
Abstract
The effect of LPS on the production of vascular endothelial growth factor (VEGF) was examined using RAW 264.7 macrophage cells. LPS induced VEGF production in RAW 264.7 cells and mouse peritoneal cells. LPS induced VEGF production via the expression of hypoxia inducible factor-1α and LPS-induced VEGF production was dependent on the activation of p38 MAPK and NF-κB activation· Transforming growth factor (TGF)-β1 augmented LPS-induced VEGF production, although TGF-β1 alone did not induce VEGF production. The augmentation of LPS-induced VEGF production by TGF-β1 was inhibited by a p38 MAPK inhibitor and was correlated with the phosphorylation of Smad3. The enhancing effect of TGF-β1 on LPS-induced VEGF production was observed in vivo in the skin lesions of mice receiving a subcutaneous injection of LPS. Taken together, it is suggested that LPS induced the VEGF production in macrophages and that it was augmented by TGF-β1 in vitro and in vivo.
Keywords
Introduction
Vascular endothelial cell growth factor (VEGF) is a mitogen for vascular endothelial cells derived from arteries, veins and lymphatics, 1 and stimulates vasculogenesis and angiogenesis. 2 VEGF is produced from not only endothelial cells, but also cancer cells and macrophages. 3 The production of VEGF is mainly regulated by hypoxia inducible factor (HIF)-1α a transcription factor, which is induced in hypoxia conditions. 4 HIF-1α is known to trigger the production of VEGF in hypoxia conditions and is degraded under normoxic conditions. 5 Thus, HIF-1α is an important regulator of the production of VEGF in macrophages.
LPS induces VEGF production in macrophages. 6 The LPS-induced VEGF production might be involved in repairing of tissue injury in endotoxemia. 7 LPS is also reported to induce VEGF production in macrophages under hypoxic conditions. 8 In hypoxic conditions transforming growth factor (TGF-β) also augments the production of VEGF via the induction of HIF-1α expression. 9 As TGF-β is well known as a post-inflammatory cytokine in LPS stimulation and endotoxemia, 10 it is of interest to characterize the collaboration of LPS and TGF-β in the production of VEGF in macrophages. In this study, we first examined if and how LPS induces the production of VEGF in the mouse macrophage-like cell line, RAW 264.7 and mouse peritoneal macrophages, and, second, how TGF-β1 affects LPS-induced VEGF production in vitro and in vivo. Here, we report that LPS induces the production of VEGF in macrophages via HIF-1α expression and that it is augmented by TGF-β in vitro and in vivo.
Materials and methods
Reagents
LPS from Escherichia coli O55:B5 was obtained from Sigma Chemicals (St Louis, MO, USA). Recombinant TGF-β1 was purchased from Peprotec (Minneapolis, MN, USA). Mouse monoclonal anti-HIF-1α Ab was obtained from Novus Biological (Littleton, CO, USA). Abs to p38 mitogen-activated protein kinase (MAPK), c-jun N-terminal kinase (JNK), p65 NF-κB, IκB kinase (IKK)-α and their phosphorylated forms, and cAMP response element binding protein (CREB) were purchased from Cell Signaling (Beverly, MA, USA). Anti-c-Rel and actin Abs were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-mouse VEGF Ab was obtained from Thermofisher Scientific (Fremont, CA, USA). Monoclonal Abs against TGF-β1, 2 and 3 were purchased from R&D systems (Minneapolis, MN, USA). SB203580 (a p38 MAPK inhibitor), SB202474 (a negative control of SB203580), BAY11-7082 (a NF-κB inhibitor), TGF-β1 R1 kinase inhibitor II, FM19G11 (a HIF-1α inhibitor), PD98059 [a extracellular signal-regulated kinase (ERK) 1/2 inhibitor] and JNK inhibitor II (a JNK inhibitor) were obtained from Calbiochem (San Diego, CA, USA). Pam3CSK4, poly I:C and CpG DNA were purchased from Invivogen (San Diego, CA, USA). Appropriate concentrations of inhibitors and Abs were determined in preliminary experiments.
Cell culture
The murine macrophage-like cell line, RAW 264.7, was maintained in RPMI 1640 medium (Sigma) containing 5% heat-inactivated FCS and antibiotics. Mouse peritoneal cells were collected by washing the peritoneum of normal untreated mice with 10 ml of RPMI 1640 medium. Cells were cultured in a 60 mm dish (2 × 106 cells/4 ml) or 96-well (2.5 × 104 cells/100 µl/well) microplates (Falcon, San Jose, California, USA) for 24 h. Mouse peritoneal cells were incubated in a 96-well microplate (30,000 cells/well) with RPMI 1640 medium supplemented with 10% FCS at 37℃ for 1 h, and non-adherent cells were removed by washing. The peritoneal adherent cells were used as macrophages.
Determination of VEGF, TNF-α, IL-6 and TGF-β1 levels
The concentrations of VEGF, TNF-α, IL-6 and TGF-β1 in the supernatant were determined with the corresponding ELISA kits, Quantikine (R&D Systems).
RT-PCR
The total RNA from cells cultured in 60-mm dishes was isolated with the RNeasy mini kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. Semi-quantitative RT-PCR was performed by using the Access Quick single tube RT-PCR system (Promega, Madison, WI, USA). Primer sequences for VEGF, HIF-1α, HIF-2α, TGF-β1 and GAPDH were 5′-AGCACAGCAGATGTGAATGC-3′ and 5′- TTTGACCCTTTCCCTTTCCT-3′;
5′-TCAAGTCAGCAACGTGGAAG-3′ and 5′- TATCGAGGCTGTGTCGACTG-3′;
5′-AGCCAAACACGGAGGATATG -3′ and 5′- GTGTGGCTTGAACAGGGATT-3′,
5′-AAGTTGGCATGGTAGCCCTT-3′ and 5′-GCCCTGGATACCAACTATTGC-3′; and
5′-AAATGGTGAAGGTCGGTGTG-3′ and 5′-TGAAGGGGTCGTTGATGG-3′, respectively. RT-PCR was performed as described previously. 11 PCR products were visualized using ethidium bromide staining after separation in 2% agarose gels.
Immunoblotting
Immunoblotting analysis was carried out as described previously. 12 Briefly, cell pellets were suspended at a concentration of 2 × 107 cells/ml in a lysis buffer. Cell lysates were diluted with an equal volume of sample buffer and boiled for 5 min. Samples were separated under reducing conditions by electrophoresis using 6% or 10% polyacrylamide gel. The membranes that proteins were transferred to were treated with various appropriately diluted Abs and HRP-conjugated second Ab. Finally, labeled Ag bands were detected with a chemiluminescence reagent, SuperSignal West Dura (Pierce, Rockford, IL, USA) and analyzed using an AE6955 light capture system with CS analyzer (Atto, Tokyo, Japan). Prestained protein markers from Gibco (Foster City, CA, USA) were used to estimate molecular mass.
Preparation of nuclear extracts
Nuclear extracts were prepared with nuclear extraction reagents (Active Motif, Carlsbad, CA, USA), according to the manufacturer’s instructions. Briefly, cells were lysed in a hypotonic buffer. After centrifugation, pellets were lysed in the nuclear extraction reagent to extract nuclear proteins. After centrifugation, nuclear proteins (40 µg) were subjected to immunoblotting for detection of HIF-1α or c-Rel. The protein concentrations were determined with the BCA protein assay reagent (Pierce) using the comparative reagents (Pierce). The CREB protein was used as a loading control of nuclear protein.
Detection of intracellular TGF-β1
For detection of intracellular TGF-β1, RAW 264.7 cells were incubated with LPS (100 ng/ml) for 24 h and then with monensin (2 µM) for 2 h. The cells were washed with PBS containing 10% FCS and fixed with the fixation buffer (eBioscience, San Diego, CA, USA) and stained with phycoerythrin-conjugated anti-TGF-β1 Ab (eBioscience) in the permeabilization buffer for 20 min at room temperature (28℃). The expression level was analyzed with a laser flow cytometer (FACS Calibur; Becton Dickinson, Palo Alto, CA, USA). The fluorescence intensity is expressed on a log scale. 13
Gene silencing with small interfering RNA
RAW 264.7 cells were plated (6 × 105 cells per 36 mm dish) 24 h prior to transfection. After the replacement of fresh culture medium, the cells were transfected with control or HIF-1α siRNA (Santa Cruz Biotechnology) using GenMute transfection reagent specific for RAW 264.7 cells (SignaGen, Gaithersburg, MD, USA) following the manufacturer’s protocol. The transfected cells were stimulated with LPS 24 h after the transfection.
In vivo administration of LPS
BALB/c mice were purchased from SLC (Hamamatsu, Japan), and used at about 6 wk of age. LPS (50 µg) was subcutaneously injected into the right inguinal region of mice. The skins of the right inguinal region were removed 48 h after LPS injection and fixed in 10% formalin. At the same time, blood samples were taken from the mice for measuring serum VEGF level with ELISA. All animal experiments were approved by the Animal Care Committee and carried out under the guide for care and use of laboratory animals, Aichi Medical University.
Immunohistochemical staining
The tissue sections were stained immunohistochemically as described previously. 13 Briefly, the endogenous peroxidase activity in deparaffinized sections was blocked by methanol containing 0.3% hydrogen peroxide. The sections were washed in PBS containing 10% normal horse serum and incubated overnight (18 h) at 4℃ with anti-VEGF Ab. HRP-conjugated goat anti-rabbit immunoglobulin Ab was used at the 1:200 dilution. Immune complexes were detected with a solution of 3,3-diaminobenzidine (0.2 mg/ml) and hydrogen peroxide in 0.05 M Tris-HCl buffer. The sections were counterstained with methyl green.
Statistical analysis
Statistical analysis was performed using a Student’s t- test, and P-values < 0.05 were considered significant. All experiments were performed at least three times. Data represent the mean value of triplicates ±SD.
Results
LPS induces the production of VEGF in macrophages
The effect of various concentrations of LPS on VEGF production was examined by culturing 2.5 × 104 RAW 264.7 cells/well for 24 h (Figure 1A). LPS at 1 ng/ml significantly induced the production of VEGF and the higher concentration of LPS led to a higher level of VEGF production. The time course of VEGF production was examined by stimulating the cells with LPS at 100 ng/ml (Figure 1B). A significant amount of VEGF production was detected 18 h after LPS stimulation. There was no significant difference in the level of VEGF between 18 h and 24 h. The expression of VEGF mRNA was examined by stimulating the cells with LPS for 4 h. LPS significantly induced the expression of VEGF mRNA (Figure 1C). In addition, LPS-induced VEGF production was examined in physiological peritoneal macrophages (Figure 1D). Mouse peritoneal macrophages produced a significant amount of VEGF production in response to LPS. For further characterization of LPS-induced VEGF production, RAW 264.7 cells at 2.5 × 104 cells/well in a 96-well microplate was stimulated with LPS (100 ng/ml) for 24 h unless otherwise stated.
Effect of LPS on the VEGF production in RAW 264.7 cells and mouse peritoneal macrophages. (A) RAW 264.7 cells at 2.5 × 104 cells/well in a 96-well microplate were cultured with various concentrations of LPS for 24 h. *P < 0.05 versus none. (B) Cells were cultured with LPS (100 ng/ml) for various lengths of time. *P < 0.05 versus 0 h. (C) Cells were cultured with LPS (100 ng/ml) for 4 h. mRNA expression was analyzed by RT-PCR. The GAPDH mRNA was used as an equal loading control. (D) Mouse peritoneal macrophages at 2.5 × 104 cells/well were cultured with LPS (100 ng/ml) for 24 h. *P < 0.05 versus none.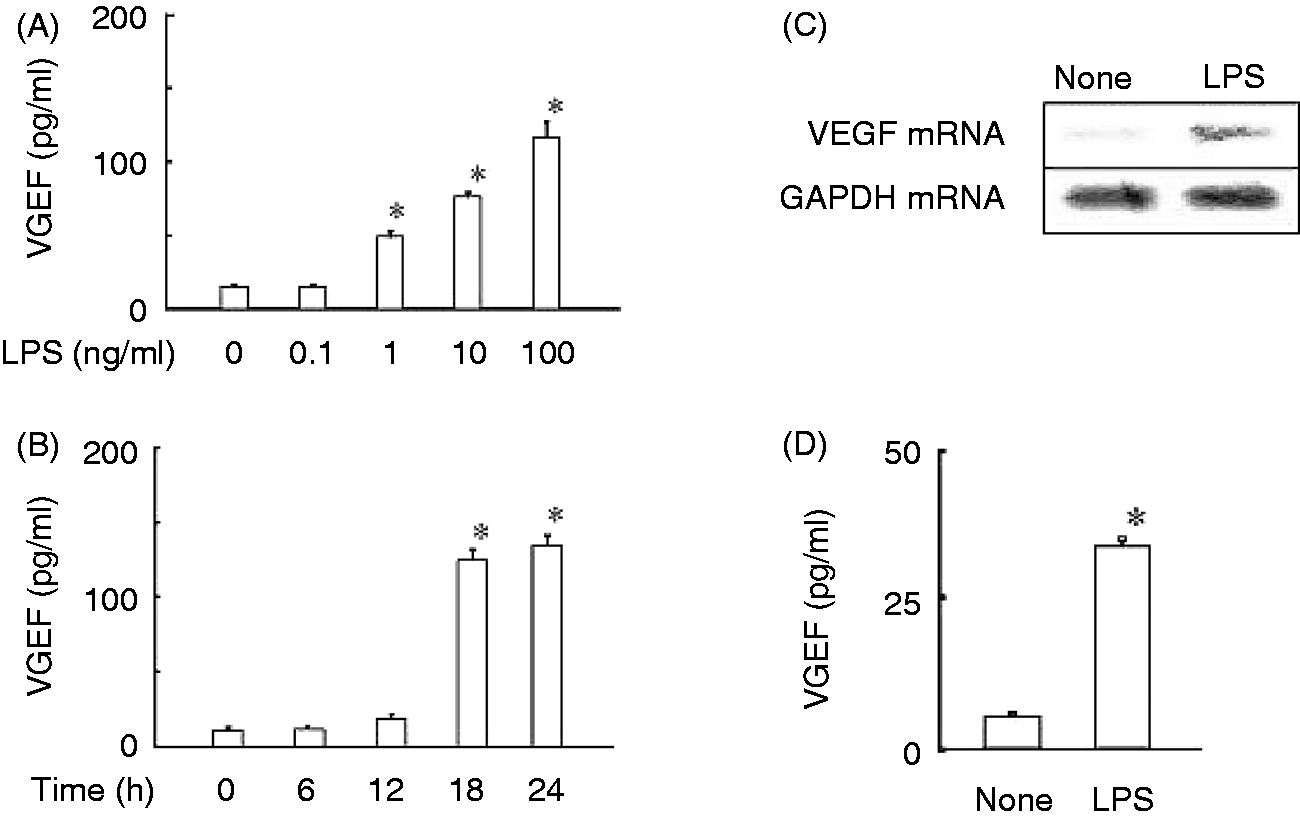
LPS induces the expression of HIF-1α
The effect of LPS on the expression of HIF-1α was examined by stimulating the cells with LPS for 8 h (Figure 2A). Although the expression of HIF-1α was not detected before LPS stimulation, it was clearly induced 4 h after LPS stimulation. The HIF-1α expression was further augmented at 8 h. However, no HIF-2α was detected in the cells stimulated with LPS (data not shown). Next, the effect of FM19G11 as an inhibitor of HIF-1α
14
on LPS-induced VEGF production was examined by stimulating the cells with LPS in the presence or absence of FM19G11 for 24 h. LPS-induced VEGF production was significantly inhibited by FM19G11 significantly (Figure 2B). Further, the effect of HIF-1α small interfering RNA (siRNA) on LPS-induced VEGF production was examined to confirm the involvement of HIF-1α. HIF-1α siRNA, but not control siRNA, inhibited LPS-induced VEGF production (Figure 2C), suggesting that the expression of HIF-1α is required for LPS-induced VEGF production.
Effect of LPS on the expression of HIF-1α. (A) RAW 264.7 cells were cultured with LPS (100 ng/ml) for various hours. The HIF-1α expression was determined by immunoblotting. (B) Cells were cultured with LPS (100 ng/ml) in the presence or absence of FM19G11 (10 µM), a HIF-1α inhibitor, for 24 h. *P < 0.05 versus LPS alone. (C) Cells transfected with control or HIF-1α siRNA were cultured with LPS (100 ng/ml) for 24 h. HIF-1αprotein expression and VEGF production was determined by immunoblotting and ELISA, respectively. *P < 0.05 versus control siRNA.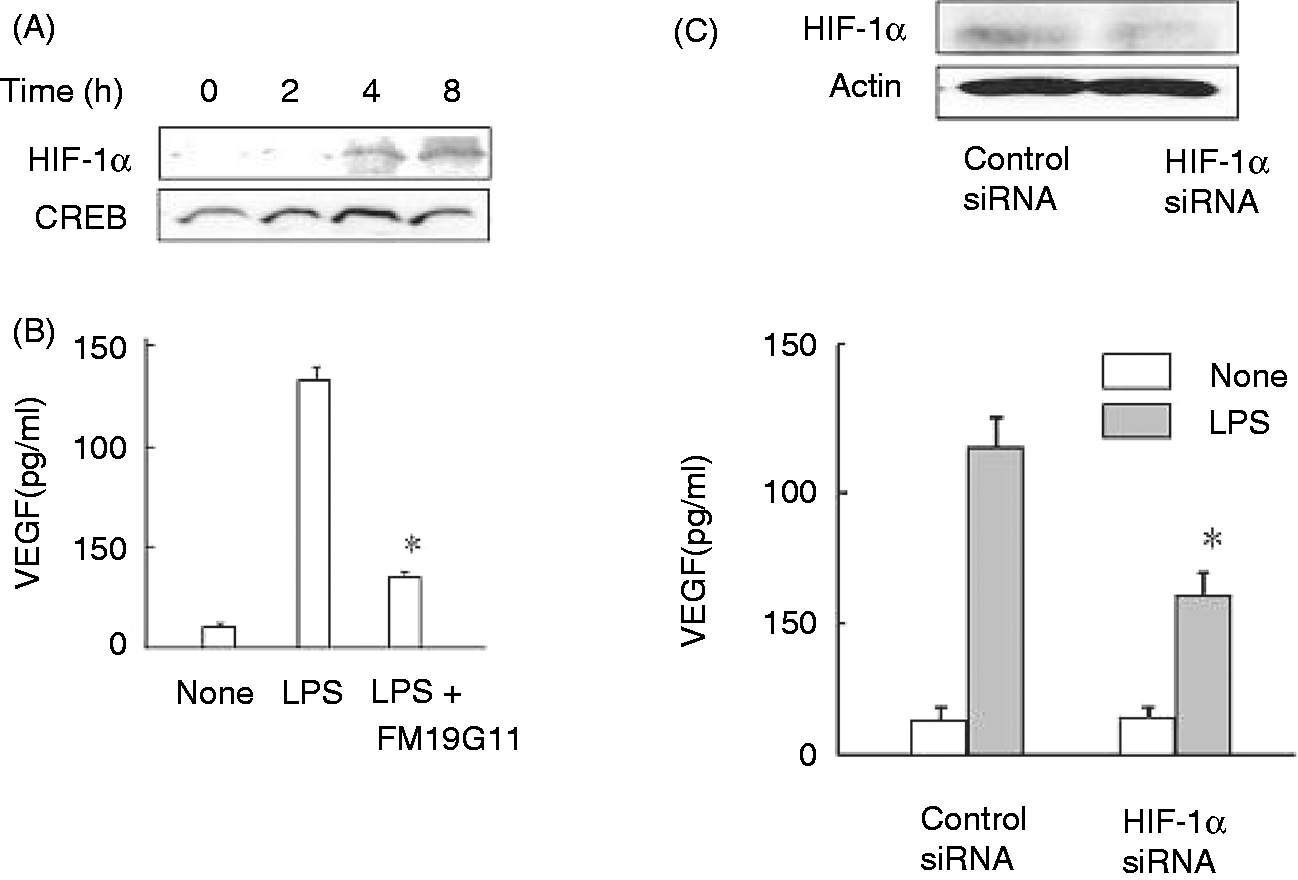
Inhibitors of p38 and NF-κB reduce LPS-induced VEGF production
To clarify the signaling molecules involved in LPS-induced VEGF production, the effect of various signaling inhibitors on LPS-induced VEGF production was examined (Figure 3A). SB203580, a p38 inhibitor, was the most effective for the inhibition of LPS-induced VEGF production among a series of MAPKs inhibitors. Bay 11-7082, a NF-κB inhibitor, was also effective for the inhibition. PD98059 as an ERK1/2 and JNK inhibitor did not significantly reduce the VEGF production. The effect of the inhibitors on LPS-induced HIF-1α expression was examined (Figure 3B). Bay 11-7082 significantly reduced LPS-induced HIF-1α expression, whereas SB203580 did not alter it.
Effect of various signaling inhibitors or TLR ligands on LPS-induced VEGF production or HIF-1α expression. RAW 264.7 cells were treated with SB203580 (10 µM), SB202474 (10 µM), PD98059 (10 µM), JNK inhibitor (10 µM) or BAY11-7082 (2 µM) for 30 min. (A) Cells were stimulated with LPS (100 ng/ml) for 24 h. *P < 0.05 versus LPS alone. (B) Cells were stimulated with LPS (100 ng/ml) for 8 h. The expression of HIF-1α protein was determined by immunoblotting. (C) RAW 264.7 cells were stimulated with LPS (100 ng/ml), Pam3CSK4 (10 µg/ml), CpG DNA (10 µM) or poly I:C (10 µg/ml) for 24 h. *P < 0.05 versus none.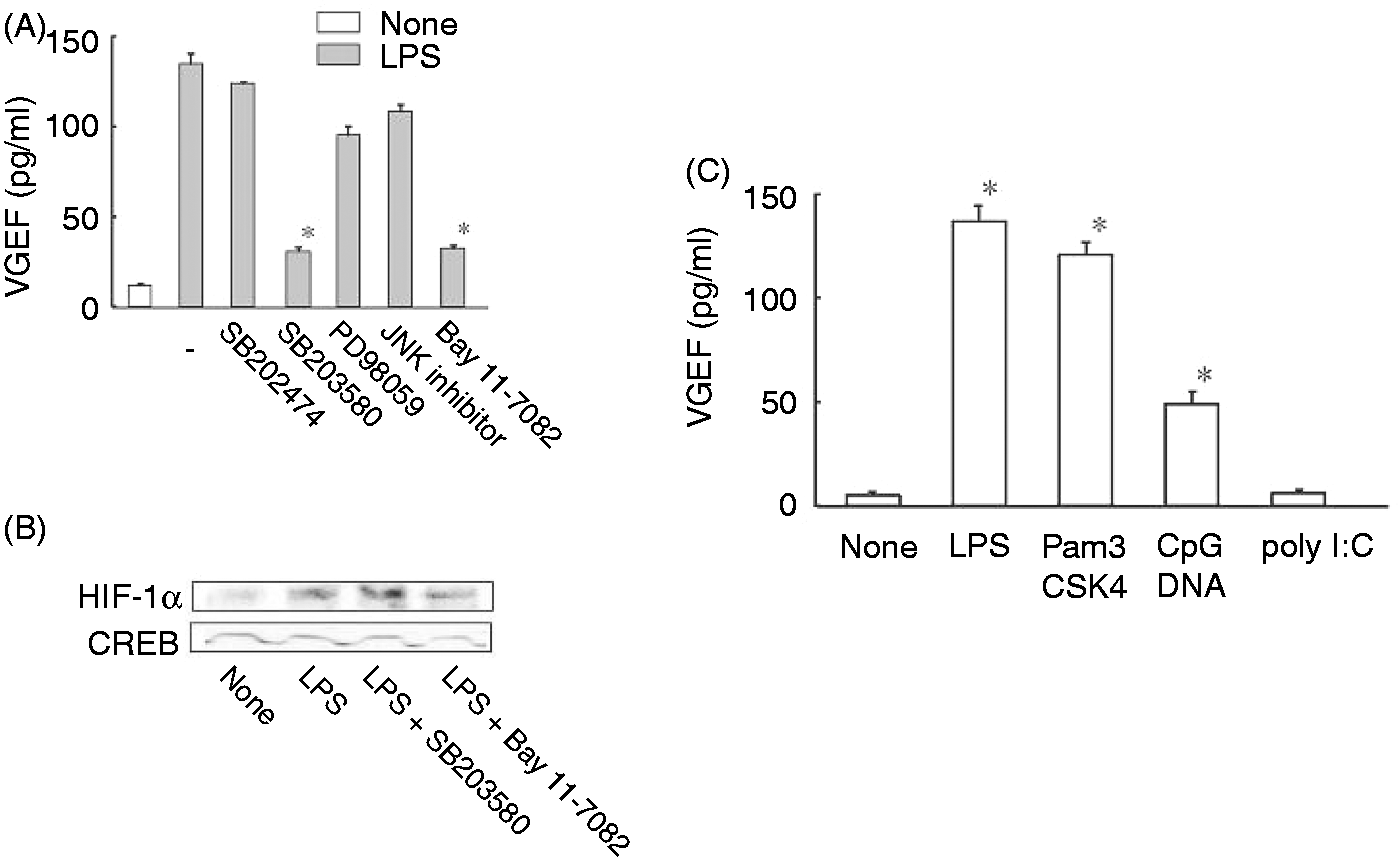
A series of TLR ligands induces the production of VEGF
The effect of a series of TLR ligands on the production of VEGF was examined. RAW 264.7 cells were stimulated with Pam3CSK4 (a TLR2 ligand), CpG DNA (a TLR9 ligand) and poly I:C (a TLR3 ligand). Pam3CSK4 and CpG DNA significantly induced the production of VEGF production, whereas poly I:C did not induce it (Figure 3C).
LPS induces the production of VEGF without the help of internal TGF-β1
TGF-β1 is well known to induce VEGF production.
9
In order to clarify the involvement of TGF-β1 in LPS-induced VEGF production, the effect of LPS on the production of TGF-β1 in RAW 264.7 cells was examined (Figure 4). RT-PCR analysis showed that a small amount of TGF-β1 mRNA was expressed in untreated RAW 264.7 cells and that LPS augmented the expression of TGF-β1 mRNA (Figure 4A). Therefore, the detection of intracellular TGF-β1 in LPS-stimulated RAW 264.7 cells was examined. FACS analysis demonstrated that intracellular TGF-β1 was detected in LPS-treated RAW 264.7 cells 24 h after the stimulation (Figure 4B). However, no TGF-β1 in the culture supernatant was detected with ELISA (Figure 4C), although a significant level of TGF-β1 was detected in the culture of 1 × 105 cells/well stimulated with LPS for 48 h. The addition of an anti-TGF-β1 neutralizing Ab or an isotype control Ab (1 µg) into the culture did not affect LPS-induced VEGF production (Figure 4D), whereas the enhancing effect of external TGF-β1 (5 ng/ml) was abolished by an anti-TGF-β1 Ab but not an isotype control Ab. LPS was suggested to induce the production of VEGF without the help of internal TGF-β1
Effect of LPS on the production of TGF-β1 mRNA and intracellular TGF-β1. (A) RAW 264.7 cells were treated with LPS (100 ng/ml) for 4 h. The TGF-β1 mRNA expression was determined by RT-PCR. The GAPDH mRNA was used as an equal loading control. (B) Cells were pretreated with TGF-β1 for 24 h and then stimulated with LPS (100 ng/ml) for 24 h. The fluorescence intensity is expressed on a log scale. (C) Cells at 2.5 × 104 or 1 × 105 cells/well were stimulated with LPS (100 ng/ml) for 24 or 48 h. TGF-β1 in the culture supernatant was determined with ELISA. (D) Cells at 2.5 × 104 were stimulated with LPS (100 ng/ml) for 24 h in the presence or absence of anti-TGF-β1 neutralizing Ab or an isotype control Ab.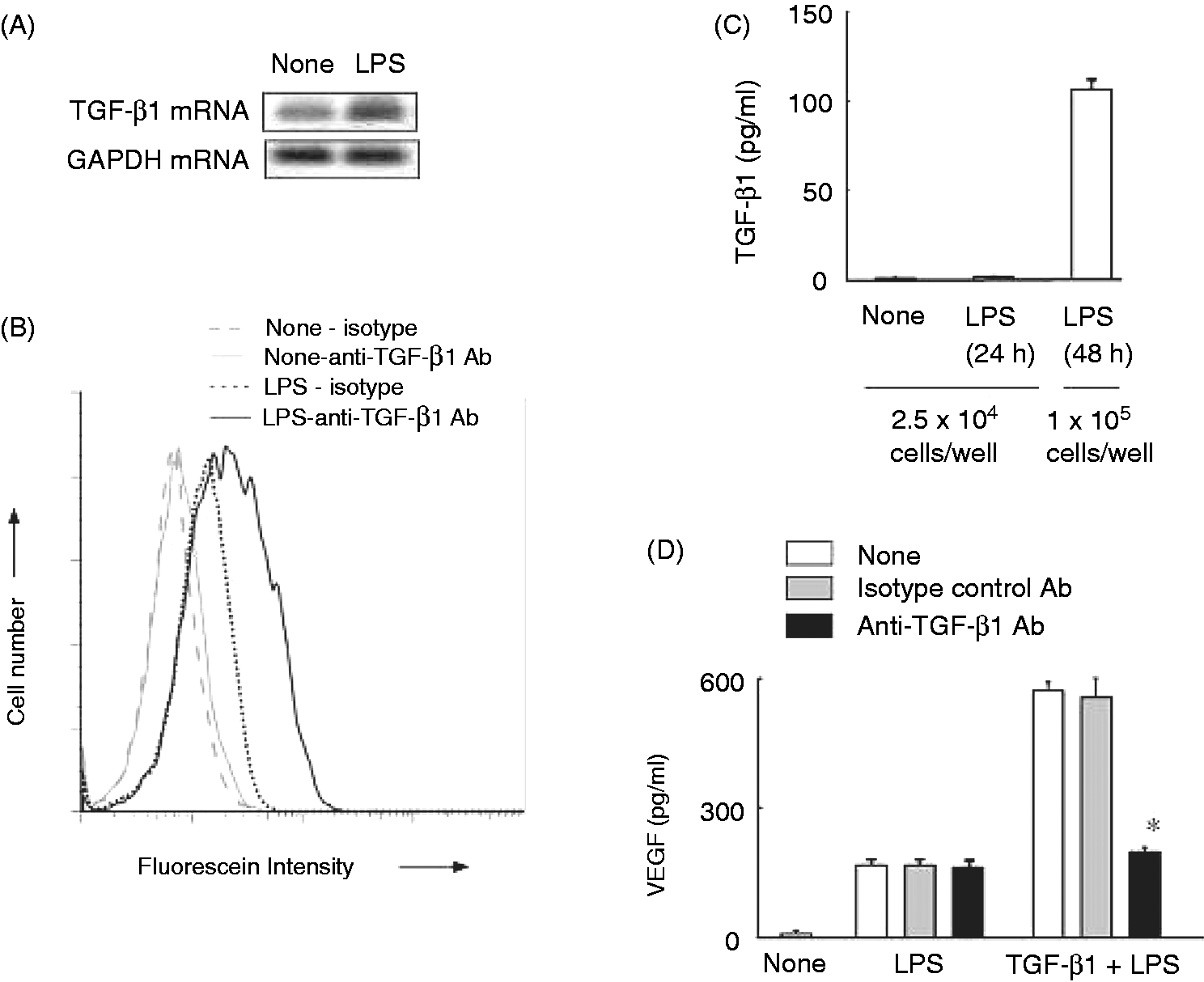
External TGF-β1 augments the production of VEGF and the expression of HIF-1α in response to LPS
The effect of external TGF-β1 on LPS-induced VEGF production was examined. RAW 264.7 cells were pretreated with various concentrations of recombinant TGF-β1 for 6 h and then stimulated with LPS for 24 h. TGF-β1 alone did not induce the production of VEGF, but enhanced LPS-induced VEGF production. The enhancing effect of TGF-β1 was strongest at 10 ng/ml among the concentration ranging from 0.1 to 100.0 ng/ml (Figure 5A). The effect of pre- and post-treatment of TGF-β1 on LPS-induced VEGF production was examined. Pretreatment of TGF-β1 significantly augmented LPS-induced VEGF production (Figure 5B). The pretreatment with TGF-β1 for 24 h led to the highest level of LPS-induced VEGF production. Post-treatment with TGF-β1 also augmented LPS-induced VEGF production, but it was much less effective than the pretreatment (P < 0.05 versus 1 h pretreatment of TGF-β1).
Effect of TGF-β1 on LPS-induced VEGF production. (A) RAW 264.7 cells were pretreated with various concentrations of TGF-β1 for 24 h and stimulated with LPS (100 ng/ml) for 24 h. *P < 0.05 versus LPS alone. (B) Cells were pre- or post-treated with TGF-β1 (10 ng/ml) for various lengths of time and then stimulated with LPS (100 ng/ml) for 24 h. *P < 0.05 versus LPS alone. (C) Cells were pretreated with TGF-β1 (10 ng/ml) for 24 h and then stimulated with LPS (100 ng/ml) for 8 h. The expression of HIF-1α protein was determined by immunoblotting. (D) Cells were pretreated with TGF-β1 (10 ng/ml) for 24 h and then stimulated with LPS (100 ng/ml). The concentrations of TNF-α and IL-6 were determined with ELISA 1 or 6 h after LPS stimulation, respectively.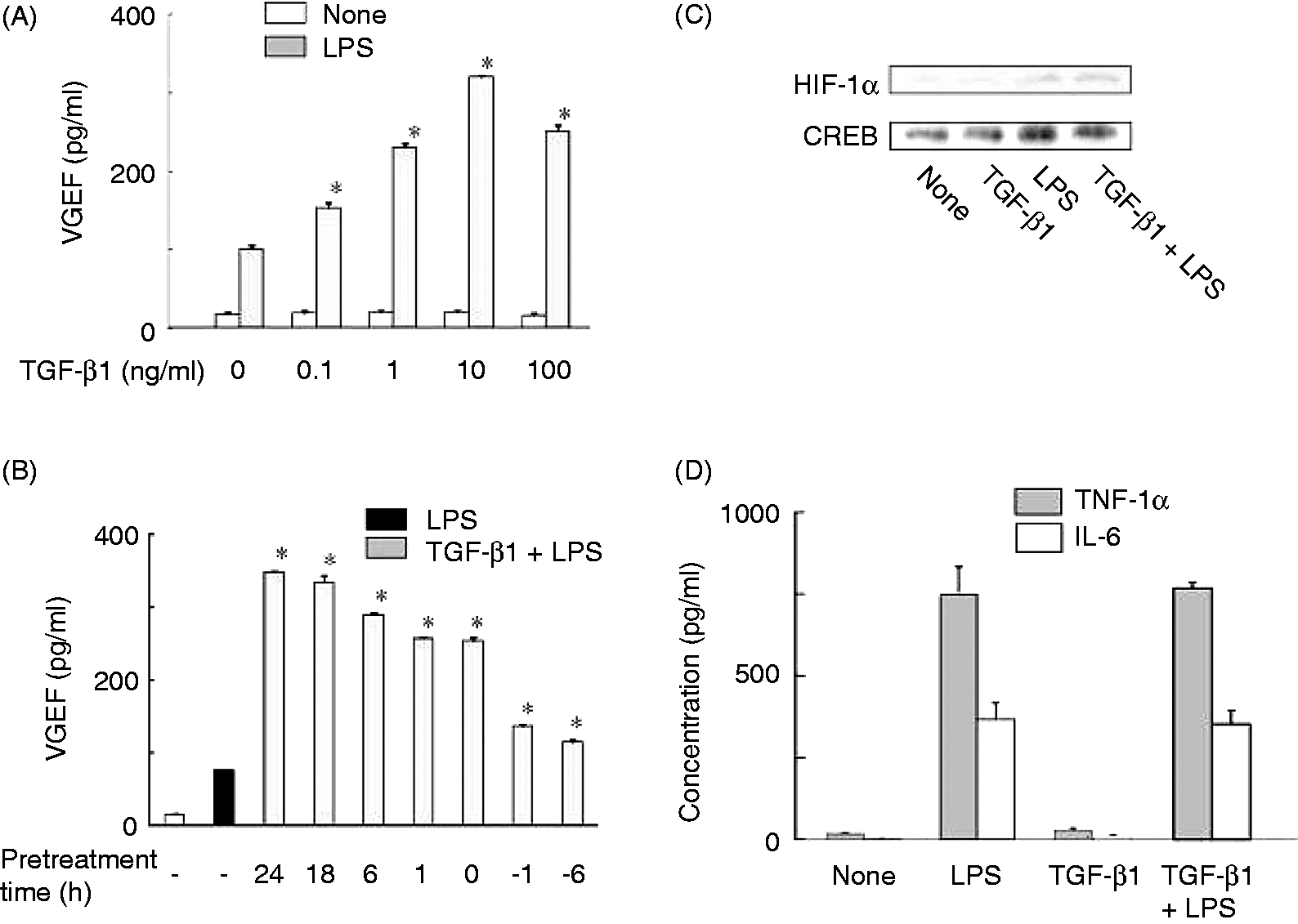
The effect of TGF-1 on LPS-induced HIF-1α expression was examined (Figure 5C). Cells were pretreated with TGF-1β for 24 h and then stimulated with LPS for 24 h. TGF-β1 pretreatment did not affect LPS-induced HIF-1α expression. Further, the effect of TGF-β1 on LPS-induced TNF-α or IL-6 production were examined. TGF-β1 pretreatment did not affect LPS-induced TNF-α or IL-6 production (Figure 5D), suggesting specific enhancement of LPS-induced VEGF production.
A p38 MAPK inhibitor abolishes the enhancement of LPS-induced VEGF production by TGF-β1
The effect of TGF-β1 on LPS-induced NF-κB and MAPK activation was examined in order to clarify the mechanism of TGF-β1-mediated enhancement of LPS-induced VEGF production (Figure 6). RAW 264.7 cells were pretreated with TGF-β1 for 24 h and then stimulated with LPS for 1 h. TGF-β1 pretreatment enhanced the p38 MAPK activation (Figure 6A), but did not alter LPS-induced activation of JNK, p65 and c-Rel NF-κB and IKK-α (Figure 6B). Therefore, the effect of a p38 inhibitor on the enhancement of LPS-induced VEGF production by TGF-β1 was examined. Cells were pretreated with each inhibitor for 30 min, treated with TGF-β1 for 24 h and then stimulated with LPS for 24 h. SB203580, a p38 inhibitor, markedly abolished TGF-β1-mediated enhancement of LPS-induced VEGF production (Figure 6C), whereas its negative control (SB202474) did not. In addition, SB 203580 markedly inhibited TGF-β1-induced Smad3 activation (Figure 6D).
Effect of TGF-β1 on LPS-induced MAPK and NF-κB activation. (A) RAW 264.7 cells were pretreated with TGF-β1 (10 ng/ml) for 24 h and then stimulated with LPS (100 ng/ml) for 30 (A, B) or 60 min (A). The phosphorylation of p38, JNK, IKKα and p65, and the nuclear expression of c-Rel were determined with immunoblotting. The nuclear protein was extracted after 2 h incubation with LPS. (C) Cells were pretreated with TGF-β1 (10 ng/ml) in the presence of SB203580 or SB202474 (10 µM) for 24 h and then stimulated with LPS (100 ng/ml) for 24 h. *P < 0.05 versus TGF-β1 + LPS. (D) Cells were pretreated with TGF-β1 (10 ng/ml) in the presence of SB203580 (10 µM) or TGF-β1 R1 kinase inhibitor II (R1 inhibitor, 10 µM) for 24 h and then stimulated with LPS (100 ng/ml) for 1 h. The phosphorylation of Smad3 was determined with immunoblotting.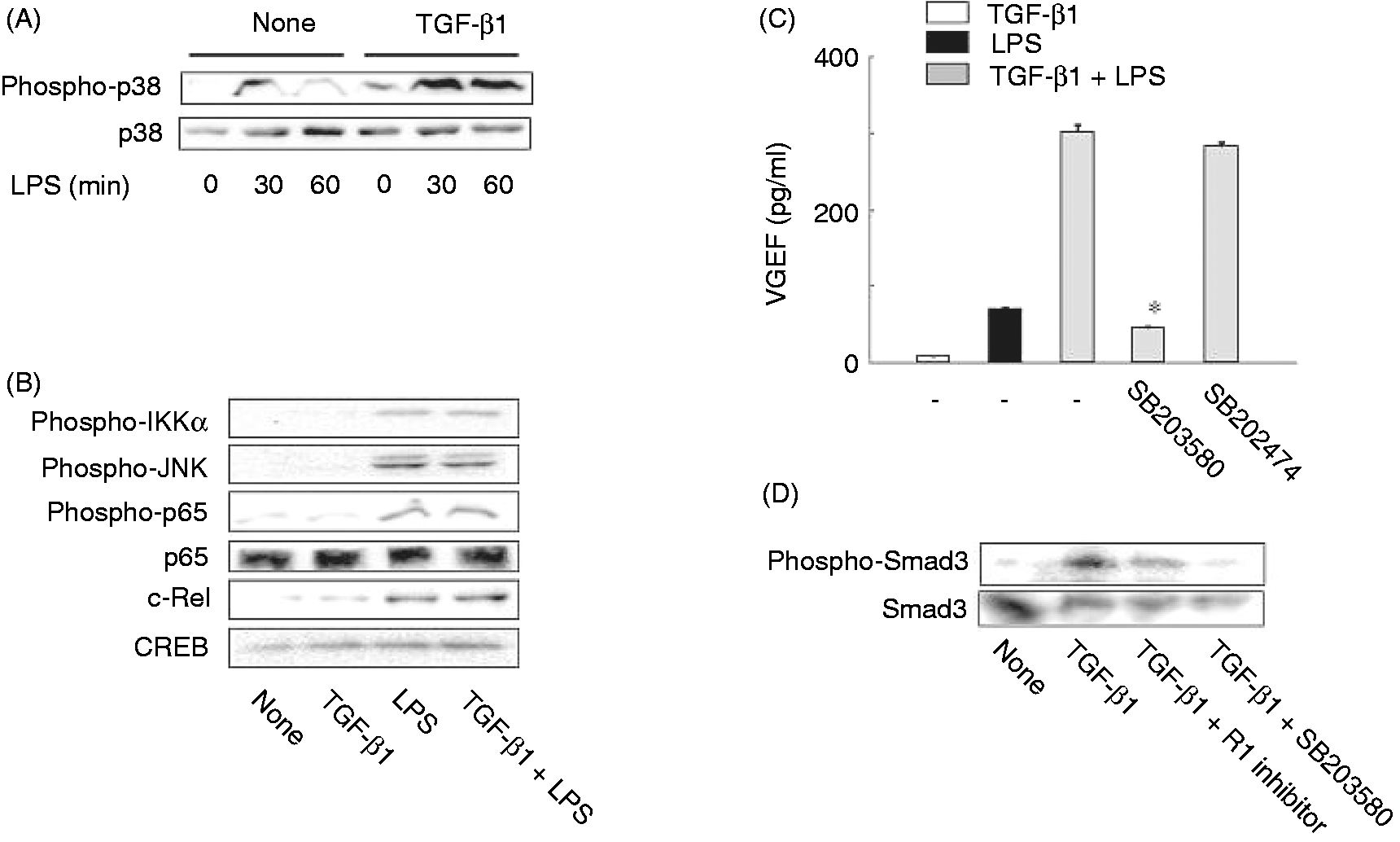
The production of VEGF is induced in mice injected with LPS and is augmented by TGF-β1
The effect of LPS on the in vivo VEGF production was examined using mice. LPS (50 µg) was injected subcutaneously into the right inguinal region of the mice and the skins were removed 48 h after LPS injection. A high level of VEGF was detected in the sera from LPS-injected mice (Figure 7A). Immunohistochemical analysis demonstrated that monocytes infiltrating were positively stained with anti-VEGF Ab in LPS-injected mice. The injection of TGF-β1 (100 ng/mouse) together with LPS significantly enhanced VEGF expression in monocytes (Figure 7B).
In vivo effect of TGF-β1 on LPS-induced VEGF production. LPS (50 µg) with or without TGF-β1 (0.1 µg) was injected subcutaneously into the right inguinal region of mice. (A) The VEGF level in the sera 24 h after LPS injection. *P < 0.05 versus none. (B) The skins were removed from the right inguinal region 48 h after the injection of TGF-β1 and/or LPS and the tissue sections were stained immunohistochemically with anti-VEGF Ab. Magnification 400×.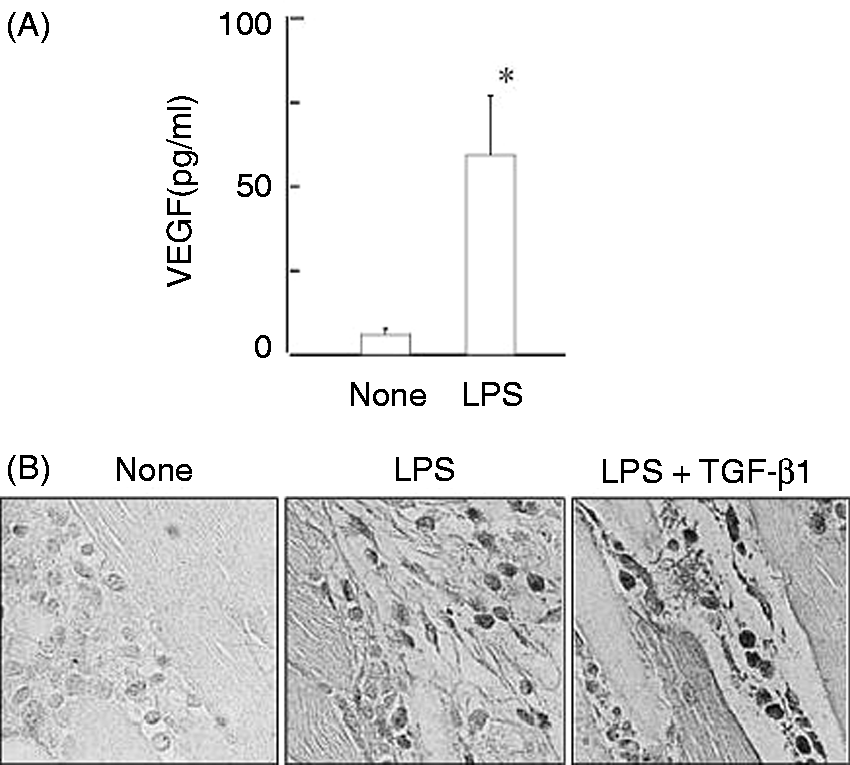
Discussion
In the present study we have demonstrated that LPS induces VEGF production in macrophages via HIF-1α expression and that TGF-β1 augments the LPS-induced VEGF production. It is reasonable that macrophages produce VEGF in response to LPS as VEGF is required for repairing of LPS-induced tissue injury via angiogenesis. Considering that LPS induces VEGF production via HIF-1α expression, LPS might lead to hypoxia conditions in the in vitro culture system and then macrophages may produce VEGF via HIF-1α expression under hypoxia conditions to supply oxygen via angiogenesis. 15 In the process, we cannot exclude the involvement of LPS-induced pro-inflammatory mediators, such as cytokines and free radicals, in LPS-induced HIF-1α expression.
TGF-β1 alone is unable to induce VEGF production in macrophages in our experimental system. However, TGF-β1 is reported to induce VEGF production under hypoxia conditions. 9 As our in vitro cultivation was not performed under hypoxic conditions, TGF-β1 was unable to induce the VEGF production. However, LPS may cause hypoxic conditions in a low cell density culture and induce the HIF-1α expression, followed by the VEGF production. Considering that LPS induces the TGF-β1 production in a high cell density culture for a longer incubation, LPS might induce the VEGF production via TGF-β1 under hypoxia conditions. Under such hypoxic conditions TGF-β1 may augment LPS-induced VEGF production via HIF-1α expression. There are several reports on LPS-induced VEGF production.5,16,17 However, they do not define either normoxia or hypoxia in the experiments. Ramanathan et al. 8 have reported that combined treatment with LPS and IFN-γ induces the production of significant levels of VEGF protein only at high cell density under conditions of pericellular hypoxia in murine peritoneal macrophages. Thus, it might be important to define whether LPS-induced VEGF production is studied under normoxic or hypoxic conditions.
LPS-induced VEGF production is inhibited by pharmacological inhibitors of p38 and NF-κB. A NF-κB inhibitor reduces LPS-induced HIF-1α expression, suggesting that NF-κB activation is required for LPS-induced HIF-1α expression and triggers VEGF production. However, poly I:C utilizing the MyD88-independent pathway does not induce VEGF production. Therefore, the MyD88-dependent pathway with NF-κB activation might be involved in VEGF production. The idea is supported by the finding that Pam3CSK4 and CpG DNA, which activate the MyD88-dependent pathway, induce the VEGF production. Further, a p38 inhibitor prevents LPS-induced VEGF production without affecting HIF-1α expression. The promoter region of the VEGF gene possesses the binding site of AP1, the expression of which is mediated by activation of p38.18,19 Therefore, p38 MAPK appears to directly affect VEGF transcription in response to LPS.
The addition of a high amount of external TGF-β1 is able to augment LPS-induced VEGF production. Therefore, the possibility is raised that LPS-induced TGF-β1 production is involved in the VEGF production in LPS-stimulated RAW 264.7 cells. In fact, LPS-stimulated RAW 264.7 cells express TGF-β1 mRNA and intracellular TGF-β1. However, TGF-β1 is undetectable in the culture supernatant of LPS-stimulated cells and anti-TGF-β1 Ab does not prevent LPS-induced VEGF production. Although RAW 264.7 cells might produce only a marginal level of TGF-β1 in response to LPS, it would be insufficient for augmentation of the VEGF production. Otherwise, intracellular TGF-β1 might not be secreted into the culture medium.
The enhancement of LPS-induced VEGF production by TGF-β1 is abolished by a p38 inhibitor. The p38 inhibitor also inhibits the phosphorylation of Smad3. Therefore, the activation of Smad3 is suggested to play an important role on the TGF-β1-mediated enhancement. In fact, the Smad family activated by TGF-β stimulation is reported to be necessary for the most effective expression of VEGF. 20
TGF-β is one of anti-inflammatory cytokines and suppresses LPS-induced pro-inflammatory cytokine production and the signaling.21,22 However, TGF-β1 does not inhibit the production of TNF-α and IL-6 in LPS-stimulated RAW 264.7 cells. TGF-β1 exclusively augments LPS-induced VEGF production. The pretreatment time of TGF-β1 for the inhibition of pro-inflammatory cytokines is usually 1 h, 23 whereas the pretreatment time for VEGF augmentation in this study was 24 h. Therefore, TGF-β1 might differentially regulate LPS actions via the different pretreatment time. Moreover, TGF-β1 is known as a post-inflammatory cytokine in response to LPS. TGF-β1 as a post-inflammatory cytokine might participate in the repair of LPS-induced tissue injury through augmenting VEGF production.
In an in vivo experimental system the injection of LPS, but not TGF-β1, induces the production of VEGF. It is consistent with the in vitro experimental result. LPS is suggested to cause VEGF production in macrophages infiltrating into the inflammatory lesions leading to angiogenesis. It might be useful for the recovery of LPS-induced inflammatory responses with newly generated blood vessels. VEGF is a pro-inflammatory cytokine and known as an important determinant of sepsis mortality. 24 However, it is a key factor for tissue healing from tissue damage in sepsis.7,25 Based on our finding, administration of TGF-β1 into patients with endotoxemia is likely to lead to tissue repairing and angiogenesis through enhancing LPS-induced VEGF production.
Footnotes
Acknowledgements
We are grateful to Kazuko Takahashi and Akiko Morikawa for the technical assistance.