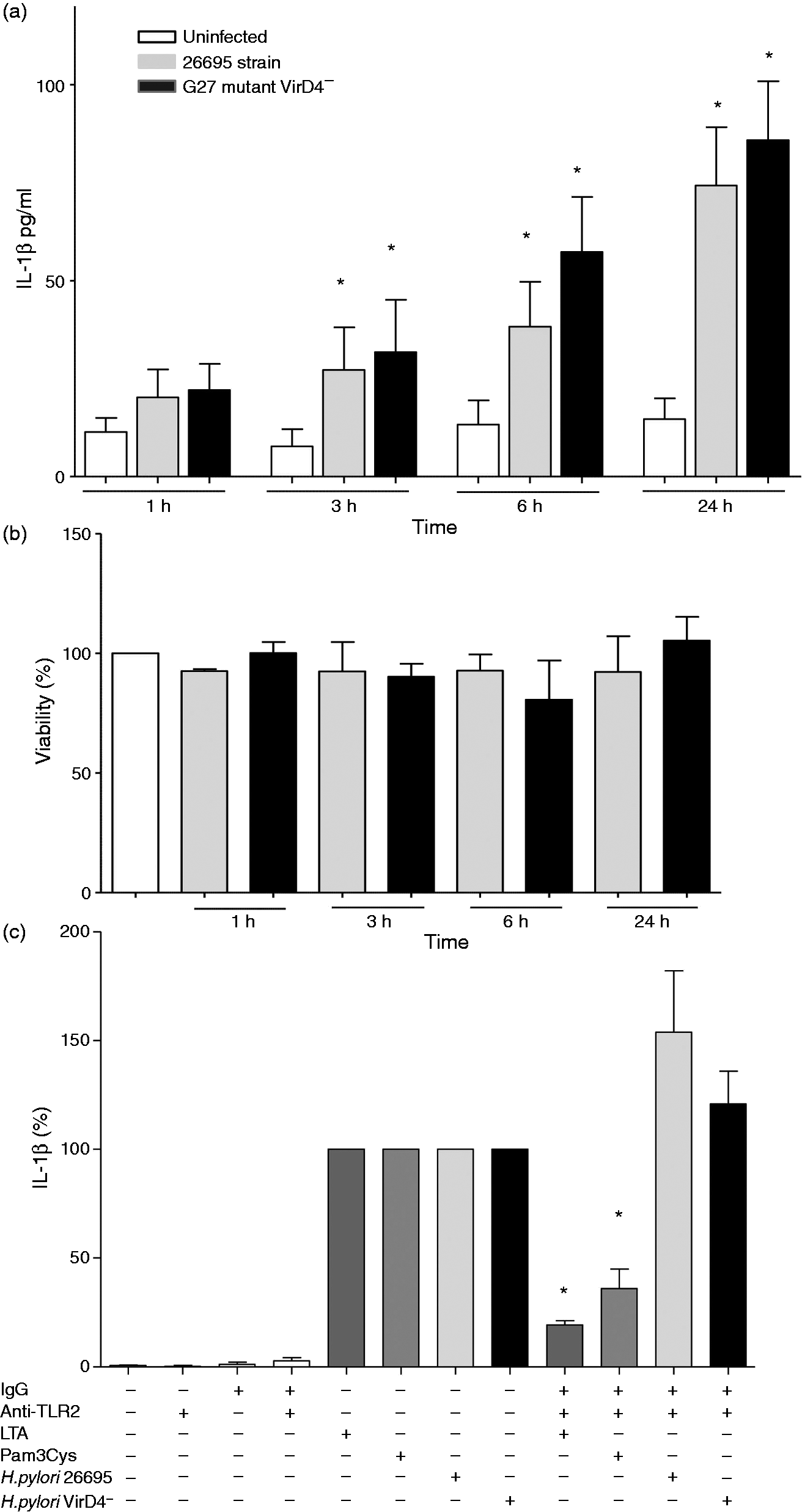
Determination of the expression of ASC and NLRP3 proteins by Western blot analysis
Neutrophils were infected with H. pylori for 1, 3, 6 or 24 h, and the expression of NLRP3 and ASC proteins was determined by Western blot analysis. Briefly, 106 infected neutrophils were re-suspended in RIPA lysis buffer (100 mM Tris-HCl pH 7.4, 300 mM NaCl, 10% NP40 and 10% sodium deoxycholate) supplemented with protease inhibitor mixture (Roche, Welwyn Garden City, UK) and 200 mM dithiothreitol.
The lysate was incubated, centrifuged and the proteins quantified using the Bradford Protein Assay Reagent (Bio-Rad, Hercules, CA, USA). Lysate protein (80 µg) was loaded onto a 6% or 12% SDS polyacrylamide gel, electrophoresed and then transferred to polyvinylidene difluoride membranes (Bio-Rad). The blots were blocked for 1 h with 5% (w/v) non-fat dried milk in phosphate-buffered saline. The membrane was incubated with rabbit anti-human ASC (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and mouse anti-human NLRP3 (Enzo Life Sciences, San Diego, CA, USA) Abs for 24 h at 4℃. Protein was detected using chemiluminescent methods with a horseradish peroxidase-linked goat anti-mouse or anti-rabbit secondary Ab (Santa Cruz Biotechnology) and excited with Western Blotting Luminol Reagent (Santa Cruz Biotechnology). THP-1 cells were included as a positive control for expression of NLRP3 protein.
Determination of the expression of ASC protein by immunofluorescence analysis
For immunofluorescence procedures, 105 uninfected and infected neutrophils were drip placed to glass clean coverslip. The cells were then fixed for 15 min at room temperature in PBS containing 4% paraformaldehyde then washed in 0.1% PBS-Tween-20. The fixed cells were incubated with an anti-ASC Ab (Santa Cruz, Biotechnology) in 0.1% PBS-Tween-20 during all night, washed twice in 0.1% PBS-Tween-20, incubated 4 h with a FITC-coupled anti-rabbit Ab and 20 min with DRAQ7 (Biostatus, Shepshed, UK). Finally, cells were washed twice in PBS-Tween. Cells were visualized using an Axiovert 100 M confocal microscope (Zeiss, Jena, Germany); each micrograph was captured at 40 × using a zoom of 1.5, 488 nm and 630 nm laser. In each experiment, pictures from different samples were taken consecutively using identical settings.
Neutrophil viability
The viability of neutrophils infected with H. pylori was determined using a CytoTox 96 non-radioactive cytotoxicity assay kit (Promega, Madison, WI, USA). Briefly, neutrophils were cultured in 96-well plates and infected with H. pylori with a MOI of 100 for 1, 3, 6 or 24 h, as described above. In each experiment, neutrophils without H. pylori infection were used as controls. After each infection time, viability was determined following the manufacturer’s instructions.
Inflammasome inhibition assays
Human neutrophils were cultured in 12-well plates. After 18 h, neutrophils were pre-incubated for 1 h with the following inflammasome inhibitors: glyburide (50 µM) (Sigma-Aldrich, St. Louis, MO, USA), which exerts inhibitory action at the ATP-sensitive potassium channel level; KCl (10 mM) (Sigma-Aldrich), which interferes with the flow of potassium ions; and Z-VAD-fmk (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone) (5 µM) (Promega), a caspase inhibitor that binds irreversibly to the catalytic site of caspase. The neutrophils were infected with H. pylori at a MOI of 100 for 24 h, after which time the supernatants were recovered and stored at –80℃ until the IL-1β concentration was measured.
Determination of caspase-1 activity (FLICA)
Neutrophils were cultured at 106 cells per well in 24-well microplates under the same conditions described above and then infected with H. pylori at a MOI of 100 for 3, 6 or 24 h. After each infection time, the cells were labeled with FLICA (FAM-YVAD-fmk caspase-1 FLICA kit; Immunochemistry, Bloomington, IN, USA) at 150 × according to the manufacturer’s instructions. The cells were then analyzed in a flow cytometer to measure the median fluorescence intensity and the percentage of positive cells.
Statistical analysis
Results are expressed as the mean ± SEM of three independent experiments. Data for the inhibition assays were analyzed using the Kruskal–Wallis test and the Student–Newman–Keuls post hoc test; and to identify differences between groups in the production of cytokines, we used two-way repeated-measures ANOVA with the Student–Newman–Keuls post hoc test. The differences in caspase-1 activity were analyzed using the Kruskal–Wallis test. P-Values < 0.05 were considered significant.
Results
H. pylori induces IL-1β production in human neutrophils independent of TLR2 and TLR4, and in the absence of a functional T4SS
Infection of human neutrophils with either the 26695 or VirD4– strain induced the production of IL-1β within 3 h of infection, and the maximum production was registered after 24 h of infection (Figure 1a). IL-1β production was higher after infection with the mutant VirD4− (which lacks a functional T4SS), although the difference was not significant (Figure 1a) between the two strains. These results indicate H. pylori does not need a functional T4SS to induce IL-1β production. We determined the viability of neutrophils after the infection and found no reduction in viability, even after 24 h of infection with either strain (Figure 1b).
We tested the possible participation of TLR2 and TLR4 in the production of IL-1β by inhibition of these receptors (Figure 1c), and found that none of the TLR Abs inhibited the production of IL-1β. These results suggest that other mechanisms are involved in the induction of IL-1β. Positive controls, LTA from E. faecalis, Pam3Cys and LPS from E. coli 0111:B4 showed the expected results (Figure 1c; Supplementary Figure 2b).
H. pylori induces expression of NLRP3 and ASC inflammasome components in human neutrophils even in the absence of a functional T4SS
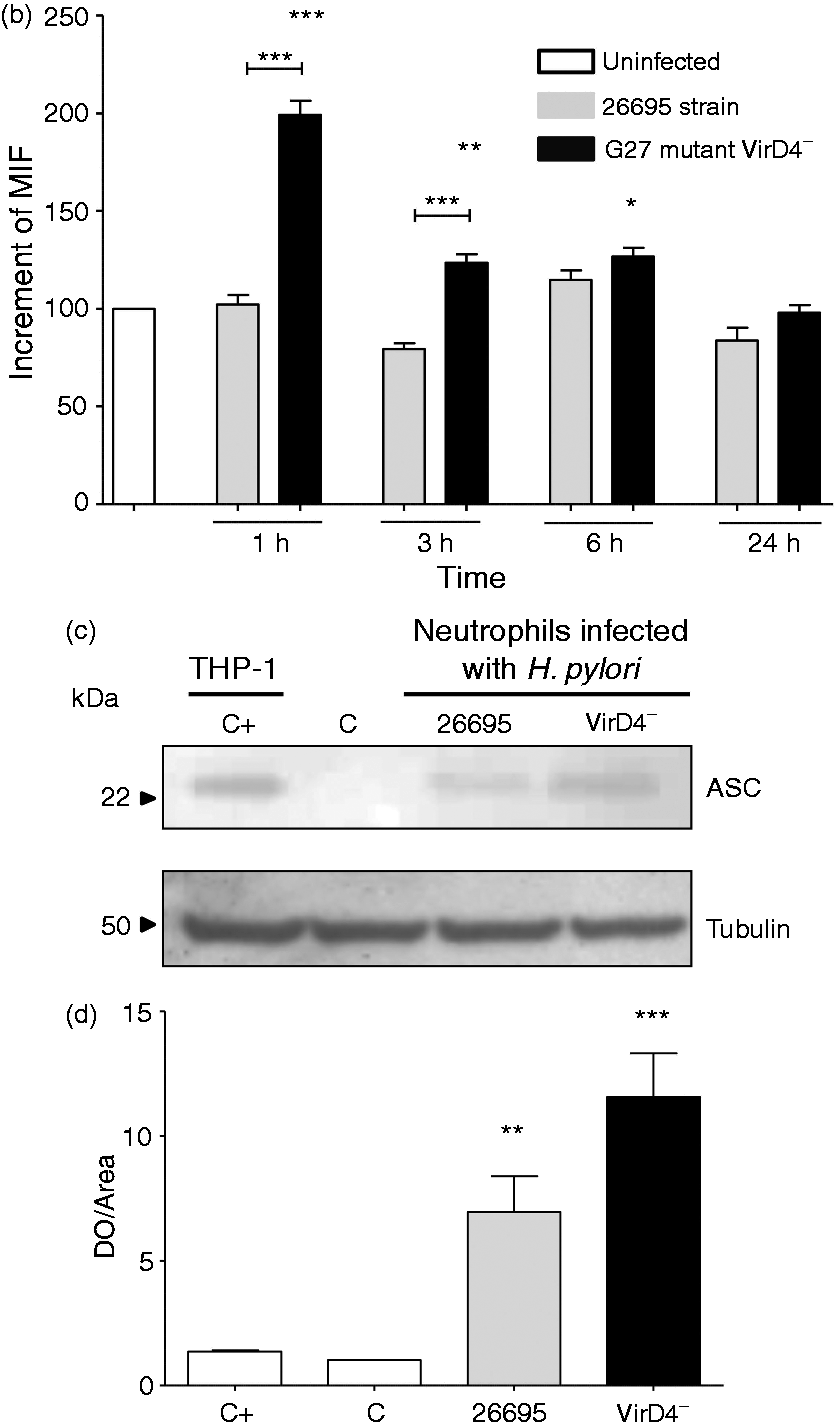
We also studied the participation of the proteins involved in the formation of the NLRP3 inflammasome as an important pathway of cell activation and in the induction of IL-1β. Neutrophils infected with H. pylori strains 26695 or VirD4− were tested to determine the expression of NLRP3 and ASC proteins after 1, 3, 6 and 24 h of infection. Both H. pylori strains induced the expression of NLRP3 after 1, 3 and 6 h of infection but not after 24 h (Figure 2a, b). In contrast, although the expression of ASC protein increased after 1, 3, 6 and 24 h of infection with both strains (Figures 3a), the increase was significant only with the mutant VirD4− but not with the 26695 strain (Figure 3b). The mutant VirD4− also induced a morphologic change after 3 h of infection, and this effect was more evident after 24 h, whereas the 26695 strain caused morphologic changes only after 6 h of infection (Figure 3a). The increased ASC protein expression was confirmed by Western blot, and although the ASC protein was expressed by uninfected neutrophils, both strains increased significantly its expression after 6 h of infection. The induction was stronger with the VirD4− than with the 26695 strain (Figure 3c, d).
H. pylori induces the expression of NLRP3. (a, b) Kinetics of the expression of NLRP3 in neutrophils infected with H. pylori (MOI 100). Detection of NLRP3 by Western blot analysis of cell lysates after 1, 3, 6 and 24 h of infection. C+: control + , lysate protein from THP-1 cells; C: control-, uninfected neutrophils; 26: 26695 strain; D4−: G27 mutant VirD4− strain. Each experiment was done in duplicate, in two independent assays. H. pylori induces the expression of ASC. (a, b) Kinetics of the expression of ASC in neutrophils infected with H. pylori (MOI 100). ASC protein (green) was detected by immunofluorescence after 1, 3, 6 and 24 h of infection. MIF was calculated from the cells after ASC staining. Results were normalized by subtracting values from uninfected neutrophils controls. ***P = 0.009, **P = 0.007, (Kruskal–Wallis statistic). (c, d) Expression of ASC in neutrophils infected with H. pylori was also detected by Western blot analysis of cell lysates after 6 h of infection. (ANOVA P = 0.0006, post Tukey’s multiple comparison test P < 0.05). Data are representative of two independent experiments. C+: control+, lysate protein from THP-1 cells; C: control-, uninfected neutrophils; 26: 26695 strain; VirD4-: G27 mutant VirD4− strain.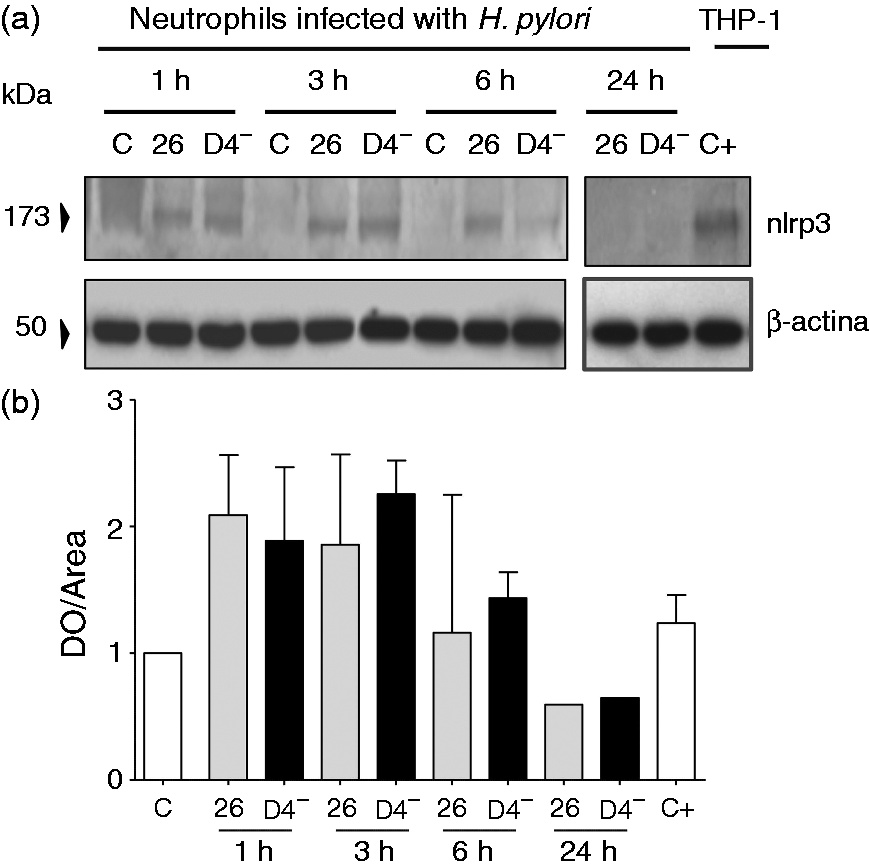
The activity of caspase-1 in human neutrophils is increased after H. pylori infection
To determine if H. pylori induces the activation of caspase-1 in the process of maturation of IL-1β, we measured the activated caspase-1 by flow cytometry in neutrophils infected with both strains of H. pylori. We observed that in uninfected neutrophils 40% of the cells expressed activity of caspase-1, which remained stable after 3, 6 and 24 h of incubation. In contrast, in infected cells, the percentage of cells exhibiting caspase-1 activity slightly decreased to 35% after 3 h of infection with the 26695 strain (P < 0.05) but increased to about 60% after 6 and 24 h of infection with either of the two strains (P < 0.05) (Figure 4a, b).
The activity of caspase-1 is increased in human neutrophils after H. pylori infection. Caspase-1 activity was quantified using the fluorescent FLICA-Casp1 reagent and then analyzed by flow cytometry. (A) Auto-fluorescent neutrophils were gated in the first log decade, and the fluorescence intensity was proportional to the level of caspase-1 activation after 3, 6 and 24 h of infection. (B) Graphical representation of the percentage of neutrophils with active caspase-1. Each experiment was done in duplicate, in two independent assays.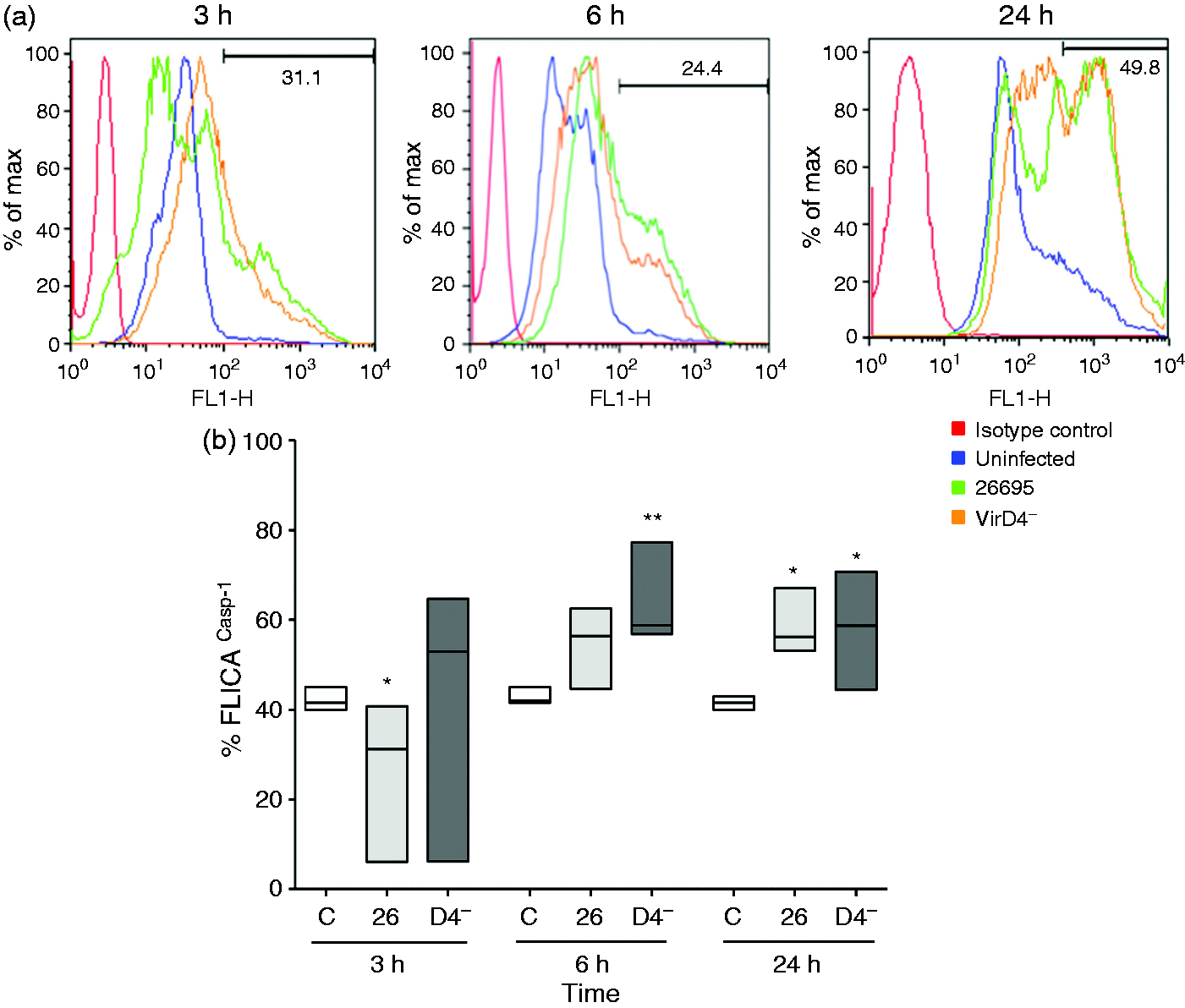
Participation of the inflammasome in the production of IL-1β by human neutrophils in response to H. pylori infection
To evaluate the participation of NLRP3 inflammasomes in the induction of IL-1β after H. pylori challenge in human neutrophils, the neutrophils were incubated with glyburide, KCl or Z-VAD-fmk before H. pylori infection. Glyburide exerts inhibitory action at the ATP-sensitive potassium channel level; KCl interferes with the flow of potassium ions; and Z-VAD-fmk is a caspase inhibitor that binds irreversibly to the catalytic site of caspase. These inhibitors decreased IL-1β production by different extents. In cells infected with 26695 strain, IL-1β production (73.0 ± 3.1 pg/ml) decreased by about 50% with KCl (36.0 ± 2.0 pg/ml) and glyburide (44.5 ± 0.7 pg/ml), and by around 60% with Z-VAD-fmk (25.5 ± 1.3 pg/ml). However, in cells infected with mutant VirD4−, IL-1β production (64.0 ± 5.5 pg/ml) decreased by 10% with KCl (59.8 ± 2.8 pg/ml), 20% with glyburide (53.7 ± 4.7 pg/ml) and 40% with Z-VAD-fmk (29.1 ± 0.99 pg/ml). These results would suggest that the involvement of the inflammasome in IL-1β production differs between strains (Figure 5).
Induction of IL-1β production by human neutrophils after H. pylori infection is inhibited by compounds that interfere with inflammasome activation. Human neutrophils were incubated for 1 h with KCl, glyburide or Z-VAD-fmk, and then infected for 24 h with the 26695 or mutant G27 VirD4− H. pylori strains. IL-1β concentration was measured by ELISA in cell-free culture supernatants. All assays were performed in duplicate, and the means ± SEMs of three independent experiments are shown. The production of IL-1β induced by each H. pylori strain was considered as 100%, and the corresponding percentage inhibition by KCl, glyburide or Z-VAD-fmk was calculated referring to this value. *P < 0.05 with respect to control (100%). (Kruskal–Wallis test and Student–Newman–Keuls post hoc test were used.)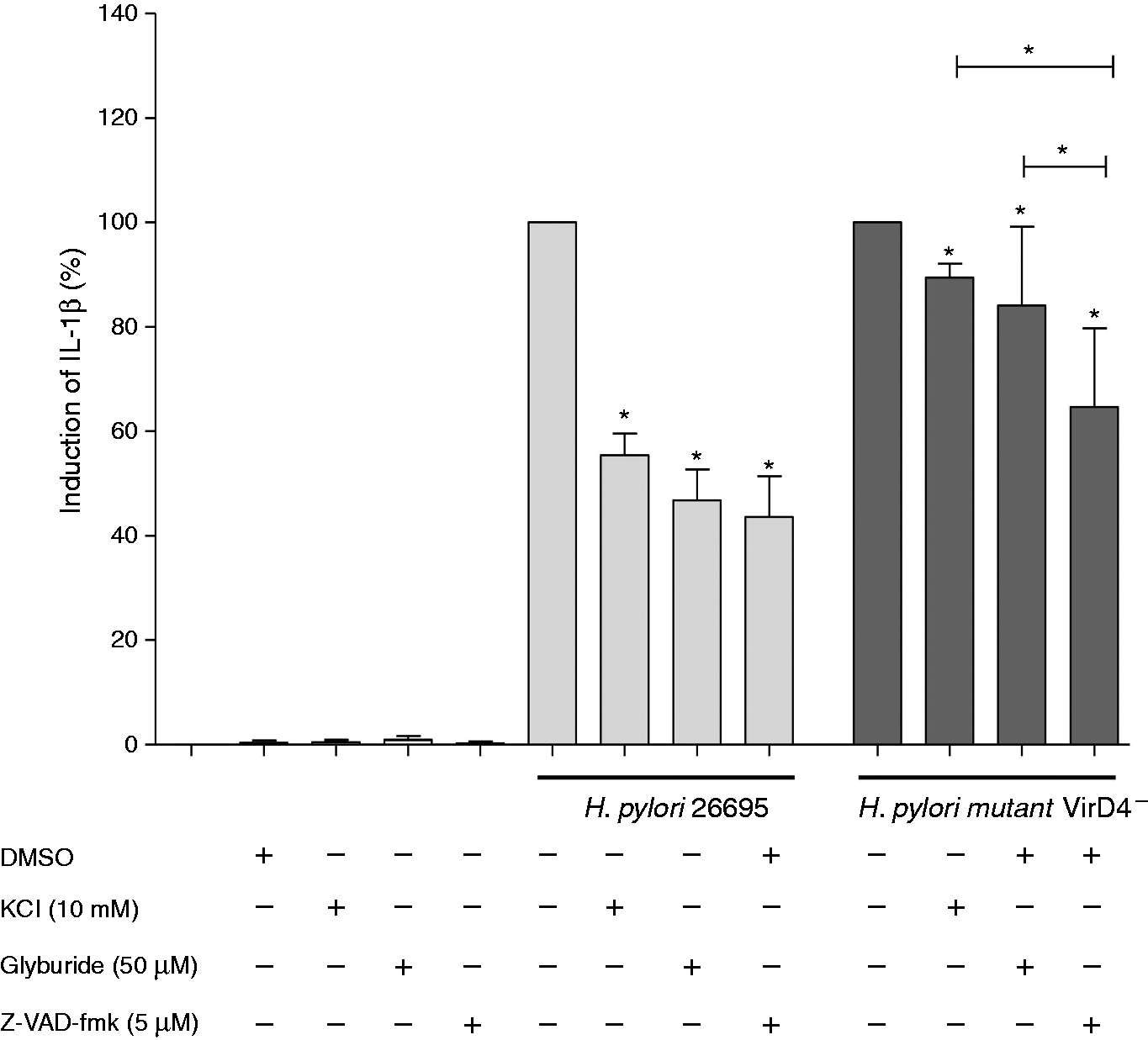
Discussion
In this study, we found that H. pylori increased the production of the pro-inflammatory cytokine IL-1β by human neutrophils, and we report for the first time that this response is independent of TLRs 2 and 4, although activation of these TLRs is necessary for induction of other cytokines such as IL-8 and IL-10. 19
We also confirmed that the production of IL-1β started early after infection with either H. pylori strain 26695 or mutant strain VirD4−. 23 Intact virD4 is essential for translocation and phosphorylation of CagA, and disruption of the T4SS causes dysfunction of the secretion system. 24 Thus, our results suggest that for induction of IL-1β production by H. pylori the presence of an intact T4SS is not essential.
The fact that the induction of IL-1β was independent of both TLRs 2 and 4 and that a T4SS is not essential, suggest that interaction of H. pylori with neutrophils probably occurs intracellularly, after phagocytosis of H. pylori by neutrophils. However, Kim et al., 17 using murine bone marrow-derived DCs, showed the production of mature IL-1β in response to H. pylori infection in a TLR2-dependent form. These discrepancies with our results may be due to the use of another cellular model, with important differences in activation mechanisms and in immune response.
Neutrophils respond to the presence of bacteria and their products with an array of receptors on their surface, such as TLRs and NLRs, which participates in an effective antimicrobial response.25,26 The activation of these cells leads to the release of inflammatory mediators that, in turn, recruit and activate other inflammatory and immune cells. The activity of neutrophils must be regulated efficiently to prevent tissue damage and therefore it is important to understand the mechanisms underlying the modulation of the activity of these cells in response to H. pylori infection.
Neutrophils express all known TLRs needed to facilitate early defense against invading microorganisms through the recognition of pathogen-associated molecular patterns and damage-associated molecular patterns.26,27 H. pylori induces the production of cytokines such as IL-8 and IL-10 but not IL-1β through TLR activation. 19 Moreover, IL-1β is also involved in the induction of the production of NO and reactive oxygen species by the H. pylori infection; these substances are normally produced in response to stress generated in the cell by the bacterium.28–30 These findings suggest that the inflammasome is assembled in neutrophils after H. pylori infection, and helps detect ‘danger signals’,15,26,31 probably when neutrophils translocate the epithelium and face H. pylori in the lumen of the gastric mucosa.
The H. pylori infection invariably leads to chronic inflammation in the gastric mucosa, which after years may lead to serious diseases such as peptic ulcer or gastric cancer in a minority of the infected population. The damaging role of mucosal inflammation as the main driving force causing gastric cancer is strongly supported by a large number of clinical and experimental reports.1,3 However, the mechanisms regulating the inflammatory and immune response to infection with H. pylori are not completely understood.
We found that glyburide and Z-VAD-fmk significantly reduced the production of IL-1β by human neutrophils after the infection with strain 26695 and mutant strain VirD4−. These substances inhibit the activation of the inflammasome by blocking recognition of NLRP3 ligands or caspase-1 activity, respectively.32–34 The inhibitory effect of both glyburide and Z-VAD-fmk was more pronounced with the 26695 strain than with the G27 H. pylori strain, suggesting some differences in the mechanisms of inflammasome activation. To further clarify if these differences might be explained by the absence of a functional T4SS, it is necessary to perform studies with isogenic mutants in the T4SS. Another possibility is that in strain 26695 inflammasome activation is caused by the T4SS, whereas in G27 genes other the T4SS are the cause. In any case, our results further confirm that the inflammasome is involved with the maturation of IL-1β in human neutrophils as a response to the H. pylori infection and that this response is elicited even in the absence of a functional T4SS.
Our results also support the important role of caspase-1 in the process eliciting IL-1β production by human neutrophils infected with H. pylori. Moreover, our study also confirms that the H. pylori infection induces the activation of the NLRP3 inflammasome and the expression of its components NLRP3 and ASC proteins, although the observed NLRP3 response occurred quickly but was sustained for a short period, suggesting that the inflammasome assembly occurs in the early hours of infecion and likely second stimuli needed to maintain or increase the expression of NLRP3. 34
However, although ASC protein was expressed in neutrophils even without infection, its expression increased significantly after incubation with both the VirD4− G27 and the 26695 H. pylori strain. This would suggest that ASC expression might be induced in the absence of the T4SS; in fact, we cannot discard the possibility that inflammasome activation by the 26695 strain might be done by the T4SS, whereas in G27 the activation could be performed by another gene. The above results also suggest that there are important differences in the mechanisms inducing expression of either nlrp3 or ASC in human neutrophils. The inducible nature of NLRP3 protein has been shown in hematopoietic myeloid cells such as dendritic cells, monocytes, macrophages and neutrophils in murine models. 34 The processing and secretion of IL-1β require the activity of caspase-1, which is activated after the assembly of the inflammasome. 35 The H. pylori infection induces the activation of caspase-1, which regulates IL-1β and IL-18 production in gastric disease models. 36
In conclusion, our study shows for the first time that the H. pylori infection induces the expression and activation of components of NLRP3 inflammasomes in human neutrophils. In addition, we corroborate that the inflammasome participates in the processing and secretion of IL-1β. Our results suggest that the production of IL-1β in human neutrophils is independent of TLR2, TLR4 and does not require functional T4SS of H. pylori.
Footnotes
Funding
This work was supported by Instituto Mexicano del Seguro Social (IMSS), grant FIS/IMSS/PROT/C2007/053. JT is a recipient of an Exclusivity Scholarship from Fundación IMSS, Mexico. Pérez-Figueroa E was a master student from Programa de Maestría en Ciencias Biológicas, Universidad Nacional Autónoma de México (UNAM) and received a fellowship from CONACYT.
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.