
Abstract
Plasmodium-induced malaria widely infects primates and other mammals. Multiple past studies have revealed that positive selection could be the main evolutionary force triggering the genetic diversity of anti-malaria resistance-associated genes in human or primates. However, researchers focused most of their attention on the infra-generic and intra-specific genome evolution rather than analyzing the complete evolutionary history of mammals. Here we extend previous research by testing the evolutionary link of natural selection on eight candidate genes associated with malaria resistance in mammals. Three of the eight genes were detected to be affected by recombination, including TNF-α, iNOS and DARC. Positive selection was detected in the rest five immunogenes multiple times in different ancestral lineages of extant species throughout the mammalian evolution. Signals of positive selection were exposed in four malaria-related immunogenes in primates: CCL2, IL-10, HO1 and CD36. However, selection signals of G6PD have only been detected in non-primate eutherians. Significantly higher evolutionary rates and more radical amino acid replacement were also detected in primate CD36, suggesting its functional divergence from other eutherians. Prevalent positive selection throughout the evolutionary trajectory of mammalian malaria-related genes supports the arms race evolutionary hypothesis of host genetic response of mammalian immunogenes to infectious pathogens.
Keywords
Introduction
Plasmodium-induced malaria is one of the most prevalent infectious diseases in the human population. In addition to humans, Plasmodium species also infect primates and non-primates;1–5 primates were reported as an adapted host of Plasmodium.1–3 Bats may have been the first mammal host of malaria syndrome. 6 Two lineages of Plasmodium genus using bats as the reservoir host are phylogenetically close to the Plasmodium parasites from rodents thereby generating rodent parasites as valuable experimental, medical and genetic models of human-form malaria.6,7 This implies the low threshold host shift of Plasmodium among mammals and divergent malaria disease susceptibility by genetic components of the mammalian immune system.8–11 In other words, different Plasmodium infections could result in different selection pressures for the evolution of the mammalian immune system.
Effective spread of adaptive alleles of a host’s immunogenes is the major strategy of disease resistance against pathogen infection.12,13 Several evolutionary models were developed for evaluating the immunogenes.14–19 Most of these evolutionary hypotheses for the host–parasite co-evolution involve positive selection as a main component for explaining the broad responses of hosts to non-specific pathogens (e.g. arms races hypothesis), in addition to the balancing selection model (trench warfare hypothesis). 20 Mechanisms for malaria resistance and the evolution model are well studied in humans.7,21 However, the evolutionary genetics of the malaria-related immunogenes in mammals is still unclear. Understanding immunogene evolution is important to understanding this infectious epidemic.
Evidence of positive selection has been found in mammalian innate immunogenes. For example, the radical amino acid changes in certain structurally important domains of the TLRs that recognize the infectious agents to initiate the innate immune response were found,22,23 and signatures of positive selection across primates and across mammals were detected.24,25 The adaptive evolution of TLRs might be more episodic in nature rather than a long-term constant phenomenon along primate evolution. 24 Another innate immunity gene that codes for the viral-sensing RIG-I-like receptors (RLRs) also represent apparent signals of positive selection along the evolutionary history of mammals.26–28 Two RLR genes, RIG-I and LGP2, even revealed parallel evolution at the same sites that are important positions for the antiviral response. 28 These studies show evidence of episodic positive selection contributing to the innate species-specific resistance/susceptibility to diverse pathogens in mammals.
The main function of the eight immunogenes during Plasmodium infection.

Materials and methods
Data collection
Accession numbers of sequences used in this study.
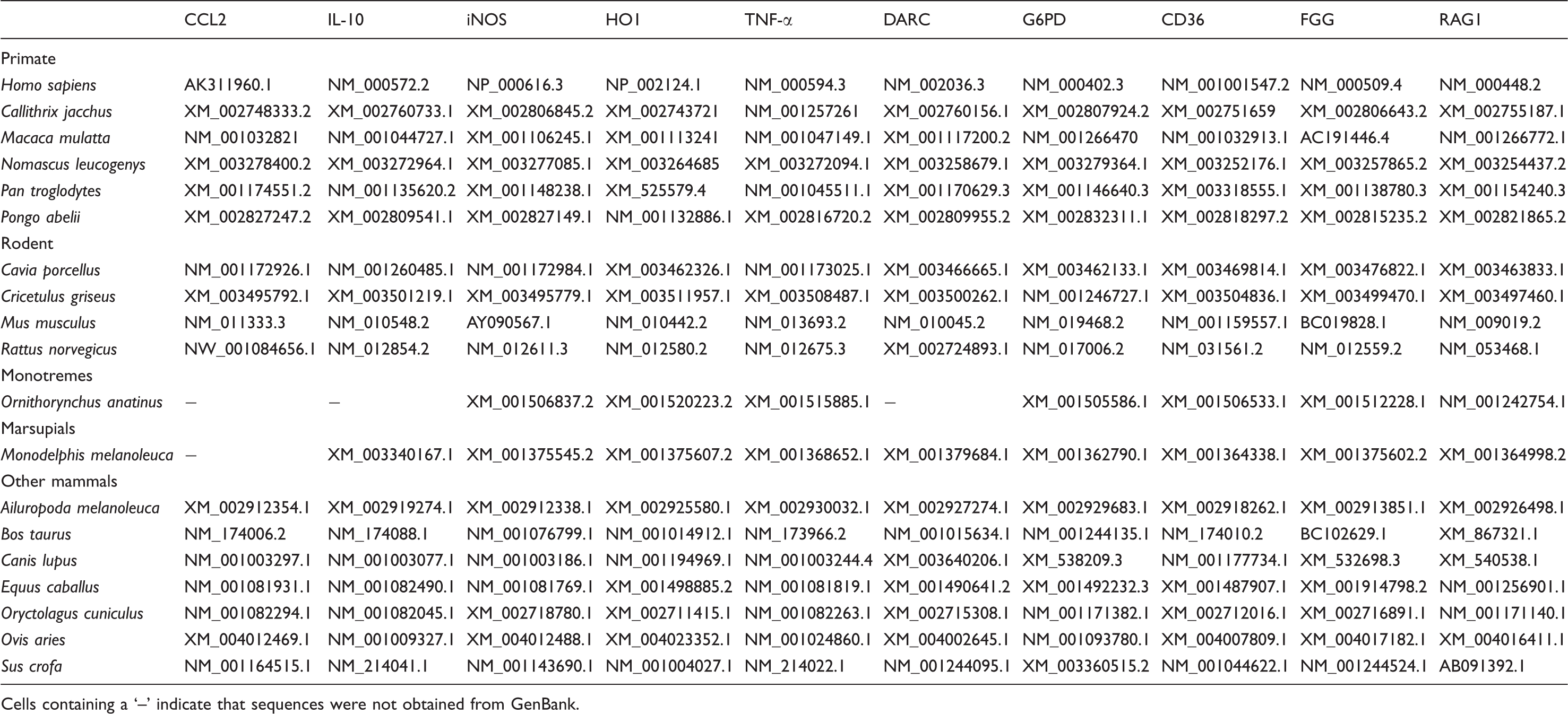
Cells containing a ‘–’ indicate that sequences were not obtained from GenBank.
Phylogenetic reconstruction
Species trees were reconstructed based on reference genes FGG and RAG1as the input tree for positive selection analysis. We chose this reference phylogeny as the input tree because it could better reflect the evolutionary relationship of selected taxa and was independent from the tested immunogenes to prevent circular arguments using the immunogene trees. 40 We used the method of Heled and Drummond to reconstruct a species tree under Yule’s pure-birth model.41,42 The general time-reversible substitution model with gamma and invariant site heterogeneity model by BEAST v. 1.7.3 were applied based on the substitution model test implemented in MEGA v. 5.05.43,44 Multiple-times independent pre-runs of 10 million generations of the length of Markov chain Monte Carlo (MCMC) were performed for obtaining better parameter priors to the last independent 10 million generations of the MCMC process. Genealogies were sampled every 1000 generations with the first 70% discarded as burning according to the simulation trajectory. Statistics of all the output values were summarized by TRACER v. 1.5. 45 Sampled trees were summarized by Tree Annotator v. 1.6.1, 43 and Fig Tree v. 1.3.146 was used to display the consensus tree. The divergent times of mammals estimated by O’Leary et al. were consulted for inferring the times of positive selection events. 47
Recombination analyses
Recombination could cause excessively long terminal branches in phylogeny owing to the introduction of apparent substitution rate heterogeneity among sites, 48 and therefore resulted in type-1 error (false positive) when detecting signals of positive selection using phylogenetic-based approaches. 49 Probable recombinant fragments were then identified by GeneConv (http://www.math.wustl.edu/∼sawyer/geneconv/), in which the segments that shared identical 5’ and 3’ ends are thought as consequences of recombination events. 50 Recombination between ancestors of two sequences (i.e. global inner fragments) was assessed by 10,000 random permutations to indicate the significance of putative recombinant regions.
Detecting genetic signatures of positive selection
The evolution of specific taxa and codons at a specific locus was tested for deviation from neutrality. We used the maximum likelihood approach to get the ratio of non-synonymous mutation rate (Ka) over the synonymous mutation rate (Ks) with higher statistical power by codon-based analysis (codeml) implemented in the PAML v. 4 package. 51 Ka/Ks (= ω) is regarded as the common approach to test the signature of molecular adaptation in response to natural selection. We used three selective models to test for positive selection in mammalian malaria-related genes: the branch model (free-ratio model), site model and branch site model.
The free-ratio model assumes independent ω in all lineages and average ω over sites. We used this model to test whether the positive selection acted on specific mammal taxa by comparison with the null model (one-ratio, M0). The likelihood ratio test (LRT) statistic, 2δL, against the χ2 distribution with degrees of freedom equal to the difference in the number of parameters was used to compare data fit for testing whether the free-ratio model provides a suitable explanation for the evolution of malaria-related genes. 52
However, the ω ratio averaged over sites may have low power to detect positive selection owing to the assumption that all sites in a sequence are under equal selective pressure, a potentially unrealistic assumption, as most amino acids in proteins are under functional constraints.
53
Therefore, site models that allow no positive selection of codon sites including M1a (the nearly neutral model) and M7 [the model allowing ω variation assumes a beta distribution over the interval (0, 1)] were tested against the nested models that allow positive selection of codon sites, including M2a (the positive selection model) and M8 (the model adds a discrete ω class to the beta model with positive selection ω >1), respectively. The M8 model was also compared to the M8a model, which has similar assumptions as the M8 model except limiting ω = 1. To perform the PAML analysis, non-eutherian mammals were removed from the analyses to prevent the high saturation in nucleotide replacement to bias the selection analysis. The input prior tree is shown as the left tree topology in Figure 1. The LRT was used to evaluate the fit of nested models to the data for positive selection.
52
We accepted the presence of positive selection when a significantly better fit of the selective model was found, and calculated the posterior probabilities of codon with ω >1 according to Bayes empirical Bayes (BEB) analysis.
54
In order to confirm the robustness of the results, we also used gene trees of each loci as inputs to repeat site model analyses to ensure the inference of positive selection.
The positively selected amino acids estimated under the site model with a posterior probability >0.8. Amino acids marked in dark background are estimated evolving with ω >1 under the M8 model.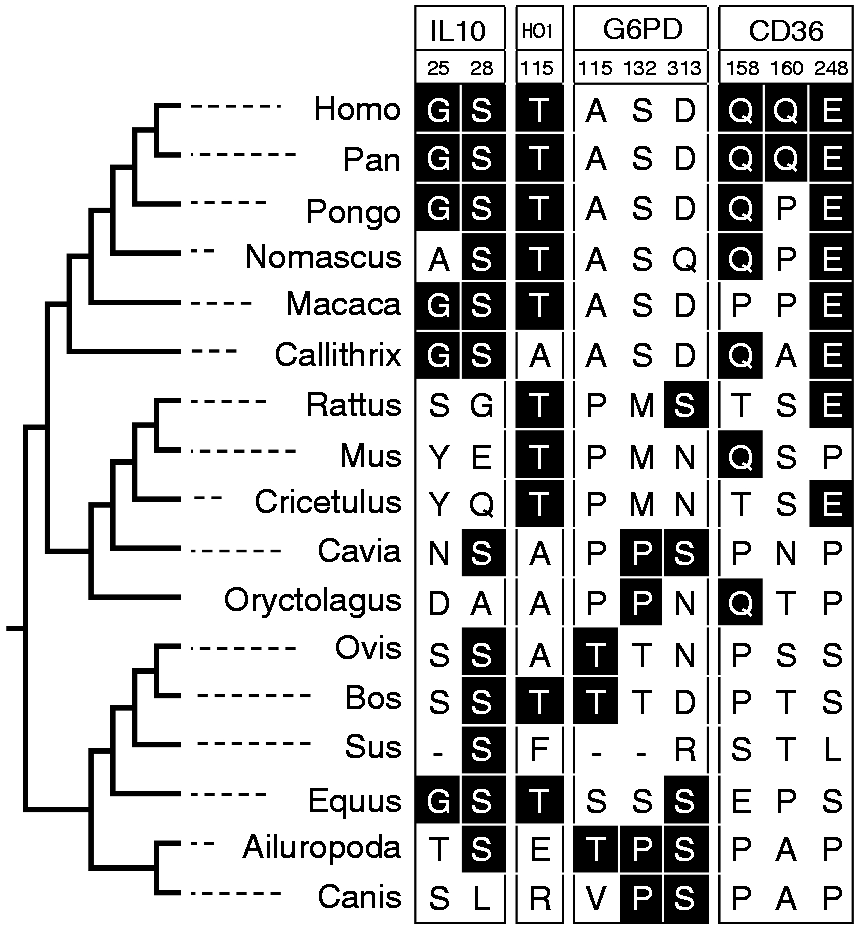
In addition, the positive selection of malaria-related genes was focused on primates and Glires (rodents + rabbit). We applied branch-site model A to the eight malaria-related genes and designated primates, Glires or primates + Glires as the foreground branches, respectively, in a series of independent tests. 55 The LRT was used to evaluate whether branch-site model A had a significantly better fit for the codon site with ω >1 in comparison with branch-site model A1, which fixes ω to 1.0 on the branches of interest. 55 If the LRT significantly rejected model A1, further tests for functional divergence between the foreground and background clades were performed by DIVERGE v.3.0 using 1000 bootstrap type-II divergence analysis. 56 For understanding the evolutionary trajectory of the change of positively selected codons in foreground clades, the ancestral states of these codons were inferred using the probabilistic-based maximum likelihood method under the M5 substitution model with the assistance of FastML.52,57
Results and discussion
Phylogenetic tree reconstruction and divergent time estimation
The Bayesian inference of evolutionary relationships of the studied species by two nuclear reference genes (left panel of Figure 1) is similar to the parsimonious analysis based on combined nuclear and phenomic data. 47 Posterior probabilities for all nodes are >0.90, indicating a reliable tree topology, except a relatively low support for the grouping of rabbits (Oryctolagus cuniculus) and rodents (posterior probability = 0.683). O’Leary et al. dated the divergent times of mammals by phenotypic and molecular data, and estimated the coalescent times of mammals, Theria, eutherians, primates, Glires and rodents as 166.2 Mya, 127.5 Mya, 64.85 Mya, 53.1 Mya, 63.4 Mya and 56.8 Mya, respectively. 47 The splitting times between old world monkey, gibbon, orang utan, chimpanzee and human are 23.3 Mya (between Oligocene and Miocene), 14.9 ± 2.0 Mya, 11.3 ± 1.3 Mya, 6.4 ± 1.5 Mya and 5.4 ± 1.1 Mya (Miocene), respectively. Comprehensive surveys on nearly 3000 fecal samples from wild chimpanzees, bonobos and gorillas in Central Africa suggested that ancestors of human-infected P. falciparum could be relatives of gorilla and chimpanzee parasites (e.g. P. falciparum and P. reichenowi, respectively).8,58 Several newly discovered Plasmodium isolates (P. gaboni, P. billbrayi and P. billcollinsi, and two unpublished isolates, P. GorA and P. GorB) were also shown to be able to infect great apes.10,11,59 The infectious history of hominid could be traced to the divergence of the P. goboni and P. falciparum/P. reichenowi lineages since 21 Mya, 9 which is roughly during the divergence period of old world monkeys and gibbons (23.3–14.9 Mya). The estimated splitting times are helpful for inferring the time of positive-selection events.
Recombination analyses
Summary of recombination (Rec.) events identified by GeneConvin of eight malaria-related immunogenes.
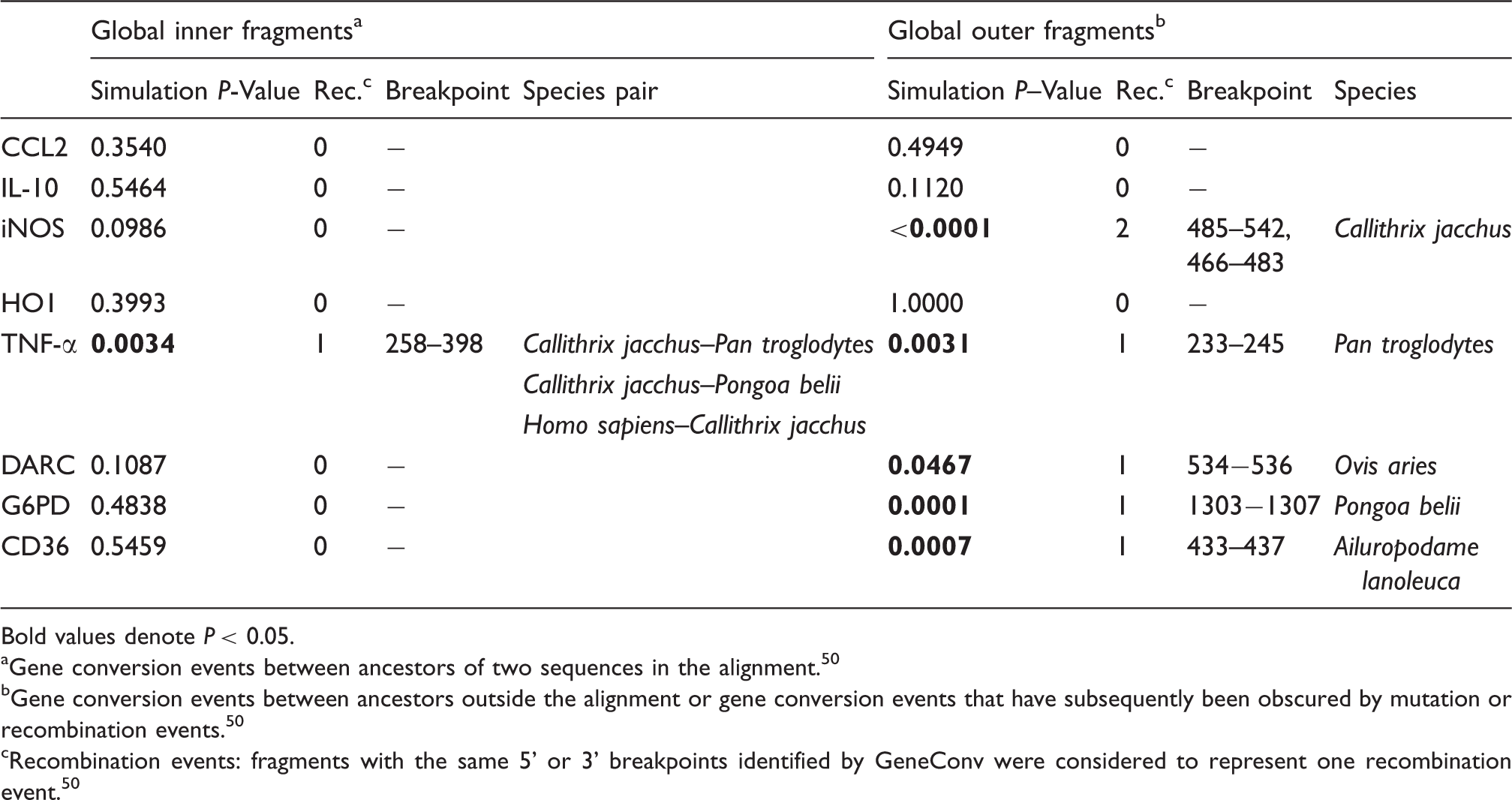
Bold values denote P < 0.05.
Gene conversion events between ancestors of two sequences in the alignment. 50
Gene conversion events between ancestors outside the alignment or gene conversion events that have subsequently been obscured by mutation or recombination events. 50
Recombination events: fragments with the same 5’ or 3’ breakpoints identified by GeneConv were considered to represent one recombination event. 50
Summaries of LRT results of PAML analyses using branch model, site models and branch site model with species tree.
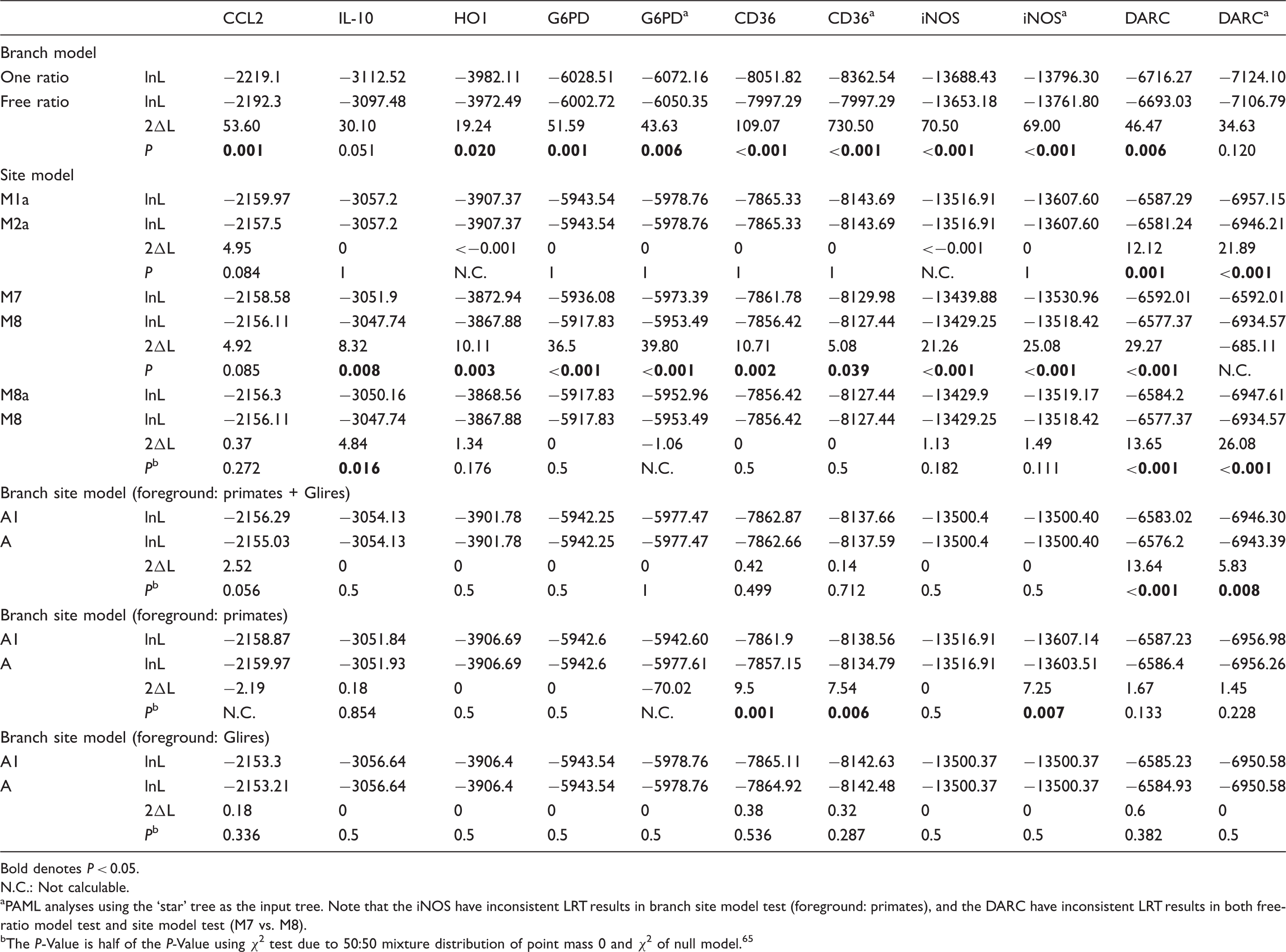
Bold denotes P < 0.05.
N.C.: Not calculable.
PAML analyses using the ‘star’ tree as the input tree. Note that the iNOS have inconsistent LRT results in branch site model test (foreground: primates), and the DARC have inconsistent LRT results in both free-ratio model test and site model test (M7 vs. M8).
The P-Value is half of the P-Value using χ2 test due to 50:50 mixture distribution of point mass 0 and χ2 of null model. 65
Gene tree vs. species tree
The gene tree of every malaria-related immunogene reconstructed by the Neighbor-Joining method revealed inconsistent tree topology with the species tree reconstructed by two reference genes (online Supplementary Figure S1). Heterogeneous tree topologies suggest different evolutionary tracks of these functionally important genes throughout the evolutionary history of eutherians. Such phylogenetic inconsistency may be signatures of natural selection, gene duplication or horizontal gene transfer. 61 To ensure the robustness of the analytical results, we used species tree and gene tree as inputs to conduct the PAML analyses.62,63 Concordant results of LRTs calculated by both analytical strategies (Table 4 and online Supplementary Table S2) are consistent with the simulation of Yang et al. that tree topology uncertainty may only be a mild effect for LRT. 64
Multiple episodes of positive selection on malaria-related genes on specific lineages and species codons
Evolutionary rates of non-synonymous sites to the synonymous sites are not constant among lineages through the evolutionary history of all the genes of interest. The one-ratio model was rejected in LRTs in CCL2, HO1, G6PD and CD36 (P < 0.05), and a fuzzy P-value (P = 0.05) in IL-10 provides moderate but not overwhelming evidence against the null one-ratio model (Table 4), but the free-ratio model was found to be a better fit for the evolution of most malaria-related immunogenes. Lineages that evolved with ω >1 are shown in Figure 2. Parameter estimates of site model and free-ratio model are listed in online Supplementary Table S1.
Summary of free-ratio model shows lineages that evolve with ω >1 (bold branches). Tree topology is inferred by two reference genes, FGG and RAG1, by Bayesian inference. The ω value of the positively selected branches is labeled beside the bold branches. The bold gray branches are the lineages with an estimated ω >100, representing relatively abundant non-synonymous variations or very rare synonymous variations.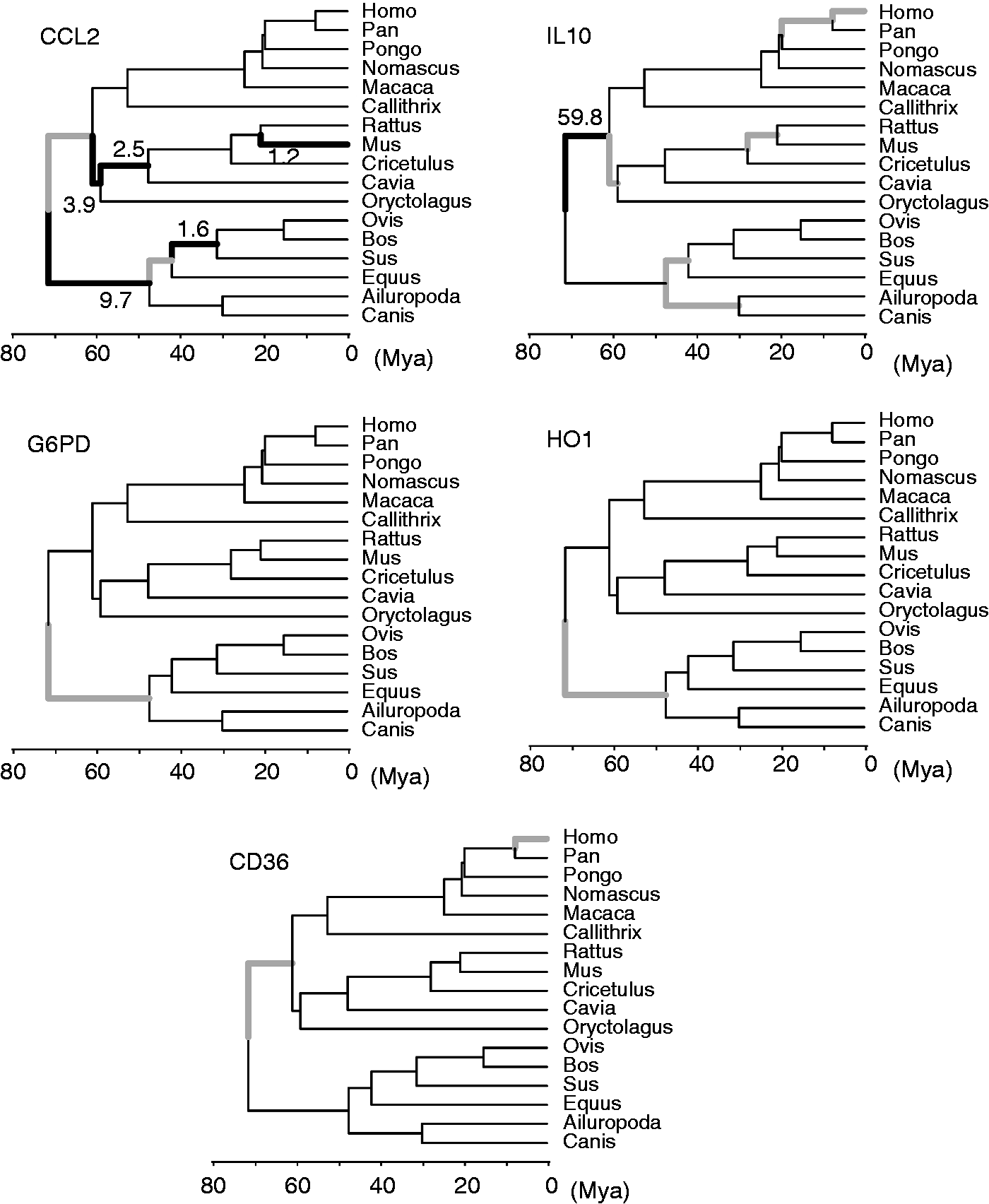
Because the M7–M8 comparison is less robust or more powerful than the M1a–M2a comparison, relatively abundant codons of a gene are able to be estimated to evolve with ω >1. 65 IL-10, HO1, G6PD and CD36 were suggested to be a better fit in the M8 model than in the null M7 model, but the nearly neutral model (M1a) cannot be rejected by the positive selection model (M2a). Among the four genes that are better fit in M8 than M7, only IL-10 could reject the null model of ω = 1 fixed (M8a) by M8, which is also consistent with the phenomenon of relatively less robust evaluation for positive selection by the M7–M8 comparison.
Cross verification of results of the molecular dating and free-ratio model analysis revealed that the timing of positive selection on malaria-related immunogenes in mammals could be divided into four phases. 47 The first phase is during Palaeocene, in which the primates + Glires and other eutherian mammals diverged at roughly 64.85 Mya (Figure 2). 47 Selective signals during this phase were detected in all genes, but these genes suffered different selective pressures on different lineages: CCL2, IL-10 and CD36 in common ancestors of primates and Glires, and CCL2, HO1 and G6PD in ancestors of other eutherians. Continued selective pressure is acted on common ancestors of Glires’ CCL2 and IL-10 at roughly 63.4 (65.0–61.7) Mya during the early Eocene (Figure 2). 47 In fact, selection signals were prevalently detected in CCL2 and IL-10 throughout evolutionary history of eutherians. The third phase describes the selection of hominidae genes, including human IL-10 and CD36. This phase describes the selective pressure on primates, especially for the apes, during the late Miocene. The fourth phase focused on the selection of other eutherians (exclusive of primates and Glires) since the Eocene, including CCL2 and IL-10 (Figure 2). Frequent detection of positive selection signals on four phases of mammalian evolutionary history is consistent with the arms races evolutionary model, a hypothesis used to explain the high variability of immunogenes.
Long-term selective pressures drove the evolution of CCL2 and IL-10
CCL2 and IL-10 are both non-specific immune-related genes. The free-ratio model showed significant higher ω in several branches in CCL2 and IL-10, revealing prevalent genetic change in response to the pathogen infections since the common ancestors of eutherians in Palaeocene (Figure 2). Frequent episodic positive selection drives the genetic change of immunogenes in responses to pathogen infections.66,67 The genetic diversity of CCL2 is associated with its receptor CCR2 regarding susceptibility to the immune-mediated disease. 68 CCR2 has been evidenced to be positively selected in placental mammals. 38 However, the site model does not reject the neutral or nearly neutral models (M1a, M7 and M8a) in CCL2 (Table 4). Signals of positive selection were detected in the branch model test but were not seen in site model analysis. This suggests that the pathogen stress could merely drive gene divergence between organisms but matter less with the structural and functional change of CCL2.
IL-10 also carried several genetic signals of positive selection throughout its evolutionary history based on the free-ratio model analysis. However, in contrast to the absence of selective signals on specific codons of CCL2, the IL-10 was detected with positive selection at its hydrophobic N terminals (25G and 28S; Figure 1) in most of the primate lineages and in certain eutherians (Table 4). The protein structure in which these two putative positively-selected amino acids are located is beyond the published crystal structure of human IL-10 (PDB: 2H24). In the genetic survey by Neves et al., 29 one codon (the 26th codon) at the N-terminal of IL-10 was also detected to be positively selected in mammals, which is consistent with our results of positive selection on N-terminal region. IL-10 plays an important function as a component of local defense (inflammation) against pathogen infections, with significantly higher ω driving the change of protein composition; its genetic divergence was apparently a genetic response to the diverse pathogens, supporting the arms race model of immune evolution.
Positive selection on HO1
Evidence for positive selection on primates and rodents HO1 was only found in the site model analysis (M8 model) but not in the free-ratio and branch-site models (Table 4, Figure 2), suggesting that the selective pressure could be pervasive in mammals, rather specifically in primates, rodents or in other specific taxa. Four codons (35R, 115T, 191I and 226P) have been estimated at ω >1 (ω = 1.773) in the M8 model but only 115T has a high posterior probability (0.943), and these putatively positively selected codons are distally located from any known ligand binding site. 69 HO1 is ubiquitously expressed and catalytically active for stress responses in multiplicity and have broad functions but are conserved along mammalian evolution.70,71 Therefore, the functionally important ligand binding sites were selectively constrained with low residual replacement rates among species, whereas non-binding regions and non-coding regions (e.g. promoters) could be more highly polymorphic, associated with the susceptibility to multiple diseases.72–74 The bending region that links to binding domains could be more flexible and has higher amino acid replacement rates. For example, the 115th codon that is located between two α-helices beside the binding domain of residues HEM_A300, HEZ_A401 and Q86_A301 has a higher replacement rate and is suggested to be positively selected in primates, rodents, cattle and horses (Figure 1). 69 In addition, a trend of identical or similar adaptive signals on HO1 and iNOS lineages (Figure 1) suggests parallel evolution at these two synergistically expressed genes,75–77 like that of the RLR genes RIG-I and LGP2, which suffer independent but parallel selective pressures at the same sites. 28
Positive selection on the primate CD36 gene
CD36 is a major receptor for the Plasmodium-infected erythrocytes for sequestering the infected erythrocytes.78,79 Recent positive selection on variations of CD36 was suggested to be responsible for differential susceptibility to P. falciparum malaria in Africa.30,32 In the present study, although we did not comprehensively survey the human population, signals of selective pressures were also detected in human lineage and the common ancestor of primates and Glires based on free-ratio model analysis (Figure 2). No other lineages showed significantly high average ω under the free-ratio model test, but signals of positive selection in primates were found by site model and branch-site model tests. This indicates that adaptive divergence of CD36 between mammals occurred only on specific codon positions.
The positive selection of CD36 is evident in primate lineages by M8 site model analysis, but the nearly neutral (M1a) model and the M2a positive-selection model failed to detect positive selection signals (Table 4). Under the M8 model analysis, three codons, 158Q, 160Q and 248E, evolving with ω >1 (posterior probability >0.80; Figure 3) were mostly found in primates, supporting the existence of selective pressures that impose rapid protein composition changes in primates. Plasmodium-infected erythrocytes can adhere to the CD36 protein and prevent parasites from adhering to neurovascular adhesion molecules (e.g. ICAM-1) in order to protect from cerebral malaria.80,81 The rapid non-synonymous evolution of CD36 genes driven by positive selection in human or in primates could not only be related to the multiple-function divergence of CD36, but also to multiple malaria parasitism with host shift through Plasmodium evolution.8–11
Posterior probability of the inferred positively selected sites estimated from site models of M2a and M8. The x-axis indicates the codon position and the y-axis is the posterior probability of the codon with ω >1.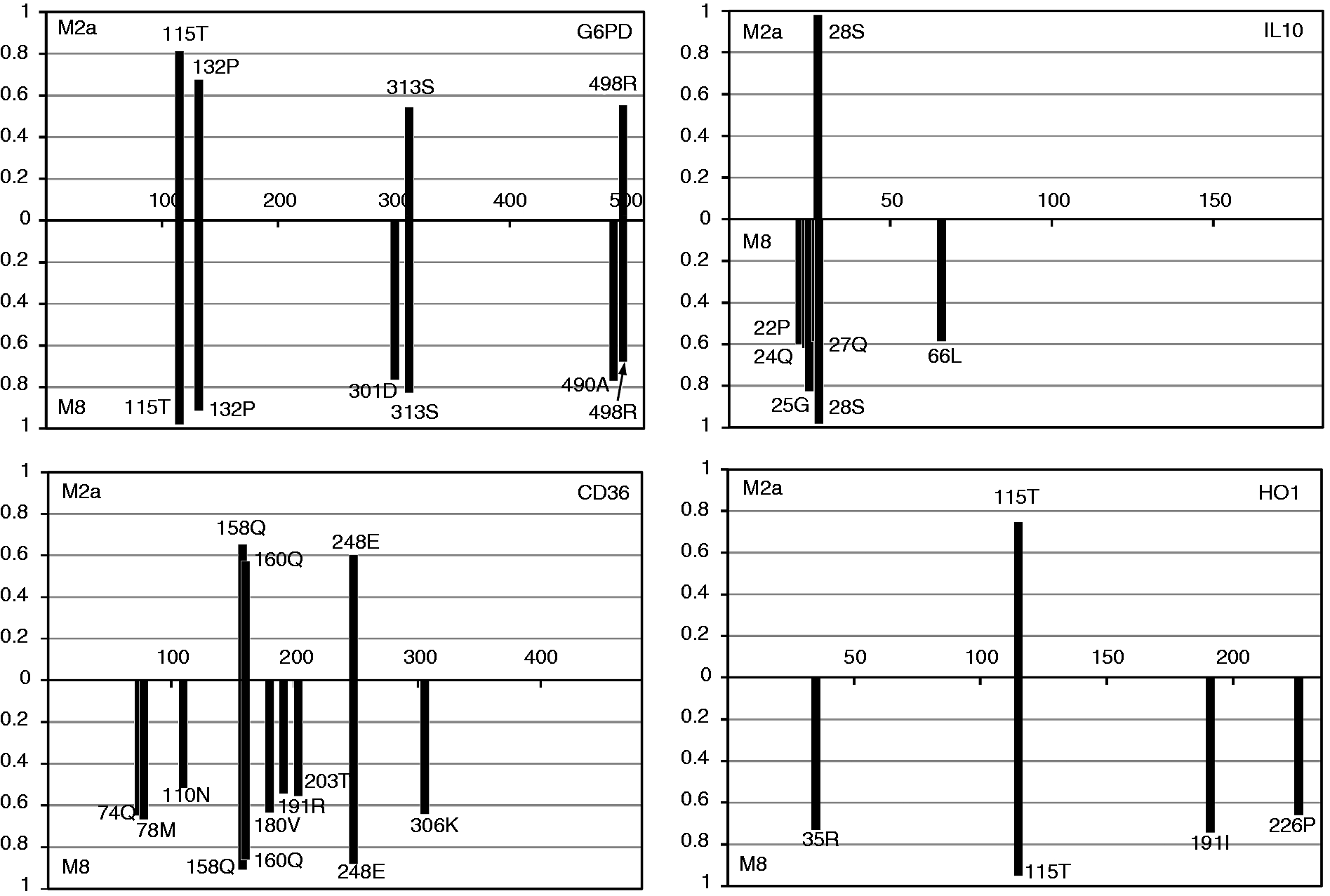
Three foreground branches were designated for the branch-site model test: primates, Glires and primates + Glires. Among all comparisons of all genes between model A1 (ωs = 1 fixed) and model A, CD36 was a better fit in model A only when the primates were designated as the foreground branch (P = 0.001, Table 4). Three codons were inferred to be ω >1with a posterior probability >0.9 in BEB analysis: 18V (probability = 0.972, for human, chimpanzee and macaque), 208L (probability = 0.953, for all primates except macaque) and 412N [probability = 0.830, for hominidae (i.e. human, chimpanzee and orang utan)]. The inter-specific test of selective pressures on specific codons indicated that the extracellular domain of CD36 might be the earlier target of positive selection since the Eocene (the MRCA of primates, 208L) and since the early Miocene (the MRCA of hominidae, 412N). 33 Selected pressure on 18V, which is located on the N-terminal transmembrane domain of CD36, happened for humans and chimpanzees during the late Miocene (c. 7.77 Mya). It should be noted that codon 18 and codon 412 of all or most background taxa are also Val and Asn, respectively, suggesting the reversible amino acid change for keeping ancestral states to be positively selected.
We further examined whether the positively selected amino acids in foreground lineages that are identical to backgrounds are a consequence of reversible evolution (i.e. identical codons) or only a coincidence (i.e. synonymous codons) based on ancestral sequence inferences. The ancestral states of the 18th codon of the common ancestor of primates, common ancestor of primates + Glires and common ancestor of all eutherians are all GTC (Val), identical to human, chimpanzee and macaque; the identical codon of the 412th Asn (AAT) was also observed between the hominidae and the common ancestors of primates + Glires and of all eutherians (Figure 4).
Ancestral states of positively selected codons of the primate CD36 gene. The codons, codon positions, amino acids and posterior probabilities of the ancestral state reconstruction (parentheses) are listed. The positively selected codons are inferred by the branch site model analysis using the primates (gray area) as the foreground branch and are indicated by asterisks (*).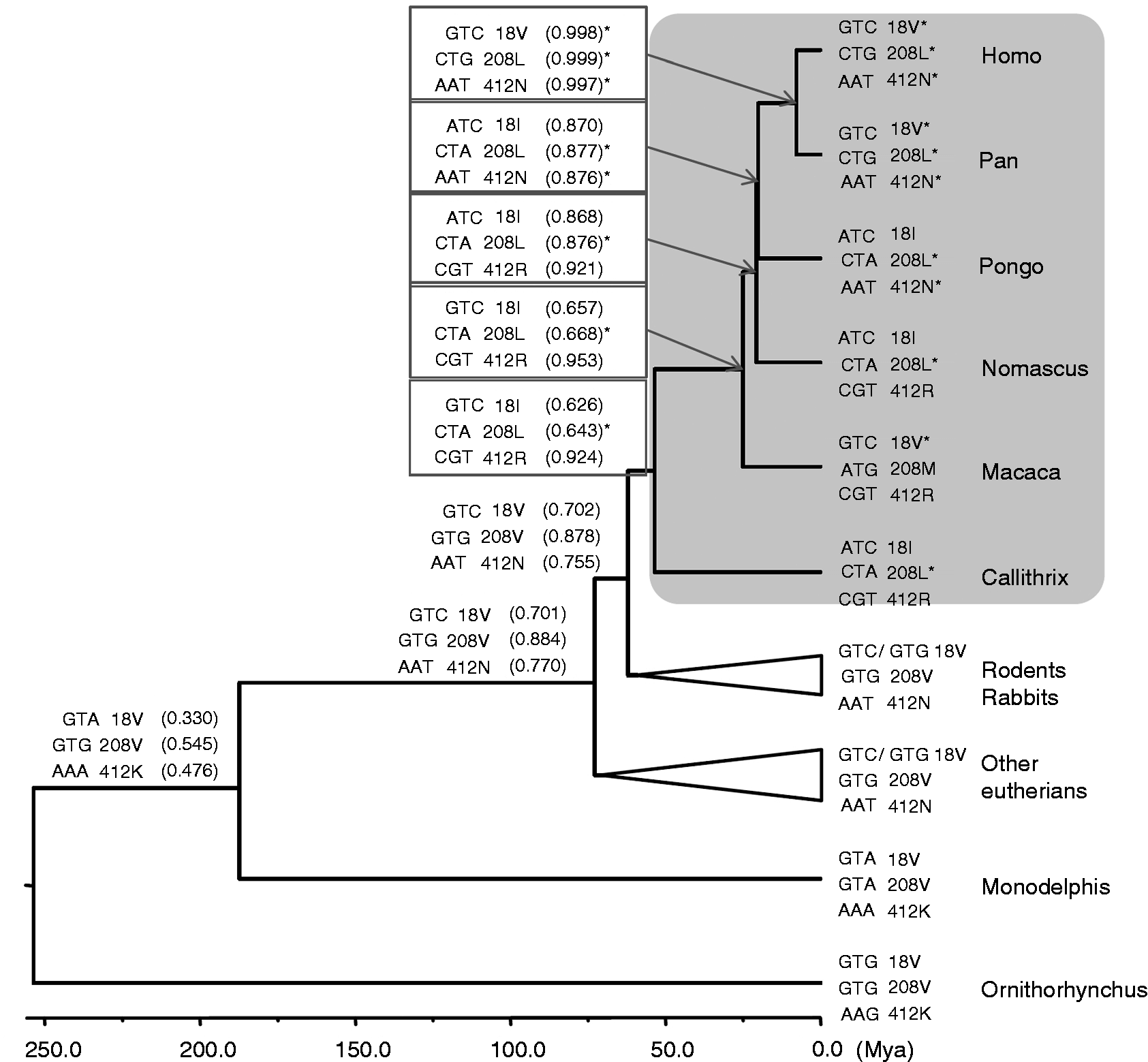
Summary of pairwise comparison of functional divergence between primates, glires and other eutherians of CD36.
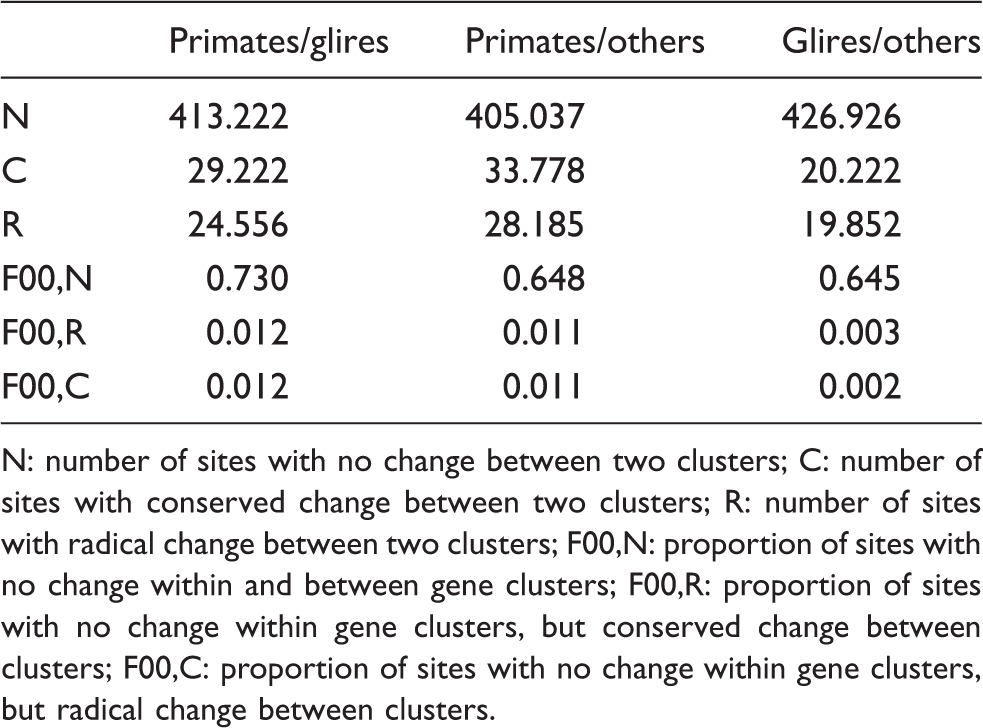
N: number of sites with no change between two clusters; C: number of sites with conserved change between two clusters; R: number of sites with radical change between two clusters; F00,N: proportion of sites with no change within and between gene clusters; F00,R: proportion of sites with no change within gene clusters, but conserved change between clusters; F00,C: proportion of sites with no change within gene clusters, but radical change between clusters.
Positive selection of non-primate–mammalian G6PD
We found evidence of positive selection in five of the eight immunogenes analyzed; G6PD is the only one gene that showed no positive selection signal in primates or the earliest ancestors. However, selective signatures G6PD were detected in ancestor lineages of other Eutheria excluding primates and Glires clade, based on free-ratio model analysis (Figure 2), and positive selection in codon sites were found in all Eutheria excluding primates, according to the site model analysis (Figure 1). Although G6PD diversity has been reported to show its positive selection regarding malaria, 30 the estimated origin of malaria-resistance alleles of G6PD is roughly from around 10,000 years ago. 31 The presumptive positive selection on G6PD in non-primate ancestors of Eutheria was approximately at 50 Mya (Figure 2), far before the origination of malaria-resistance alleles. Therefore, the positive selection at G6PD detected in this study is probably unrelated to malaria but it can be driven by other factors.
Conclusions
Plasmodium-induced malaria parasitism found in most mammals implies the long term inter-relationship between the Plasmodium parasite and mammalian immune system. In this study, primates showed the highest rate of evolution in CD36 than that of other mammals. A higher ratio of radical amino acid changes suggests that primate CD36 might have undergone functional divergence from other eutherians in response to the blood-borne infectious diseases, such as malaria. In addition to CD36, other immunogenes were also detected to be under positive selection in different lineages and on several codons. For example, multiple lineages of CCL2 and IL-10 were evolved with a higher proportion of non-synonymous mutations, revealing a prevalent evolutionary response against infections. Ancient recombination detected in the pro-inflammatory cytokine gene TNF-α among primates is likely an alternative mechanism in maintaining gene polymorphism. Genetic signatures of positive selection during evolutionary trajectories of mammalian immune-related genes indicate the prevalence of a particular type of adaptive mutations offering protection against malaria, which clues the arms race co-evolution of infectious Plasmodium and eutherians.
Footnotes
Funding
This research was financially supported by the National Science Council in Taiwan (NSC 102-2621-B-003-005-MY3).
Conflicts of interest
The authors do not have any potential conflicts of interest to declare.
Acknowledgements
Our gratitude goes to the Academic Paper Editing Clinic, NTNU.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.