
Abstract
Background:
Inflammation plays a crucial role in the pathophysiology of myocardial ischemia/reperfusion (I/R) injury. A clinical trial has recently reported a smaller infarct size in a cohort of patients with ST-segment elevation myocardial infarction (MI) treated with a short colchicine course. The mechanism underlying colchicine-induced cardioprotection in the early MI phase remains unclear. We hypothesized that a short pretreatment with colchicine could induce acute beneficial effects by protecting the heart against inflammation in myocardial I/R injury.
Methods and Results:
Rats were subjected to 40-minute left anterior descending coronary occlusion, followed by 120-minute reperfusion. Colchicine (0.3 mg/kg) or a vehicle was administered per os 24 hours and immediately before surgery. Infarct size was significantly reduced in the colchicine group (35.6% ± 3.0% vs 46.6% ± 3.3%, P < .05). The beneficial effects of colchicine were associated with an increased systemic interleukin-10 (IL-10) level and decreased cardiac transforming growth factor-β level. Interleukin-1β was found to increase in a “time of reperfusion”-dependent manner. Colchicine inhibited messenger RNA expression of caspase-1 and pro-IL-18. Interleukin-1β injected 10 minutes prior to myocardial ischemia induced greater infarct size (58.0% ± 2.0%, P < .05) as compared to the vehicle. Colchicine combined to IL-1β injection significantly decreased infarct size (47.1% ± 2.2%, P < .05) as compared to IL-1β alone, while colchicine alone exhibited a significantly more marked cardioprotective effect than the colchicine-IL-1β association.
Conclusion:
The cardioprotection induced by a short colchicine pretreatment was associated with an anti-inflammatory effect in the early reperfusion phase in our rat MI model.
Introduction
The introduction of early reperfusion therapy for the clinical management of acute myocardial infarction (AMI) has resulted in decreased mortality and improved cardiac function. However, reperfusion induces irreversible damages within the injured myocardium. While reducing infarct size by limiting the wave front phenomenon of ischemic cell death, reperfusion also triggers a sterile inflammatory response that limits the reperfusion benefits. There is increasing evidence that nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is involved in the pathophysiology of myocardial ischemia/reperfusion (I/R) injury. 1 Interleukin-1β (IL-1β) is a key molecule and early mediator of inflammation in I/R injury. 2,3 Interleukin-1β is initially expressed within the injured myocardium and orchestrates sterile inflammation including leukocyte recruitment. 4 The IL-1β inhibition decreased myocardial damage in an I/R injury model and improved adverse cardiac remodeling, 5 thus supporting the critical role of IL-1β in I/R injury development.
While displaying potent anti-inflammatory properties, colchicine has been initially used for the treatment of acute gout and arthritis. 6 In addition to its anti-inflammatory effect on neutrophil recruitment into the heart, 7 it has also been shown to suppress NLRP3 inflammasome monocyte activation in patients with acute coronary syndrome (ACS). 8 Spatial arrangement of mitochondria was found to promote activation of the NLRP3 inflammasome, and colchicine was able to abolish this phenomenon. 9 Moreover, recent experimental studies 10,11 demonstrated the beneficial effects of colchicine on improving remodeling, survival, and cardiac function. Increasing evidence indicates that colchicine therapy suppresses local and systemic inflammatory cytokine production in patients with ACS. 12
In the present study, we hypothesized that (1) NLRP3 inflammasome-associated IL-1β aggravates I/R myocardial injury and that (2) a short colchicine pretreatment could induce acute beneficial effects by protecting the heart against inflammation in myocardial I/R injury.
Materials and Methods
Animal Preparation
All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85 (23), revised in 1996). The protocol was approved by our regional animal care and use committee.
Chemicals
Pentobarbital was obtained from Ceva Sante Animal (Angers, France). Colchicine was purchased from Sigma-Aldrich (St Louis, Missouri) and recombinant rat IL-1β protein from R&D Systems, Inc (Minneapolis, Minnesota).
Surgical Procedure
Male Wistar rats aged 8 to 10 weeks and weighing ∼250 to 300 g were used for all experiments. Experimental AMI was induced by transient myocardial ischemia for 40 minutes followed by reperfusion, as described in previous publications. 13 -15 Briefly, all animals were anesthetized by intraperitoneal injection of sodium pentobarbital (60 mg/kg) and endotracheally intubated with 16-gauge tube. Animals were ventilated using a small animal ventilator (SAR-830 AP; CWE Inc, Ardmore, Pennsylvania). Body core temperature was continuously monitored throughout the surgical procedure and maintained at 37°C using a homoeothermic blanket connected to a temperature control unit (HB101/2 RS; Bioseb, Vitrolles, France). The chest was opened via a median sternotomy. After removing the pericardium, the heart was exposed, and a 7-0 monofilament suture (Premio 7.0; Peters Surgical, Bobigny, France) was placed around the proximal portion of the left anterior descending (LAD) coronary artery and passed through a short piece of tubing (PE50) in order to create a reversible snare. Following cardiac stabilization, coronary occlusion was initiated by clamping the snare onto the epicardial surface directly above the coronary artery. Ischemia was confirmed by epicardial cyanosis below the suture, along with dyskinesis of the ischemic region. After 40-minute occlusion, reperfusion was achieved by removing the snare and confirmed by the disappearance of cyanosis, with a vascular blush observed in the myocardium. A sham surgery was performed, without the final ligation of the coronary artery.
Drug Preparation and Administration
Colchicine was dissolved in distilled water (45 mg/mL) and administered per os. Rats were pretreated twice with colchicine (0.3 mg/kg body weight) or vehicle, that is, 24 hours and immediately before the surgical procedure of AMI (Figure 1).
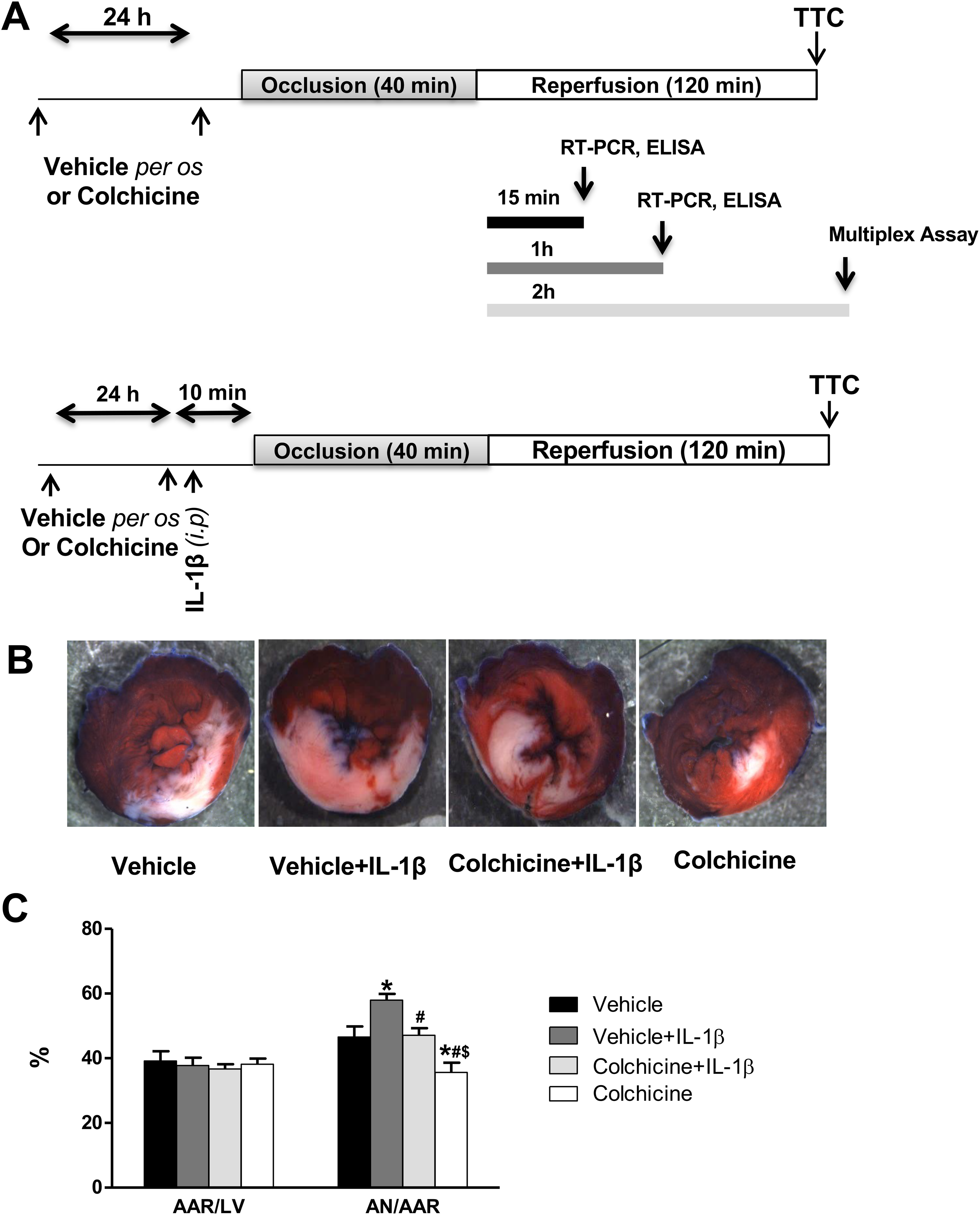
A, Experimental protocols. All groups were subjected to 40-minute coronary artery occlusion followed by either 120-minute reperfusion for infarct size and inflammatory cytokine measurements or 15-minute and 1-hour reperfusion for real-time polymerase chain reaction (RT-PCR) and enzyme-linked immunosorbent assay (ELISA) analysis. Colchicine treatment was administered per os 24 hours as well as immediately before the surgical procedure. Interleukin 1β was injected 10 minutes prior to myocardial ischemia. B, Representative sections of triphenyltetrazolium chloride (TTC)-stained heart following 40-minute ischemia and 120-minute reperfusion. C, Bar graph showing area of necrosis (AN) expressed as a percentage of area at risk (AAR), and AAR as a percentage of total left ventricular area (LV). All data were expressed as mean ± standard error of the mean (SEM). *P < .05 versus vehicle, #P < .05 versus vehicle + IL-1β, $P < .05 versus colchicine + IL-1β. n = 8 for vehicle, n = 6 for vehicle + IL-1β, n = 7 for colchicine + IL-1β, and n = 10 for colchicine group.
Study Groups and Experimental Design
For myocardial infarct size analysis, rats were subjected to 40-minute LAD coronary artery occlusion, followed by 2-hour reperfusion. Rats were randomly assigned to one of the following groups (Figure 1): vehicle (no further intervention, n = 8), colchicine (n = 10), vehicle + IL-1β (n = 6), and colchicine + IL-1β (n = 7). Rats received intraperitoneal injections of sterilized phosphate-buffered saline (PBS) or rat recombinant IL-1β (0.1 μg/kg) 10 minutes before ischemia. For mechanistic investigations, blood and left ventricular tissue samples were collected at different times of reperfusion (15 minutes, 1 hour, and 2 hours) and stored at −80°C, and a hypothesis-driven selection of protein or messenger RNA (mRNA) targets was investigated. Thus, 6 further groups were added to the study: sham + vehicle (n = 4), sham + colchicine (n = 4), vehicle for 15-minute reperfusion (n = 7), colchicine for 15-minute reperfusion (n = 7), vehicle for 1-hour reperfusion (n = 7), and colchicine for 1-hour reperfusion (n = 8).
Area at Risk and Infarct Size Determination
At the end of the 2-hour reperfusion period, infarct size was measured in the first 4 groups (Figure 1). The heart was removed, and the LAD coronary artery was occluded using the monofilament suture kept in place. The heart was then retrogradely perfused with Evans blue (1%) so as to delineate the area at risk (AAR). Next, the heart was cut into 6 slices from apex to base, followed by incubation at 37°C in a 1% solution of phosphate-buffered 2,3,5-triphenyltetrazolium chloride to delineate the infarcted myocardium. Slices were subsequently fixed in 4% formalin, and infarct size was quantified by computerized planimetry using ImageJ software. The area of necrosis (AN) was expressed as a percentage of the AAR and the AAR as a percentage of the total left ventricular area.
Analysis of Systemic and Local Inflammatory Response
In additional experiment sets, rat hearts underwent 40-minute coronary occlusion and were collected after 2-hour reperfusion for analysis of systemic and local inflammatory cytokines. Rats were randomly assigned to one of the 2 following groups: vehicle (n = 12) and colchicine (n = 11). The analyses were performed on myocardial tissue from the AAR.
Blood Sampling and Serum Preparation
The serum used for multiplexed bead-based immunoassays was prepared by collecting blood into a Vacutainer serum separator tube (Becton Dickinson, Franklin Lakes, New Jersey), allowing it to clot for 15 minutes at room temperature. Then, it was centrifuged (1000g) for 15 minutes at +4°C. Serum was collected and centrifuged for a second time (10 000g) for 10 minutes before being stored at −80°C until analysis.
Left Ventricular Tissue Sample Collection and Preparation
Hearts were removed and placed in cold (2°C-4°C) PBS (10 mM; pH 7.4). There, the ventricular tissue at risk was excised and frozen on dry ice before being stored at −80°C. For analysis, tissue samples were thawed in 500 µL of cell lysis buffer (from the cell lysis kit; Bio-Rad, Hercules, California) containing protease inhibitor cocktail and 3 µL of a stock solution containing 500 mM phenylmethylsulfonyl fluoride in dimethyl sulfoxide (both from Sigma-Aldrich, St Louis, Missouri). Then, the samples were homogenized and sonicated with an ultrasonic dismembrator (EMAG Technologies, Inc, Ann Arbor, Michigan) at +4°C using 3 rapid pulses. The homogenate was then centrifuged at 4500g for 15 minutes at 4°C, and the supernatants were collected and stored at −80°C. The total protein concentration was determined using a DC Protein Assay Kit (Bio-Rad), according to the manufacturer’s instructions. All tissue samples were brought to a final total protein concentration of 9000 µg/mL.
Measurement of Inflammatory Cytokines
The concentrations of Erythropoietin (EPO), IL-1β, IL-1α, IL-2, IL-4, IL-5, IL-6, IL-7, IL-10, IL-12 (p70), IL-13, IL-17A, IL-18, Granulocyte colony-stimulating factor (G-CSF), Keratinocyte chemoattractant humain growth-regulated oncogene (GRO/KC), Macrophage colony-stimulating factor (M-CSF), Granulocyte-macrophage colony-stimulating factor (GM-CSF), Interferon-gamma (IFNγ), Monocyte chemoattractant protein-1 (MCP-1), Macrophage inflammatory protein-1 alpha (MIP-1α), MIP-3α, Regulated on activation normal, T cell expressend and secreted (RANTES), Tumor necrosis factor-alpha (TNF-α), Vascular endothelial growth factor (VEGF), and transforming growth factor-β1 (TGF-β1) were determined in sera and tissue homogenates by fluorescent-bead-based multiplex immunoassay (Luminex 200, Austin, Texas, Bio-Rad) using a commercially available Luminex screening assay kit (Bio-Plex Pro Rat Assay; Bio-Rad). The serum samples were 4-fold diluted with Diluent Kit before being incubated with specific antibody-coated fluorescent beads, according to the manufacturer’s instructions. Tissue homogenates were pretreated with a Cell Lysis Kit 16 prior to the experimentations. For TGF-β1 detection, serum samples were diluted to 1:16. Both serum and tissue samples were activated by adding HCl (to 1:5) followed by NaOH (vol/vol), as recommended by the manufacturer. Standard curves and samples were immediately processed using the Bio-Plex Manager software 6.1.
RNA Isolation and Determination of mRNA Expression Levels of Inflammatory Cytokines and Inflammasome Components
Total RNA was isolated from the ischemic area of the left ventricular tissue using the RNeasy Mini Kit (Qiagen, Venlo, The Netherlands), according to the manufacturer’s protocol. Then, 1000 ng of RNA extract was used to synthesize complementary DNA using the QuantiTect Reverse Transcription Kit (Qiagen). Quantitative real-time polymerase chain reaction (RT-PCR) was performed with the SYBR Select Master Mix (Applied Biosystems, Illkirch, France) using a LightCycler 480 real-time PCR system (Roche, Basel, Switzerland). Thermal cycling conditions were as follows: 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. The expression levels of NLRP3, pro-IL-1β, pro-IL-18, caspase-1, and TGF-β1 in the ventricular tissue at risk were quantified using RT-PCR and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Gusb, and Hprt housekeeping genes. Primer pairs were designed using Primer-Blast (NCBI-NIH), with those with a single dissociation peak and efficacy comprised between 1.85 and 2 retained. Results were expressed as 2(Ct target gene − Ct mean of ref genes). The primers used for RT-PCR are listed in Supplemental Table 1.
Measurement of Inflammasome Activation
Both IL-1β and IL-18 levels, along with caspase-1 activity, were determined in clarified homogenates of the ischemic tissue at 15 minutes and 1 hour postreperfusion and in the sham group, treated with colchicine or vehicle, and immediately frozen in liquid nitrogen. The frozen samples were homogenized on ice in 0.5 mL ice-cold lysis buffer containing 30 mM hydroxyethyl piperazine ethane sulfonic acid (HEPES), 20 mM KCl, 2.5 mM egtazic acid (EGTA), 2.5 Mm EDTA, 40 Mm sodium fluoride, 4 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 10% glycerol, 1% Nonidet P-40, a phosphatase inhibitor cocktail (Sigma, St Louis, Missouri), as well as a protease inhibitor cocktail (Complete Mini; Roche Applied Science, Mannheim, Germany). The homogenate was centrifuged at 13 000 rpm at 4°C for 1 hour, and the resulting supernatant was collected. Protein concentration was determined using the Bio-Rad DC protein assay kit (Bio-Rad), according to the manufacturer’s instructions. Protein extracts were brought to a final total protein concentration of 4500 µg/mL for IL-1β (R&D Systems) and IL-18 (Life Technologies, Carlsbad, California) assessment using a rat enzyme-linked immunosorbent assay kit, according to the manufacturer’s instructions.
Caspase-1 Activity
Caspase-1 activity was determined by cleavage of a luminogenic substrate using Caspase-Glo 1 Inflammasome Assay Kit (Promega, Madison, Wisconsin, USA). From each sample, 75 µg of proteins was used for the assay, according to the supplier’s instructions. Bioluminescence was measured 60 minutes later and expressed as relative luminescence units (RLUs) per 1 µg of sample (RLU/µg of protein).
Statistics
Statistical analyses were performed using SPSS version 17.0 (Chicago, Illinois). All values were expressed as mean ± standard error of the mean (SEM), unless stated otherwise. One-way analysis of variance (ANOVA) was conducted to assess the differences between groups, followed by post hoc Fisher least significant difference-corrected multiple comparison test. Two-way ANOVA was applied to assess IL-1β, IL-18, and caspase-1 activity measurements at several time points. The nonparametric Mann-Whitney test was used when the normal distribution between 2 variables could not be reasonably assessed. The significance level was set at P < .05.
Results
Colchicine Protected the Myocardium Against I/R Injury
First, we assessed the cardioprotective effect of a therapeutic colchicine dose (0.3 mg/kg body weight) administered per os prior to myocardial ischemia in the rat MI model. The AAR/LV ratio (AAR/LV) did not differ among the groups (Figure 1). Colchicine was associated with less myocardial injury as compared to the vehicle (AN/AAR = 35.6% ± 3.0% vs 46.6% ± 3.3%, P < .05; Figure 1C).
Colchicine Increased IL-10 and Decreased TGF-β1 Levels
The benefit of colchicine on infarct size was observed after 2 hours of reperfusion; hence, its effect on systemic and local inflammatory responses was assessed at this time point. Among the 25 cytokines analyzed by multiplexed bead-based immunoassay, only 2 proved significantly altered by colchicine at this time point: Median of the circulating level of the anti-inflammatory cytokine IL-10 was higher following colchicine treatment as compared to vehicle (492 [454-552] pg/mL vs 428 [382-473] pg/mL, P < .05; Figure 2A), whereas median of the myocardial level of TGF-β1 in the infarcted area was lower following colchicine treatment as compared to vehicle (294 [173-750] pg/mg vs 2004 [539-3147] pg/mg of protein, P < .01; Figure 2B).

Anti-inflammatory effects of colchicine at 2-hour reperfusion following myocardial infarction (MI). A, Systemic interleukin-10 (IL-10) levels in the rat serum. B, Cardiac transforming growth factor-β (TGF-β) levels in myocardial homogenates. Heart extracts were obtained from the ischemic tissue area of rats treated with either vehicle (n = 12) or colchicine (n = 12). Data were expressed as median and interquartile range. *P < .05 and **P < .01 versus vehicle.
Myocardial IL-1β Level Was Increased in the Early Reperfusion Phase
We assessed the effect of colchicine on inflammasome activation by measuring the levels of the pro-inflammatory cytokines IL-1β and IL-18 after 15 minutes and 1 hour of reperfusion. In the infarcted area, IL-1β level was increased in a time-dependent manner in vehicle- and colchicine-treated groups, reflecting the NLRP3 inflammasome activation in the early reperfusion phase (15 minutes; Figure 3A). However, colchicine did modulate neither the IL-1β nor IL-18 levels at the studied reperfusion times (Figure 3A and B).
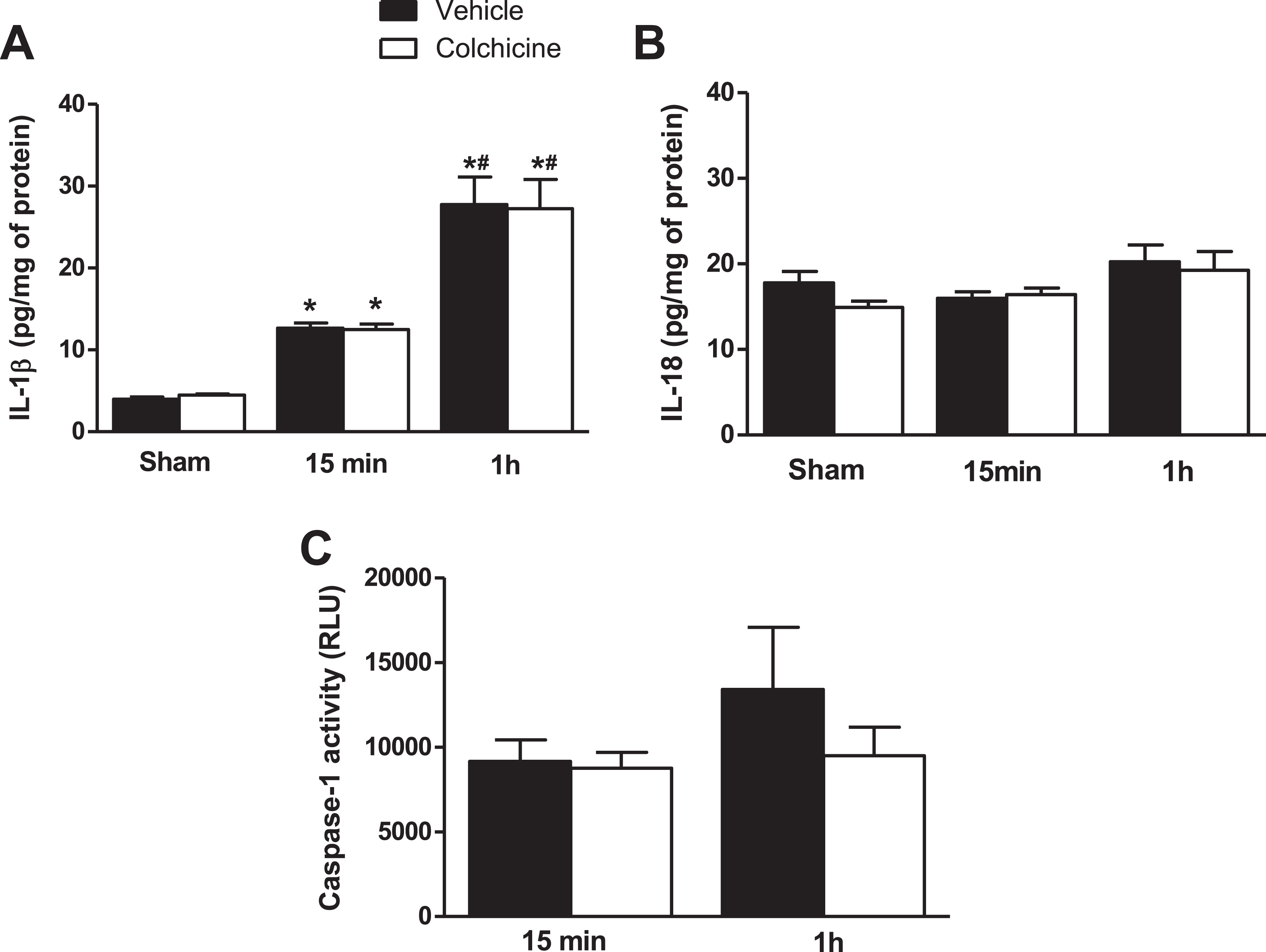
Inflammatory cytokine expression and caspase-1 activity measurement. Heart samples were obtained from control (sham) and ischemic area of rats at 15-minute and 1-hour reperfusion after myocardial ischemia/reperfusion (I/R) injury, treated either with vehicle or colchicine. Myocardial interleukin-1β (IL-1β; A), IL-18 (B) levels, and caspase-1 activity (C) were assessed. Data were expressed as mean ± standard error of the mean (SEM; n = 4 for sham group, n = 7 for 15-minute reperfusion group, n = 6-7 for IL-1β assessment at 1-hour reperfusion, and n = 4 for IL-18 assessment at 1-hour reperfusion). *P < .05 versus sham in the same group and #P < .05 versus 15-minute reperfusion in the same group.
Impact of Colchicine on Caspase-1 Activity
In order to determine whether colchicine impacts caspase-1 activity, we evaluated the enzymatic activity of caspase-1 at early reperfusion. Colchicine did not affect the caspase-1 activity whatever the time of reperfusion (Figure 3C).
Colchicine Attenuated IL-1β-Induced Myocardial Injury
Since IL-1β levels were increased in the infarcted area at early reperfusion suggesting their involvement in the pathophysiology of I/R injury, we assessed the effect of a single dose of IL-1β prior to myocardial ischemia, as well as the protective effect of colchicine against IL-1β-induced damages. As compared to the vehicle, IL-1β injection induced significantly greater infarct size (AN/AAR = 58.0% ± 2.0%, P < .05; Figure 1C). Interestingly, colchicine attenuated the IL-1β-induced myocardial injury as compared to the group that received only IL-1β (47.10% ± 2.2%, P < .05).
Impact of Colchicine on the NLRP3 Inflammasome in the Early Reperfusion Phase
Colchicine exhibited anti-inflammatory properties in our MI model. Thus, we evaluated its effect on inflammasome expression in the early reperfusion phase. At 15 minutes of reperfusion, RT-PCR revealed that colchicine inhibited the mRNA expression of NLRP3 inflammasome component caspase-1, but not NLRP3 in the infarcted area (Figure 4). In addition, colchicine inhibited the mRNA expression of TGF-β1 and that of the pro-inflammatory cytokine pro-IL-18, while pro-IL-1β remained unaffected. Inflammasome expression was not affected by colchicine at 1-hour reperfusion (data not shown).

Effects of colchicine on messenger RNA (mRNA) expression of nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome components. Myocardial mRNA levels of pro-interleukin-1β (IL-1β), pro-IL-18, caspase-1, NLRP3, and transforming growth factor-β (TGF-β) were assessed at 15-minute of reperfusion after acute myocardial infarction (AMI) by real-time quantitative polymerase chain reaction (qPCR) analysis. Data were expressed as mean ± standard error of the mean (SEM; n = 7 for each group). *P < .05 versus vehicle.
Discussion
Recent studies have reported that colchicine can limit infarct size and inhibit the increased expression of inflammatory cytokines after MI. 10,11 However, these studies focused on myocardial inflammation hours and days after the occurrence of MI and long-term cardiac remodeling. Here, we sought to analyze the effect of a colchicine pretreatment on infarct size and its potential impact on myocardial inflammation within minutes (15, 60, and 120 minutes) after reperfusion. Our main findings were as follows: (1) early activation of NLRP3 inflammasome and IL-1β release occurred after I/R, (2) a single IL-1β injection prior to I/R increased infarct size, (3) a short colchicine treatment course given 24 hours and immediately before I/R reduced infarct size observed 2 hours after reperfusion, (4) colchicine pretreatment was associated with increased systemic IL-10 levels and decreased cardiac TGF-β levels, and (5) colchicine attenuated the deleterious effect of IL-1β injection on infarct size.
In human, Deftereos et al 17 recently reported on lower serum creatine kinase and troponin levels, as well as on a smaller infarct size assessed by magnetic resonance imaging in a patient cohort treated with colchicine in the acute ST-Elevation Myocardial Infarction (STEMI) phase. The mechanism involved in this acute protection remained unclear. In our rat model, colchicine treatment increased systemic IL-10 levels. Interleukin-10 is pro-angiogenic factor induced in the reperfused myocardium and associated with an anti-inflammatory response that modulates the reaction to I/R injury. 18 Neutrophil infiltration plays a key role in the inflammatory process post-MI at early reperfusion while worsening the infarct size. 19 In a knockout mice IL-10−/− model, myocardial injury was exacerbated and accompanied by neutrophil infiltration into the myocardium after I/R. 20 Administration of exogenous IL-10 was associated with a decreased infarct size, along with an inhibition of neutrophil accumulation within the injured myocardium. 21 Similarly, a preclinical study showed that remote ischemic preconditioning (RIPC) was associated with upregulation of both serum and cardiac IL-10 levels, while a loss of these genes was linked to a loss of RIPC-induced cardioprotection. 22 Due to its anti-inflammatory properties, colchicine is known to decrease neutrophil infiltration within the injured myocardium. 23 -25 This drug could thus exert its cardioprotective effects by enhancing systemic IL-10 levels, thereby inhibiting neutrophil infiltration into the myocardium after MI. We then suggest a link between colchicine and the anti-inflammatory cytokine IL-10 as a potential cardioprotective mechanism of the drug at the acute phase of MI.
In addition to its acute IL-10 modulation, colchicine also decreased RNA expression and TGF-β levels in the infarcted tissue. Because of its crucial role in cardiac injury, repair, and remodeling, 26,27 the TGF-β system may be a promising therapeutic target for patients with heart failure. 28 Recently, the beneficial effects of a single dose of colchicine on cardiac remodeling have been demonstrated in a mouse MI model, resulting in less cardiac fibrosis and improved hemodynamic parameters. 10 Moreover, this antifibrotic effect was also demonstrated in a rabbit heart failure model, resulting in reduced left atrial fibrosis and additional antiarrhythmic effects by suppressing atrial fibrillation. 29 Its antifibrotic effect has also been proven in the pericarditis setting, 30 as well as in several organs such as the liver, 31 lung, 32 and kidneys. 33 Although the exact mechanism by which colchicine exerts its antifibrotic effect remains unclear, it may be linked to its anti-inflammatory effect.
Inflammasome plays an essential role in the pathophysiology of myocardial I/R injury and thus constitutes a novel therapeutic target for preventing myocardial damages. 1 Several experimental studies reported the beneficial effects of inhibiting the NLRP3 inflammasome in AMI. 34,35 Beyond colchicine’s anti-inflammatory effect on the injured myocardium, the mechanism by which it protects the heart remains to be elucidated. Using a mouse model of permanent coronary artery ligature, Fujisue et al showed that colchicine attenuated the mRNA expression of NLRP3 inflammasome components after MI. 11 In the current study, we have explored the therapeutic window for NLRP3 inflammasome inhibition by colchicine during early reperfusion. Among the 25 cytokines analyzed at 2 hours of reperfusion, IL-10 and TGF-β1 only were altered by colchicine. Furthermore, IL-1β was increased in a time-dependent manner in the early reperfusion phase, but colchicine did modulate neither the IL-1β nor IL-18 levels. In these specific experimental conditions, the cardioprotective mechanism of colchicine could have been more certainly mediated by systemic elevation of IL-10 levels and TGF-β1 system downregulation in the infarcted myocardium than by inhibition of NLRP3 inflammasome activation.
A recent clinical study hypothesized that NLRP3 inflammasome and associated pro-inflammatory cytokine IL-1β release may constitute cardiovascular risk biomarkers, particularly in post-AMI patients with otherwise low risk scores. 36 In AMI, IL-1β is initially released by ischemic endothelial cells and cardiomyocytes during ischemia and, at a later time point, by leukocytes infiltrating the myocardium. 4 Our results showed that IL-1β levels increased in the infarcted area at reperfusion onset. Additionally, we clearly showed that IL-1β levels increased with reperfusion in a time-dependent manner, reflecting the NLRP3 inflammasome activation at early reperfusion. Although IL-1β levels are associated with infarct healing due to leukocyte recruitment, these cytokines also promote cell death in cardiomyocytes and cardiac fibroblasts. 4 Several studies have indeed reported the usefulness of blocking IL-1β in the context of AMI. A recent clinical trial showed that IL-1 blockade with anakinra proved safe, favorably affecting LV remodeling in patients with ST-segment elevation in AMI. 37 In another study, the same authors had already previously reported that the recombinant human IL-1 receptor antagonist anakinra improved cardiac remodeling after a large anterior wall AMI in an experimental mouse model while improving survival. 37 Moreover, mice with IL-1 type I receptor (IL1RI−/−) deletion were protected against adverse cardiac remodeling, 38 thus further supporting the critical role of IL-1 activity in AMI. In this context, we showed that a single dose of rat recombinant IL-1β was able to increase the infarct size after MI, resulting in an inflammatory injury. Colchicine administration reduced the infarct size by limiting the IL-1β-induced inflammatory injury. This is in accordance with Toldo et al, 35 who demonstrated, in a mouse AMI model, that pharmacological inhibition of the NLRP3 inflammasome within 1 hour of reperfusion was able to limit the secondary inflammatory injury and infarct size. 35 We then confirm the interest of pharmacological inhibition of inflammation at early reperfusion. However, the effect of colchicine on myocardial IL-1β and IL-18 levels remains a missing key in the relationship between colchicine treatment and the attenuation of inflammasome activation in our experimental model of AMI.
Contrarily, although experimental studies in mice with genetic caspase-1 deletion have reported caspase-1 inhibition to be a potential therapeutic target for pharmacologic intervention in the AMI setting, 39,40 in our experimental model, colchicine did not affect the caspase-1 activity, whatever the time of reperfusion.
Taken together, these data enhance our understanding of the mechanism of action of the drug and confirm that colchicine protects the ischemic heart against myocardial I/R injury through an anti-inflammatory effect. In the acute reperfusion onset, anti-inflammatory effect might be other than NLRP3 inflammasome inhibition. It might be conducted through an elevation of IL-10, the attenuation of the Toll-like receptor (TLR 4/9) activation, and thus TGF-β system downregulation in the infarcted myocardium. From a clinical perspective, it would be relevant to test an acute colchicine treatment given as early as possible before reperfusion to target IL-10 and TGF-β expression, in combination with a prolonged colchicine treatment to limit the secondary inflammatory injury through the NLRP3 inflammasome inhibition.
Limits
This study presents several limitations. We have reported the cardioprotective effects of a colchicine pretreatment initiated 24 hours before and repeated immediately prior to myocardial I/R. Because oral treatment administration was not easily feasible during coronary occlusion or immediately after in animals under deep anesthesia, we decided to work with a pretreatment. We initiated the pretreatment 24 hours before the onset of coronary occlusion because colchicine anti-inflammatory effects appear 24 to 48 hours after ingestion. 41 This timing was chosen to assess whether a short pretreatment colchicine course may affect I/R injury by modulating inflammation response. However, these results cannot be extrapolated to the clinical AMI scenario.
Another limitation of the study lies in the timing of studying IL-1β and IL-18 protein expression to assess the colchicine effect on inflammasome activation at early reperfusion. We chose early reperfusion times (15-minute and 1-hour reperfusion) for assessing inflammasome activation and colchicine’s beneficial effects, given that the cardioprotective role of colchicine was observed at 2-hour reperfusion. Although colchicine inhibited the mRNA expressions of caspase-1, TGF-β1, and IL-18 at early reperfusion, it did not affect the protein expression of inflammasome components. Further studies are thus required to determine the precise timing of the colchicine inhibition effect on inflammasome activation.
Although this study was focused on colchicine’s effect on the ischemic heart, it is not clear yet whether colchicine exerts its inhibitory effects on either infiltrated leukocytes (neutrophils) or resident cell populations. Lastly, animals used for experimental AMI were mostly young and healthy, whereas patients suffering from AMI tend to be elderly with comorbidities such as diabetes that may highly influence inflammation response to I/R.
Conclusion
In our experimental model, a short colchicine pretreatment course was effective in decreasing reperfusion injuries, accompanied by systemic anti-inflammatory and local antifibrotic effects.
Supplemental Material
SUPPLEMENTARY_DATA_(1) - Cardioprotective Role of Colchicine Against Inflammatory Injury in a Rat Model of Acute Myocardial Infarction
SUPPLEMENTARY_DATA_(1) for Cardioprotective Role of Colchicine Against Inflammatory Injury in a Rat Model of Acute Myocardial Infarction by Oussama Bakhta, Simon Blanchard, Anne-Laure Guihot, Sophie Tamareille, Delphine Mirebeau-Prunier, Pascale Jeannin, and Fabrice Prunier in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Acknowledgments
The authors would like to thank the “Service Commun d’Animalerie Hospitalo-Universitaire,” the University Hospital Joint Animal Care Service, for taking care of the animals.
Authors’ Contribution
Oussama Bakhta contributed to conception and design, contributed to acquisition, analysis, and interpretation, and drafted the manuscript. Simon Blanchard and Anne-Laure Guihot contributed to acquisition and analysis. Sophie Tamareille contributed to acquisition. Pascale Jeannin contributed to design. Fabrice Prunier contributed to conception and design, critically revised the manuscript, and gave final approval. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Oussama Bakhta was supported by a fellowship from the French Ministry of Education and Research.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.