
Abstract
The effect of TGF-β1 on CpG DNA-induced type I IFN production was examined by reconstituting a series of signaling molecules in TLR 3 signaling. TGF-β1 inhibited CpG DNA-induced IFN-α4 productivity in HeLa cells. Transfection of IFN regulatory factor (IRF)7 but not TNF receptor-associated factor (TRAF)6 and TRAF3 into cells triggered IFN-α4 productivity, and TGF-β1 inhibited IRF7-mediated type I IFN production in the presence of TRAF6. TGF-β1 induced ubiquitination of TRAF6, although CpG DNA did not induce it. Moreover, TGF-β1 accelerated the ubiquitination of TRAF6 in the presence of CpG DNA. TGF-β1 ubiquitinated TRAF6 at K63 but not K48. TGF-β1 also induced ubiquitination of IRF7. Further, TGF-β1 did not impair the interaction of IRF7 and TRAF6. CpG DNA induced the phosphorylation of IRF7 in the presence of TRAF6, whereas TGF-β1 inhibited the IRF7 phosphorylation. Blocking of TRAF6 ubiquitination abolished the inhibition of CpG DNA-induced type I IFN production by TGF-β. Taken together, TGF-β was suggested to inhibit CpG DNA-induced type I IFN production transcriptionally via ubiquitination of TRAF6.
Keywords
Introduction
TGF-β1 has a multifunctional action on cell proliferation, differentiation, migration, apoptosis and carcinogenesis in many cell types. 1 It acts as a negative autocrine growth factor for immune cells.2,3 Therefore, TGF-β1 secreted by most immune cells plays an important role in controlling the immune system. 4 TLRs are the major cell surface initiators of inflammatory responses to pathogens and recognize PAMPs.5,6 TLRs 3, 7, 8 and 9 have the ability to recognize viral nucleic acids and induce anti-viral responses through producing the type I IFN. 5 In particular, TLR9 is expressed in numerous immune cells and recognizes unmethylated CpG sequences in DNA molecules.7–9 TLR9, reactive to CpG DNA, is expressed intracellularly within the endosomal compartments and functions to alert the immune system to viral and bacterial infections. 10 Interaction between CpG DNA and TLR9 causes the complex formation consisting of MyD88, IL-1 receptor-associated kinase (IRAK)1, TNF receptor-associated factor (TRAF)3, TRAF6, IκB kinase (IKK) and IFN regulatory factor (IRF)7, and triggers the production of type I IFN via activation of TRAF6 and IRF7.11,12 The type I IFNs produced are the first line of innate immunity against viral and bacterial infections. 13 There are several reports on the inhibition of CpG DNA-induced type I IFN production by TGF-β1.14,15 However, the precise mechanism of the inhibitory action of TGF-β1 is not fully characterized. In the present study, we characterized the molecular mechanism of the inhibitory action of TGF-β1 on CpG DNA-induced type I IFN production by in vitro reconstitution of a series of signaling molecules involved in TLR9 signaling. Here, we report that TRAF6 plays a pivotal role in the inhibition of CpG DNA-induced type I IFN production by TGF-β1.
Materials and methods
Reagents and Abs
Recombinant human TGF-β1 was obtained from R&D Systems (Minneapolis, MN, USA). CpG ODN2216 as CpG DNA was obtained from Invivogen (San Diego, CA, USA). Abs to MyD88 (N-19 and HPL296), TRAF6 (H-274), TRAF3 (H-122), actin (I-19) and IRF7 (H-246) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Abs to HA and FLAG were purchased from Nacalai tesque (Kyoto, Japan). Abs to K48R or K63R linkage-specific ubiquitin, phosphorylated IRF7 (S471/472) and p38 were purchased from Cell Signaling Technology (Beverly, MA, USA).
Cell culture
HeLa and 293 T cells were cultured in D-MEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated FBS and antibiotic cocktail from Sigma (St. Louis, MO, USA) at 37℃ under 5% CO2.
Constructs
Plasmid DNA used in transfection experiments was isolated with an endotoxin-free Plasmid Maxi kit (Qiagen, Valencia, CA, USA). A mouse IFN-α4, IFN-α6 and IFN-β promoter luciferase reporter plasmid were obtained from Dr. T. Sugiyama (Gifu Pharmaceutical University, Gifu, Japan). 16 The K43 or K63 mutant plasmid was obtained from Dr. Y. Kitagawa (Shiga University of Medical Science, Shiga, Japan).17,18 The IFN-stimulated response element (ISRE) and pRL-TK Renilla luciferase control reporter vectors were purchased from Promega (Fitchburg, WI, USA).
Transfection and luciferase assay
HeLa and 293 T cells were transfected as described elsewhere. 17 Briefly, the total amount of cDNA transfected was kept constant. After overnight transfection, 293 T and HeLa cells were stimulated with CpG DNA (3 μM) for 0–8 h in the presence or absence of TGF-β1 (30 ng/ml) for 1 h. The pRL control vector (10 ng) was used as an internal transfection control. Following 5 h incubation, cells were washed once in PBS and lysed, and the luciferase activities were measured as described previously. 17
Immunoprecipitation
Cells were lysed for 30 min on ice with lysis buffer (5 mM EDTA, 150 mM NaCl, 0.2 mM orthovanadate, 0.5% NP40, 5 mM DTT, each of the protease inhibitors pepstatin, leupeptin, aprotinin, antipain and chymostatin, 30 µg/ml TPCK and 1 mM PMSF). After cell lysis, cell debris was removed by centrifugation. The supernatants collected were adjusted at 1 mM sodium orthovanadate and 0.1 mM quercitin, and stored at –80℃ until use. The protein concentration was determined by a Bradford assay (Pierce, Rockford, IL, USA). Two µg of the corresponding Ab was added to each cellular extract (130 µg) and the mixtures were incubated at 4℃ for 3–5 h on a rotator. Then, 30 µl of protein G and protein A agarose (Calbiochem, La Jolla, CA, USA) was added to each sample, followed by incubation for 2 h at 4℃. The precipitates were washed three times with lysis buffer and the samples were eluted by boiling for 3 min in 2× sample buffer. The immunoprecipitates were used for immunoblotting analysis.
Immunoblotting
The samples immunoprecipitated (130 µg) were run with SDS-PAGE under reducing conditions. Separated proteins were electrically transferred onto PVDF transfer membranes (Millipore, Damstadt, Germany). The membranes were blocked with 5% skimmed milk for 3 h and then treated with appropriately diluted Abs for 3 h. Bands were detected with chemiluminescence using supersignal west dura extended duration substrate (Thermo Scientific Pierce, Loughborough, UK) according to the manufacturer’s recommendations. The chemiluminescence was analyzed by a light capture system analyzer AE6955 (Atto, Tokyo, Japan).
ELISA
The IFN concentration in the supernatants was determined with the VeriKine human IFN-α ELISA kit (PBL Assay Science, Piscataway, NJ, USA).
Statistical analysis
The data shown are the mean ± SD of a typical experiment of three independent experiments. The statistical significance of differences between mean values was determined by Student’s t-test and ANOVA in Excel (Microsoft, Redmond, WA, USA). A P-value < 0.05 was considered significant.
Results
TGF-β1 down-regulates the production of type I IFN in response to CpG DNA
Inhibition of CpG DNA-induced type I IFN production by TGF-β1 was confirmed using an IFN-α4 luciferase activity assay (Figure 1). HeLa cells were transiently transfected with IFN-α4 luciferase assay plasmid, treated with TGF-β1 (30 ng/ml) for 1 h,
17
and then stimulated with CpG DNA (3 μM) for 1 h.
18
CpG DNA alone markedly enhanced the IFN-α4 luciferase activity. However, pretreatment with TGF-β1 reduced the luciferase activity in response to CpG DNA. In the following experiments, cells were treated with TGF-β1 at 30 ng/ml and/or CpG DNA (3 μM) unless otherwise stated.
Effect of TGF-β1 on CpG DNA-induced type I IFN production. HeLa cells were transiently transfected with a plasmid with IFN-α4 luciferase activity. Cells were pretreated with TGF-β1 (30 ng/ml) for 1 h and stimulated with CpG DNA (3 μM) for 1 h * P < 0.01 vs. untreated control.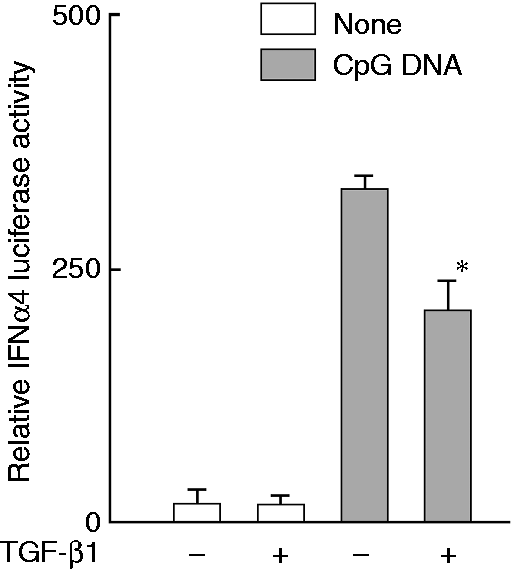
TGF-β1 inhibits IRF7-mediated type I IFN productivity in the presence of TRAF6
CpG DNA-induced type I IFN production was triggered by IRF7 as a transcriptional factor via the complex formation consisting of MyD88, IRAK1, TRAF3 and TRAF6 and IKKβ.
19
Therefore, the inhibitory action of TGF-β1 on CpG DNA-induced type I IFN production was studied by reconstituting those molecules in vitro. First, the involvement of TRAF3 and TRAF6 in the TGF-β1-mediated inhibition was examined (Figure 2a). HeLa cells were transiently transfected with plasmids for IFN-α4 luciferase activity, TRAF6, TRAF3 or IRF7, and then treated with TGF-β1 for 4 h. Transfection of IRF7 augmented the IFN-α4 luciferase activity, whereas that of TRAF6 or TRAF3 did not. TGF-β1 inhibited IRF7-mediated enhancement of the luciferase activity in the presence of TRAF6 but not TRAF3. TGF-β1 alone did not significantly reduce the IRF7-mediated luciferase activity, suggesting that the inhibition by TGF-β1 required the presence of TRAF6.
Effect of TGF-β1 on type I IFN productivity in cells reconstituted with various signal molecules. HeLa cells were transiently transfected with a luciferase assay plasmid of (a) IFN-α4 or (b) IFN-β, and indicated plasmids for overnight, and then stimulated with TGF-β1 (30 ng/ml) for 4 h. (c) HeLa cells were transfected with plasmids of IFN-β, TRAF6 (µg/ml, treated with or without TGF-β1 (30 ng/ml) for 1 h and then stimulated with CpG DNA (3 μM) for 4 h. **P < 0.01, **P < 0.005 vs. untreated control.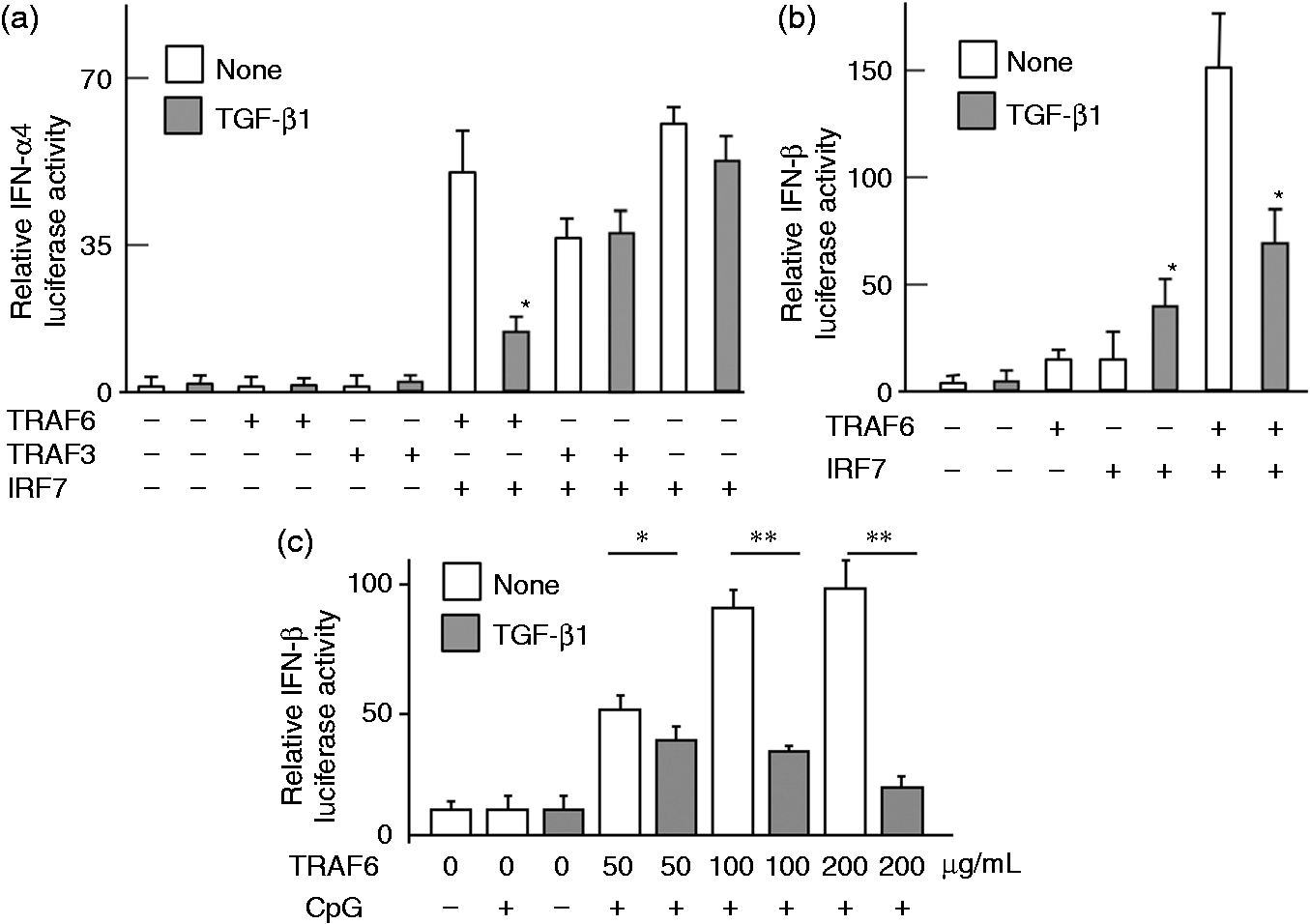
The involvement of TRAF6 and IRF7 on the IFN-β productivity was also examined (Figure 2b, c). HeLa cells were transiently transfected with plasmids for IFN-β luciferase assay, TRAF6 and IRF7, and then treated with TGF-β1 for 4 h. Although the IFN-β luciferase activity was markedly augmented in the presence of TRAF6 and IRF7, TGF-β1 reduced it. However, TGF-β1significantly enhanced the IFN-β luciferase activity in the presence of IRF7 alone. TRAF6 enhanced the IFN-β luciferase activity in a dose-dependent manner (Figure 2c). In addition, neither IRF7 nor TRAF6 significantly augmented the IFN-β productivity.
TGF-β1 induces ubiquitination of TRAF6
As TGF-β1 inhibited IRF7-mediated type I IFN production in the presence of TRAF6, the effect of TGF-β1 on the ubiquitination of TRAF6 was examined (Figure 3). First, the time course of TGF-β1-induced TRAF6 ubiquitination was examined (Figure 3a). HeLa cells were transfected with TRAF6 plasmid and then treated with TGF-β1 for various lengths of time. TRAF6 was immunoprecipitated with anti-TRAF6 Ab and the ubiquitinated TRAF6 in immunoprecipitates was detected with immunoblotting using anti-ubiquitin Ab. The ubiquitinated TRAF6 was detected 0.5 h after TGF-β1 treatment. Its expression increased up to 1–2 h and then decreased 3–4 h after the treatment. Next, HeLa cells transfected with TRAF6 plasmid were stimulated with or without CpG DNA for 2 or 4 h. Although CpG DNA did not affect TRAF6 ubiquitination, TGF-β1 induced the ubiquitination of TRAF6 2 or 4 h after the stimulation and the TRAF6 ubiquitination at 2 h in the presence of CpG DNA was more than that in the absence of it (Figure 3B). The TRAF6 ubiquitination by TGF-β1 was accelerated on stimulation with CpG DNA.
Effect of TGF-β1 on ubiquitination of TRAF6. (a) HeLa cells were treated with TGF-β1 (30 ng/ml). (b) HeLa cells were transfected with TRAF6 plasmid and then stimulated with CpG DNA (3 μM) and/or TGF-β1 (30 ng/ml) for 2 or 4 h. (c) 293 T cells were transfected with plasmids of TRAF6, Myc-tagged wild type ubiquitin, lysin (K) 63 or K48-mutated ubiquitin overnight and then treated with TGF-β1 (30 ng/ml) for 8 h. The expression of ubiquitinated TFAF6 was analyzed with immunoblotting.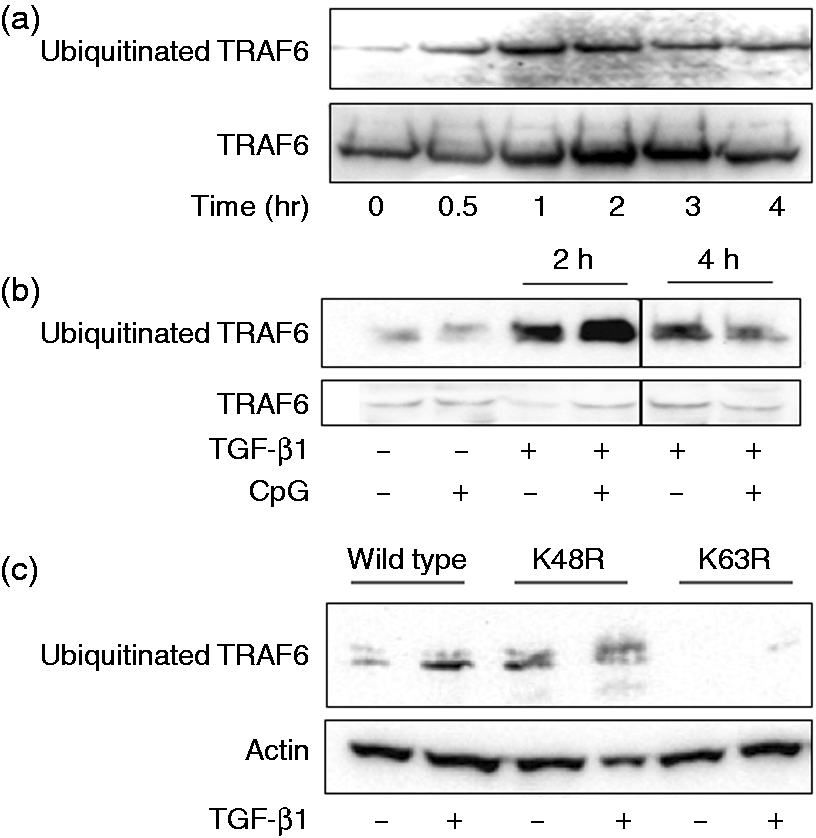
There are two ubiquitination sites at lysin (K) 48 and K63 in TRAF6. 20 K48-linked-ubiquitination promotes proteasomal degradation whereas K63-linked one regulates the activation of proinflammatory signaling.21–24 To determine whether TGF-β1-induced ubiquitination is linked to K48 or K63, 293 T cells were transfected with Myc-tagged K48 or K63 mutant ubiquitin plasmid and then stimulated with TGF-β1. Immunoblotting analysis demonstrated that the ubiquitination was detected in K48R but not K63R-mutant TRAF6-transfected cells, suggesting that TGF-β1-induced TRAF6 ubiquitination was linked to K63 (Figure 3c).
TGF-β1 induces ubiquitination of IRF7, as well as TRAF6
In the preceding section, TGF-β1 was determined to induce the K63 ubiquitination of TRAF6. Therefore, the effect of TGF-β1 on the K63 ubiquitination of IRF7 was examined (Figure 4). HeLa cells transfected with IRF7 plasmid were treated with TGF-β1 for various lengths of time. TGF-β1 induced the ubiquitination of IRF7 at K63 1–2 hr after the treatment.
Effect of TGF-β1 on the ubiquitination of IRF7. HeLa cells were treated with TGF-β1 (30 ng/ml) for various h. The K63 ubiquitination was determined with immunoblotting using anti-K63 ubiquitin Ab.
CpG DNA induces the interaction between IRF7 and TRAF6
First, the interaction between TRAF6 and IRF7 in response to CpG DNA was examined (Figure 5). HeLa cells were transiently transfected with TRAF6 and IRF7 plasmids, and then stimulated with CpG DNA for 4 h. In order to confirm the complex formation between IRF7 and TRAF6, the presence of IRF7 in the TRAF6 fraction immunoprecipitated by anti-TRAF6 Ab was detected with immunoblotting using anti-IRF7 Ab. Immunoblotting analysis demonstrated the presence of IRF7 in the TRAF6 immunoprecipitates, suggesting the physical interaction between IRF7 and TRAF6. Further, the presence of ubiquitinated IRF7 and TRAF6 was examined with anti-ubiquitin Ab. CpG DNA did not significantly induce the ubiquitination of TRAF6 and IRF7.
Effect of CpG DNA on the interaction of IRF7 and TRAF6. HeLa cells were transiently transfected with both TRAF6 and IRF7 and then stimulated with 3 μM CpG DNA for various lengths of time. The TRAF6 fraction was immunoprecipitated with an anti-TRAF6 Ab and the presence of TRAF6 and IRF7 in the TRAF6 immunoprecipitates was determined with immunoblotting.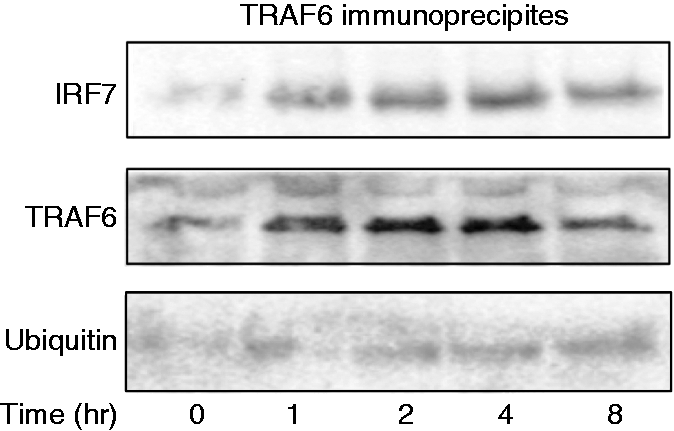
TGF-β1 does not impair the interaction of TRAF6 and IRF7
The effect of TGF-β1 on the interaction between TRAF6 and IRF7 was examined (Figure 6a). HeLa cells transfected with TRAF6 and IRF7 plasmids were treated with TGF-β1 for various lengths of time. TRAF6 was marginally detected in the IRF7 fraction immunoprecipitated before TGF-β1 treatment. The presence of TRAF6 in the immunoprecipitates increased 1 h after TGF-β1 treatment but it declined at 4 h. Further, the physical interaction between TRAF6 and IRF7 was confirmed using FLAG protein (Figure 6b). The analysis with FLAG also confirmed that TGF-β1 induced the complex formation of TRAF6 and IRF7, suggesting no effect of TGF-β1 on the interaction between TRAF6 and IRF7.
Effect of TGF-β1 on the interaction of IRF7 and TRAF6. (a) HeLa cells were transiently transfected with TRAF6 and IRF7, and then treated with TGF-β1 (30 ng/ml) for various lengths of time. The IRF7 fraction was immunoprecipitated with anti-IRF7 Ab and the presence of TRAF6 in immunoprecipitates were determined with immunoblotting using anti-TRAF6 Ab. (b) HeLa cells were transiently transfected with FLAG-tagged TRAF6 and HA-tagged IRF7. Transfected cells were treated with TGF-β1 (30 ng/ml) for various lengths of time. The FLAG fraction was immunoprecipitated with anti-Flag Ab and the presence of HA in the immunoprecipitates were determined with immunoblotting using anti-HA Ab. (c) HeLa cells were transiently transfected with TRAF3, RIP1 and IRF7, and treated with TGF-β1 (30 ng/ml) for various lengths of time. The IRF7 fraction was immunoprecipitated with anti-IRF7 Ab and the presence of TRAF3 or RIP1 in the immunoprecipitates was determined with immunoblotting using anti-TRAF3 or RIP1 Ab.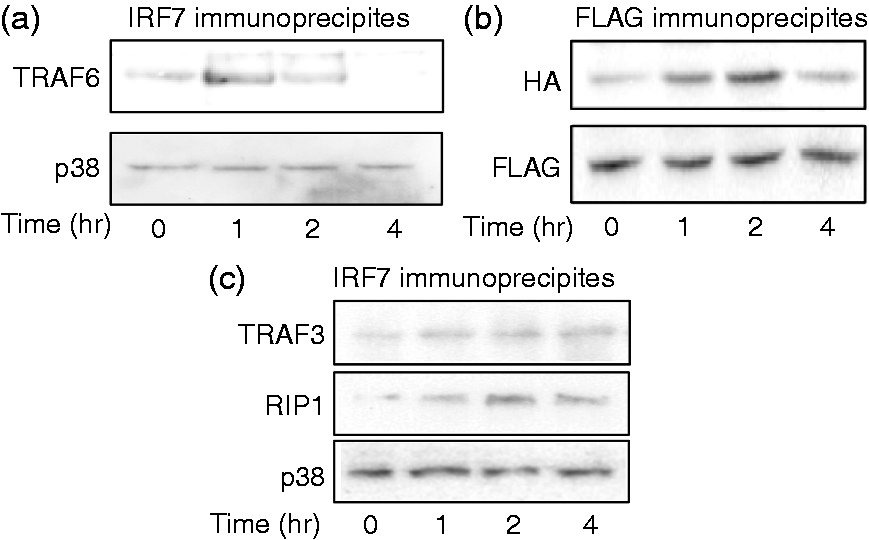
The effect of TGF-β1 on the interaction of IRF7 with TRAF3 or RIP1 was examined (Figure 6c). Neither TRAF3 nor RIP1 was marginally detected in IRF7 immunoprecipitates and the interaction was not significantly changed by TGF-β1 treatment.
TGF-β1 inhibits IRF7 phosphorylation in response to CpG DNA
First, the effect of CpG DNA on phosphorylation of IRF7 was examined. HeLa cells transfected with IRF7 plasmid were stimulated with CpG DNA and the phosphorylation of IRF7 was determined with immunoblotting (Figure 7a). CpG DNA induced the phosphorylation of IRF7 1 h after the treatment and a high level of phosphorylated IRF7 expression was detected 2 and 4 h after CpG DNA treatment. Next, the effect of TGF-β1 on CpG DNA-induced IRF7 phosphorylation was examined (Figure 7b). HeLa cells transfected with IRF7 plasmid were stimulated with CpG DNA in the presence or absence of TGF-β1. The expression of phosphorylated IRF7 was determined 2 and 4 h after stimulation with immunoblotting. CpG DNA clearly induced the phosphorylation of IRF7 4 h after the stimulation. However, TGF-β1 inhibited CpG DNA-induced IRF7 phosphorylation at 4 h.
Effect of TGF-β1 on the phoshorylation of IRF7 in response to CpG DNA. (a) HeLa cells were transiently transfected with IRF7 and treated with 3 μM CpG DNA for various lengths of time. (b) HeLa cells were pretreated for 1 h with TGF-β1 (30 ng/ml) and then stimulated with CpG DNA (3 μM) for 2 or 4 h. The IRF7 fraction was immunoprecipitated with anti-IRF7 Ab and the presence of phosphorylated (phospho) IRF7 in the immunoprecipitates was determined with immunoblotting.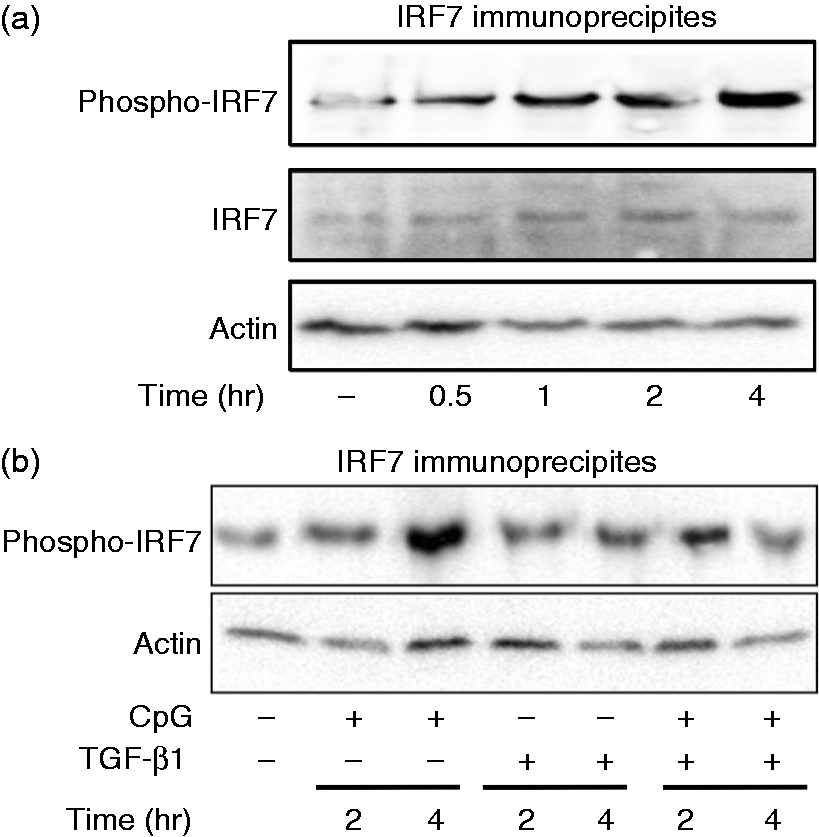
TGF-β1 inhibits CpG DNA-induced type I IFN production via TRAF6 K63 ubiquitination
The effect of K63 ubiquitination of TRAF6 on ISRE and IFN-β activities in response to CpG DNA was examined (Figure 8a, b). HeLa cells were transfected with wild type or K63R mutant ubiquitin plasmid and further plasmid for ISRE or IFN-β luciferase assay, and stimulated with or without CpG DNA for 4 h. Both ISRE and IFN-α4 luciferase activities increased in wild type ubiquitin plasmid-transfected cells. However, K63R mutant did not augment them. Next, the involvement of K63 ubiquitination in the inhibition by TGF-β was examined (Figure 8c). HeLa cells were transfected with wild type or K63R mutant ubiquitin plasmid, treated with or without TGF-β1 for 1 h and then stimulated with CpG DNA. The inhibition of CpG-DNA-induced type I IFN production by TGF-β was detected in wild type plasmid but not K63R-mutant one-transfected cells, suggesting that K63 ubiquitination might be required for inhibition of the type I IFN production by TGF-β.
Effect of K63R mutant on CpG DNA-induced type I IFN production and the inhibition by TGF-β. HeLa cells were transfected with plasmids of TRAF6, wild type (wt) ubiquitin, or K63-mutated ubiquitin overnight, transfected with luciferase assay plasmid of (a) ISRE or (b) IFN-α4, and then stimulated with or without CpG DNA (3 μM) for 5 h. *P < 0.01, **P < 0.05 vs. untreated control. (c) HeLa cells were transfected with plasmids of TRAF6, wild type ubiquitin, and K63-mutated ubiquitin overnight, treated with or without TGF-β1 (30 ng/ml) for 1 h and then stimulated with or without CpG DNA (3 μM) for 4 h. *P < 0.05 vs. wild type.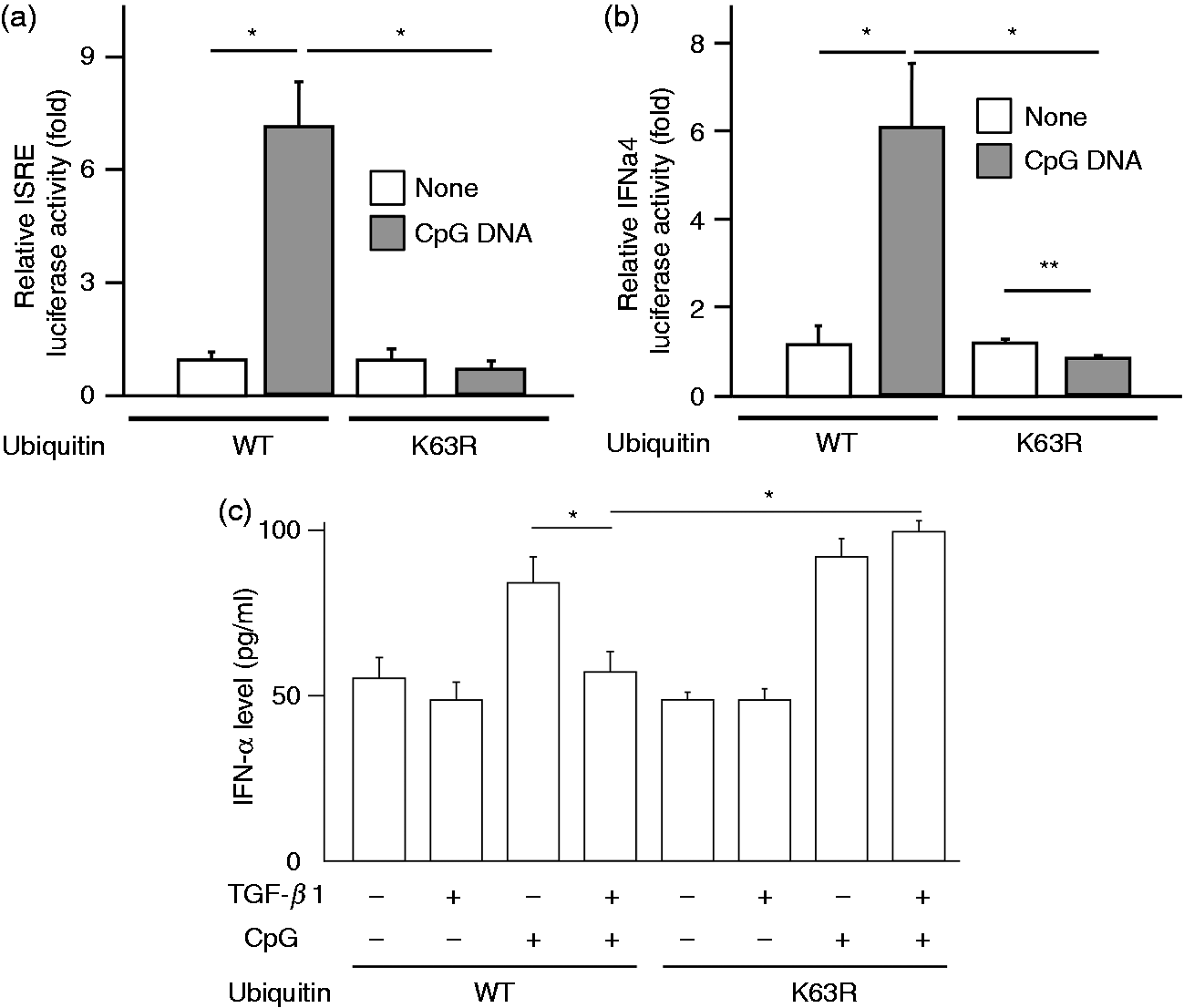
TGF-β1 does not affect the expression of MyD88 in response to CpG
As TGF-β1 induces degradation of MyD88 in response to LPS, 17 the effect of TGF-β1 on the expression of MyD88 in response to CpG DNA was examined by immunoblotting (Supplementary Figure 1). HeLa cells transfected with a MyD88 plasmid were stimulated with CpG DNA and TGF-β1 for various lengths of time. The MyD88 expression did not alter within 8 h in response to TGF-β1, excluding involvement of MyD88 degradation by TGF-β1 in the inhibition.
Discussion
In the present study, we have demonstrated that TRAF6 is involved in the inhibition of CpG DNA-induced type I IFN production by TGF-β1. Our finding shows that CpG DNA as a TLR9 ligand causes the physical interaction between TRAF6 and IRF7, induces the activation of IRF7 and leads to the type I IFN production. However, TGF-β1 but not CpG DNA induces ubiquitination of TRAF6 and IRF7. However, TGF-β1 does not impair the interaction of ubiquitinated TRAF6 and IRF7. Under the interaction of ubiquitinated TRAF6 and IRF7, the phosphorylation of IRF7 is down-regulated in response to CpG DNA. Based on the fact that TGF-β1 augments IRF7-mediated IFN production in the absence of TRAF6, TGF-β1 is suggested to inhibit the phosphorylation of IRF7 in response to CpG DNA via TRAF6 ubiquitination. Blocking of TRAF6 ubiquitination is shown to abolish the inhibition of CpG DNA-induced type I IFN production by TGF-β. Once again, TGF-β1 is suggested to inhibit CpG DNA-induced type I IFN production via TRAF6 ubiquitination.
We, for the first time, demonstrate that TRAF6 is ubiquitinated by TGF-β1. TRAF6 has an E3 ubiquitin ligase activity that plays an important role on the NF-κB activation in the cell surface-expressed TLR signaling. 25 Our finding demonstrates that the ubiquitination of TRAF6 by TGF-β1 is K63-linked ubiquitination, which regulate the activation of proinflammatory signaling but not the degradation.26–28 It is still unclear how TRAF6 ubiquitinated at K63 causes reduces IRF7 phosphorylation. As TLR9 stimulation with CpG DNA results in the formation of IRAK1, TRAF3 and TRAF6, IKKβ and IRF7 complex under MyD88, 29 the ubiquitination of TRAF6 with TGF-β1 might not develop the functional complex formation leading to IRF7 phosphorylation. The precise mechanism of the inhibition of IRF7 phosphorylation by ubiquitinated TRAF6 is still a matter of speculation.
TGF-β1 induces the ubiquitination of IRF7, as well as TRAF6. To our knowledge, there is no report on TGF-β1-induced IRF7 ubiquitination. Although TGF-β1 induces the ubiquitination of IRF7, it does not inhibit IRF7-mediated type I IFN production. Therefore, the IRF7 ubiquitination may not be involved in the inhibition of CpG DNA-induced IRF7 phosphorylation by TGF-β1. Further, TGF-β1 does not inhibit the interaction of IRF7 and TRAF6. Collectively, the ubiquitination of IRF7 may not be involved in reduced IRF7 phosphorylation by TGF-β1. Interestingly, the ubiquitin ligase activity of TRAF6 is reported to participate in the ubiquitination of IRF7. 30 Therefore, the TRAF6 ubiquitination might be a key event in the inhibition of CpG DNA-induced type I IFN production by TGF-β1.
Qing et al. 31 have reported that TGF-β1 activates IRF7 via SMAD3 and leads to IRF7-mediated induction of IFN-β expression. We also demonstrate that TGF-β1 leads to phosphorylation of IRF7 and augments IFN-β production in the presence of IRF7. However, the intensity of TGF-β1-induced IFN-β production is much lower than that of CpG DNA-induced IFN-β production. TGF-β1 inhibits CpG DNA-induced type I IFN production whereas it enhances IRF7-mediated IFN-β production. The difference might be dependent on the stimulation with or without CpG DNA. TGF-β1 alone may lead to activate IRF7, followed by type I IFN production. However, it may down-regulate the TLR signaling leading to the strong type I IFN production through impairing activation of signal molecules, such as TRAF6 in TLR9 signaling. TGF-β1 as a post-inflammatory cytokine might be reasonable to terminate excessive cytokines production in response to a series of TLR ligands.
TGF-β is reported to inhibit the type I IFN production in plasmacytoid dendritic cells.13,14 However, the precise mechanism of the inhibition is not clear. In this study, we characterized the inhibitory mechanism of TGF-β by reconstituting a series of signaling molecules as we also failed it in physiologic cells. Artificially reconstituted cells are not necessarily the same as physiologic cells. Therefore, the inhibitory mechanism shown in our study may not be applied to physiologic cells. In addition, a possibility that a small amount of endogenous signaling molecules may affect our experimental result cannot be excluded.
We previously reported that TGF-β signaling interacts directly with TLR4 signaling in a MyD88-dependent manner, and that TGF-β1 decreases the level of MyD88 proteins by a mechanism that involves ubiquitination-dependent proteolysis of MyD88. 17 In the present study, however, we could not find degradation of MyD88 in the inhibition of CpG DNA-induced type I IFN production by TGF-β1 (Supplementary Figure 1). It might be due to the overexpression of MyD88 with transfection. MyD88 degradation is unlikely to participate in the inhibitory action of TGF-β1 against CpG DNA signaling.
In summary, TGF-β1 is suggested to inhibit phosphorylation of IRF7 via ubiquitination of TRAF6, and down-regulate the production of type I IFN in response to CpG DNA. It might be an immunoregulatory mechanism of TGF-β1 on TLR9-mediated innate immunity.
Footnotes
Acknowledgements
We are grateful to Drs T. Sugiyama and S. Vogel for providing materials. We thank K. Takahashi for technical assistance.
Funding
This work was supported, in part, by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan and a grant of MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2011–2015(S1101027). Conflict of interest
The authors do not have any potential conflicts of interest to declare.