
Abstract
SR-A/CD204 and CD36 are major receptors responsible for oxidized lipoproteins uptake by macrophages in atherosclerotic plaques. Both receptors also share the role as receptors for different pathogens, but studies on their signaling have been hampered by the lack of selective ligands. We report that, upon specific ligation by Ab, SR-A does not induce cytokine production, but mediates inhibition of LPS-stimulated production of IL-6 and IL-12/23p40, enhancement of IL-10 release, and has no effect on TNF-α and RANTES production in murine macrophages. In contrast, anti-CD36 Ab alone stimulated production of all these cytokines, with IL-10 production being exceptionally high. Effects of anti-CD36 Ab, except of IL-10 production, were mediated by CD14 and TLR2, whereas those of SR-A ligation by heterotrimeric Gi/o proteins and by phosphatidylinositol 3-kinases. Surprisingly, we found that LPS uptake by macrophages was mediated in part by CD36 cooperating with CD14, whereas SR-A was not involved in this process. Finely, during in vitro Ag presentation to naïve CD4+ lymphocytes, pre-incubation of macrophages with anti-CD36 Ab enhanced IFN-γ production in the co-culture, but exerted the opposite effect under conditions enabling IL-10 accumulation. In contrast, anti-SR-A Ab was ineffective alone, but reversed the Th1-polarizing effect of LPS.
Introduction
The hallmark of atherosclerosis is the accumulation within the arterial intima of lipid-laden macrophages, known as foam cells. The non-saturating uptake of oxidized lipoproteins by macrophages, leading to their transformation into foam cells, is mediated by a heterogeneous group of receptors called scavenger receptors (SR). Although the ability to bind oxidized low-density lipoprotein (LDL) is the defining feature of all members of this artificial family, only two SR—SR-A/CD204 and CD36—account almost entirely for this ligand uptake by macrophages. 1 SR-A and CD36 are members of, respectively, class A and class B families of SR. In addition to binding oxidized LDL, both receptors also share the ability to mediate adhesion to, or uptake of, a very wide range of other ligands, including extracellular matrix components, sulfated polysaccharides, apoptotic cells, amyloid-β fibrils, proteins modified with advanced glycation end products, unopsonized yeasts, bacteria and their products, such as LPS and lipoteichoic acid (LTA).2–7 This ability is the basis for the involvement of both receptors in cellular adhesion and migration, innate immunity, as well as in the pathogenesis of Alzheimer’s disease and diabetes-associated pathologies. In addition, by binding long-chain fatty acids CD36 participates in lipid sensing and metabolism, mediates the anti-angiogenic effect of proteins containing thrombospondin type I repeats, as well as, by serving as a receptor for glycosylphosphatidylinositol (GPI) anchors from Plasmodium falciparum and P. falciparum-infected erythrocytes, contributes to innate immunity to blood-stage malaria.8–10 In turn, other functions of SR-A, which do not seem shared with CD36, include its role as a receptor for nucleic acids and inorganic environmental particles.11–13
Despite the demonstration of SR-A and CD36 involvement in so many physiological and pathological processes, studies on their signaling abilities have been hampered by the lack of selective ligands. For instance, oxidized LDL not only binds to all SR, but oxidation of LDL also generates biologically active mediators from its lipid component, which act through separate sets of receptors, including platelet-activating factor receptor 14 and nuclear peroxisome proliferator-activated receptors. 15 Sulfated polysaccharides [dextran sulfate (DS), fucoidan, carrageenan] also bind to many, if not all, SR, as well as to receptors not classified into SR, such as selectins 16 and the mannose receptor. 17 Another problem associated with the use of SR ligands is their contamination with microbial products having high biological activity, such as LPS or lipopeptides. For instance, the results of Kim et al. 18 indicated that contamination with microbial products was responsible for the observed effects of fucoidan on macrophages, as these effects were abolished in CD14-deficient macrophages, whereas SR-A-deficient macrophages responded similarly to wild type (WT) cells.
The most frequently applied method in studies of SR signaling involves deduction of SR-specific effects from the comparison of responses of WT and receptor-deficient cells to non-selective SR ligands. However, this methodological approach has also been challenged by recent reports showing that SR-A-deficient cells or mice exhibit generalized enhancement of immune responses, which seems, in large part, caused by compensatory changes in the expression of other receptors 19 and/or by the removal of tonic inhibitory effects exerted by intracellular SR-A on signal transduction from receptors belonging to the TLR/IL-1R family, 20 rather than by the loss of receptor functions of SR-A.
In our previous studies we overcame the problems associated with the use of non-selective ligands by using specific Ab to selectively stimulate class A SR on the surface of macrophages.21,22 In this study, using a similar approach, we compared signaling abilities of SR-A and CD36. Our results reveal that despite sharing several ligands and being involved in largely overlapping physiological and pathological processes, SR-A and CD36 differ significantly in their effects on pro-inflammatory and immunoregulatory functions of macrophages.
Materials and methods
Reagents
Anti-mouse SR-A mAb (MαSR-A, clone 2F8) was obtained from AbD Serotec (Oxford, UK); anti-human/mouse TLR2 (clone T2.5) and mouse IgG1 isotype control mAb (clone P3.6.2.8.1) from eBioscience (San Diego, CA); anti-mouse CD36 mAb (MαCD36, clone CRF D-2712) from Hycult Biotech (Uden, the Netherlands); anti-mouse TLR4/MD-2 complex (clone MTS510) from BioLegend (San Diego, CA); anti-mouse CD14 (clone 4C1/CD14), anti-mouse-CD11b (clone M1/70), rat IgG2b isotype control (clone A95-1), neutralizing anti-mouse IL-10 (clone JES5-16E3) and mouse IgA isotype control (clone M18-254) mAb from BD Biosciences (San Jose, CA). The above listed Ab were functional grade purified (low endotoxin/no azide).
The synthetic triacylated lipopeptide Pam3CSK4 (TLP), LTA from Staphylococcus aureus, ultrapure LPS from Escherichia coli K-12 strain and biotinylated LPS from E. coli 0111:B4 were purchased from InvivoGen (San Diego, CA); DS (∼ 500 ku), and chondroitin sulfate (CS) A sodium salt from bovine trachea from Sigma-Aldrich (St. Louis, MO). Low endotoxin acetylated LDL (AcLDL) was obtained from Biomedical Technologies (Stoughton, MA), and BODIPY- or Alexa Fluor 488-labeled LPS (AF488-LPS) from E. coli serotype 055:B5 from Invitrogen (Eugene, OR).
Inhibitors of mitogen-activated protein kinase kinase [an upstream activator of extracellular signal-regulated kinases (ERK)]—PD 98059 (ERKi), p38 kinase–SB 203580 (p38i), JNK−SP600125 (JNKi), adenyl cyclase−MANT-GppNHp, and of Src family protein tyrosine kinases (STK)−PP2 were obtained from Calbiochem (Darmstadt, Germany). Wortmannin was from Invitrogen and pertussis toxin, cytochalasin D and dynasore from Sigma-Aldrich.
Mice
Breeding pairs of SR-A-deficient mice and OT-II transgenic mice, both on the C57BL/6 background, as well as C3H/HeJ mice, WT C57BL/6 and Balb/c mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were housed in our facility in microisolator cages with filter tops on a 12-h light/dark cycle. The studies were reviewed and approved by an appropriate institutional review committee.
Cells
Twelve to sixteen-wk-old female mice were quickly euthanized by overdosing of halothane vapors (Halocarbon Products, River Edge, NJ), followed by cervical dislocation. Inflammatory peritoneal cells, elicited with 1.5 ml of aged, 3% thioglycollate (Difco Laboratories, Detroit, MI), injected i.p. 4 d earlier, were washed out with PBS. The cells were re-suspended in FCS-RPMI [RPMI 1640 medium with HEPES supplemented with 10% FCS, 2 mM stable
Alveolar macrophages (AM) were isolated by bronchoalveolar lavage, as described previously. 21
Dendritic cells (DC) were differentiated from bone marrow cells of 4-wk-old female mice under the influence of 20 ng/ml recombinant granulocyte-macrophage colony-stimulating factor (eBioscience), as described previously. 23 CD11c-positive DC were purified using magnetic beads (Miltenyi Biotec, Auburn, CA).
CHO-K1 cells were transfected with human type I SR-A and cultured, as described previously. 12
Stimulation of cytokine production
Unless otherwise indicated, PEM were treated for 4 h (TNF-α and RANTES production) or 22 h (IL-12/23p40, IL-6 and IL-10 production) with 100 ng/ml LPS, 10 µg/ml LTA, 200 ng/ml TLP, 100 µg/ml DS, CS or AcLDL, 5 µg/ml MαCD36, 20 µg/ml MαSR-A or the same concentrations of isotype-matched control mAb in 0.2 ml of FCS-RPMI (four wells for each treatment). To block TLR2 or CD14 receptor, cells were pre-incubated for 30 min at room temperature (22 ± 2℃) with double-concentrated solutions (40 µg/ml) of anti-TLR2, anti-CD14 or isotype-matched control mAb before the same volumes of double-concentrated solutions of SR-specific mAb or TLR agonists were added. In experiments with pharmacological inhibitors, PEM were pre-incubated for 40 min at 37℃ with 20 μM ERKi, 10 µM p38i, 5 µM JNKi, 500 nM wortmannin, 5 µM PP2, 10 µM MANT-GppNHp, 12 µM cytochalasin D, 50 µM dynasore or for 2 h with 1 µg/ml pertussis toxin, and then stimulated with mAb and/or TLR agonists in the continuous presence of inhibitors. Neutralizing anti-IL-10 mAb and isotype-matched control rat IgG2b were used at 4 µg/ml.
Effects of treatments on cell viability were assessed by the MTT reduction assay, as described previously. 24
IL-6, IL-4, IL-12/23p40, IL-12p70 and TNF-α concentrations in culture supernatants were determined by standard sandwich ELISA, using matched pairs of capture and biotinylated, detection mAb and standards of recombinant cytokines from eBioscience or BD Biosciences, streptavidin-HRP conjugate from Vector Laboratories (Burlingame, CA), and TMB Substrate Reagent Set (BD Biosciences) as the HRP substrate. RANTES, IFN-γ and IL-10 determinations were performed with the use of ELISA sets, obtained from R&D Systems (Minneapolis, MN), eBioscience and BD Biosciences, respectively.
Effects of receptor ligation on PEM on the polarization of primary immune response in vitro
Peritoneal exudate cells were plated at 1.4 × 105/well in 0.1 ml of FCS-RPMI and, following 1.5 h incubation, non-adherent cells were removed. Adherent PEM were incubated for 2 h in 0.1 ml of FCS-IMDM [IMDM medium (Cytogen) supplemented with 25 mM HEPES, 10% FCS, 1 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol (Invitrogen) and gentamycin] with 0.02 µM OVA323-339 peptide (InvivoGen), receptor-specific or control mAb, with or without 200 ng/ml LPS. Subsequently, 2 × 105 of CD4+ lymphocytes, isolated from spleens of naïve OT-II mice with the use of magnetic beads (Miltenyi Biotec), were added in 0.1 ml of FCS-IMDM. Concentrations of cytokines in the culture medium were determined after 1–3 d of co-culture.
In the second type of experiments, after 2 h pre-incubation, OVA323-339, LPS and mAb were removed by washing, before OT-II cells were added.
Assessment of surface protein expression
Adherent C57BL/6 PEM in 96-well Optilux Black/Clear Bottom plates (BD Biosciences) were treated with mAb and LPS as described in the previous subsection, except that OT-II lymphocytes were not added. The effects of these treatments on CD40, CD86 and MHC class II (I-Ab) expression were assessed as follows. In order to block non-specific binding, PEM were pre-incubated for 30 min on ice with 60 µl of FCS-RPMI containing 20% normal mouse serum, 0.1 mg/ml mouse IgG and 20 µg/ml anti-FcγRII/III 2.4G2 mAb (BD Biosciences). All subsequent incubations were performed on ice in 20% mouse serum. CD86 and MHC class II molecules were labeled by 1 h incubation with 7 µg/ml of phycoerythrin (PE)-conjugated specific mAb (BD Biosciences). CD40 was labeled with 7 µg/ml of biotinylated anti-CD40 mAb, followed by 7 µg/ml PE–streptavidin conjugate (BD Biosciences). Following thorough washing, fluorescence of mAb bound to cells was measured with the use of a fluorescence plate reader (Infinite M200 PRO; Tecan, Männedorf, Switzerland). Values of receptor-specific binding were obtained by subtracting values of binding of the same concentrations of control mAb (BD Biosciences).
Expression of SR-A on the surface of PEM [detached with 15 mM lidocaine (Sigma-Aldrich) plus 5 mM EDTA in PBS], DC or freshly-isolated AM was assessed by flow cytometry (FACSCalibur; BD Biosciences) using 3 µg/ml MαSR-A or control rat IgG2b mAb, followed by 5 µg/ml PE-conjugated F(ab’)2 fragment of goat anti-rat IgG Ab (Invitrogen). Expression of TLR4/MD-2, CD14, MARCO and CD11b (CR3) on adherent PEM, as well as binding of different concentrations of MαSR-A to these cells, were determined by cellular ELISA, as described previously. 22 Primary mAb were used at 10 µg/ml, except of anti-MARCO (clone ED31) and isotype-matched rat IgG1 control mAb (Serotec) that were used at 20 µg/ml. HRP-conjugated F(ab’)2 fragments of goat anti-rat IgG Ab (Rockland Immunochemicals, Gilbertsville, PA) at 5 µg/ml was used as the secondary Ab. Amounts of HRP-conjugated Ab bound to PEM were read from standard curves prepared using different amounts of this Ab.
PEM-mediated LPS clearance from the culture medium
LPS concentrations in culture supernatants of PEM were determined with the use of Endpoint Chromogenic LAL Assay QCL-1000 (Lonza, Basel, Switzerland), according to the manufacturer’s instruction.
Uptake of fluorescently-labeled LPS
CHO-K1 cells, transfected or not with human SR-A, were incubated for 1.5 h with AF488-LPS in 10% FCS-containing F12 medium (Mediatech, Herndon, VA). At the end of incubation, cells were washed four times with HBSS, and cell-associated LPS was quantified by measuring its 488 nm/530 nm fluorescence in a fluorescence plate reader (Spectrafluor Plus; Tecan).
In the LPS uptake assay by PEM we used BODIPY-labeled LPS instead of AF488-LPS because the labeling level of AF488-LPS was insufficient for this application. Monolayers of PEM in 24-well plates were pre-incubated for 30 min at room temperature with mAb before BODIPY-LPS was added for 40 min or 2 h incubation at 37℃. Unbound BODIPY-LPS was removed by washing, PEM were detached with lidocaine/EDTA and cell-associated fluorescence was determined by flow cytometry.
Binding of biotin-conjugated LPS
Adherent PEM were incubated for 1 h on ice with different concentrations of biotinylated LPS in FCS-RPMI. Subsequently, cells were washed three times with 0.5% BSA (Roche Diagnostics, Basel, Switzerland) in calcium- and magnesium-containing PBS (BSA-PBS) and incubated for another hour with 5 µg/ml streptavidin-HRP in BSA-PBS. Following thorough washing, the enzymatic reaction for peroxidase was performed. In experiments in which effects of receptor-blocking mAb was studied, PEM were pre-incubated with mAb for 40 min on ice before biotinylated LPS was added.
Data analysis
Experimental groups were compared with control groups with the Student’s t-test (GraphPad Prism software) and P-Values < 0.05 were assumed to denote statistically significant differences. The GraphPad Prism program was also used to calculate receptor binding parameters by non-linear regression curve fitting.
Results
Responses to non-selective SR ligands are preserved in SR-A-deficient PEM
Although activation of cells by LPS and LTA is mediated mainly by TLR4/MD-2 and TLR2, respectively, both these major constituents of bacterial cell walls have also been reported to be ligands of SR-A.25,26 We therefore assessed effects of SR-A deficiency on cytokine production stimulated in PEM by LTA and LPS, as well as by TLP, an agonist of TLR2, which seems unable to bind to SR.
27
In an average from several experiments, all these ligands stimulated similar production of TNF-α, RANTES, IL-10 and IL-6 in WT and SR-A−/− PEM (Figure 1A and data not shown). Consistent with similar production of inflammatory cytokines in response to LPS, WT and SR-A−/− PEM expressed very similar levels of TLR4/MD-2 and CD14 (Supplementary Figure S1A). However, LPS, but not LTA or TLP, stimulated significantly lower IL-12/23p40 production in WT than in SR-A−/− PEM (Figure 1A).
Responses to non-selective SR ligands are preserved in SR-A-deficient PEM. PEM isolated from WT and SR-A-deficient (SR-A-/-) mice were treated with indicated TLR agonists (A) or co-treated with 100 ng/ml LPS and 100 µg/ml DS, CS or AcLDL (B) and cytokine concentrations were determined in culture supernatants by ELISA. The graphs show averages (+ SEM) of 5–8 independent experiments (A) or results (means + SEM) of a representative experiment out of three such experiments performed. *A significant difference between WT and SR-A-/- PEM (A) or a significant effect versus the control stimulated with LPS only (B).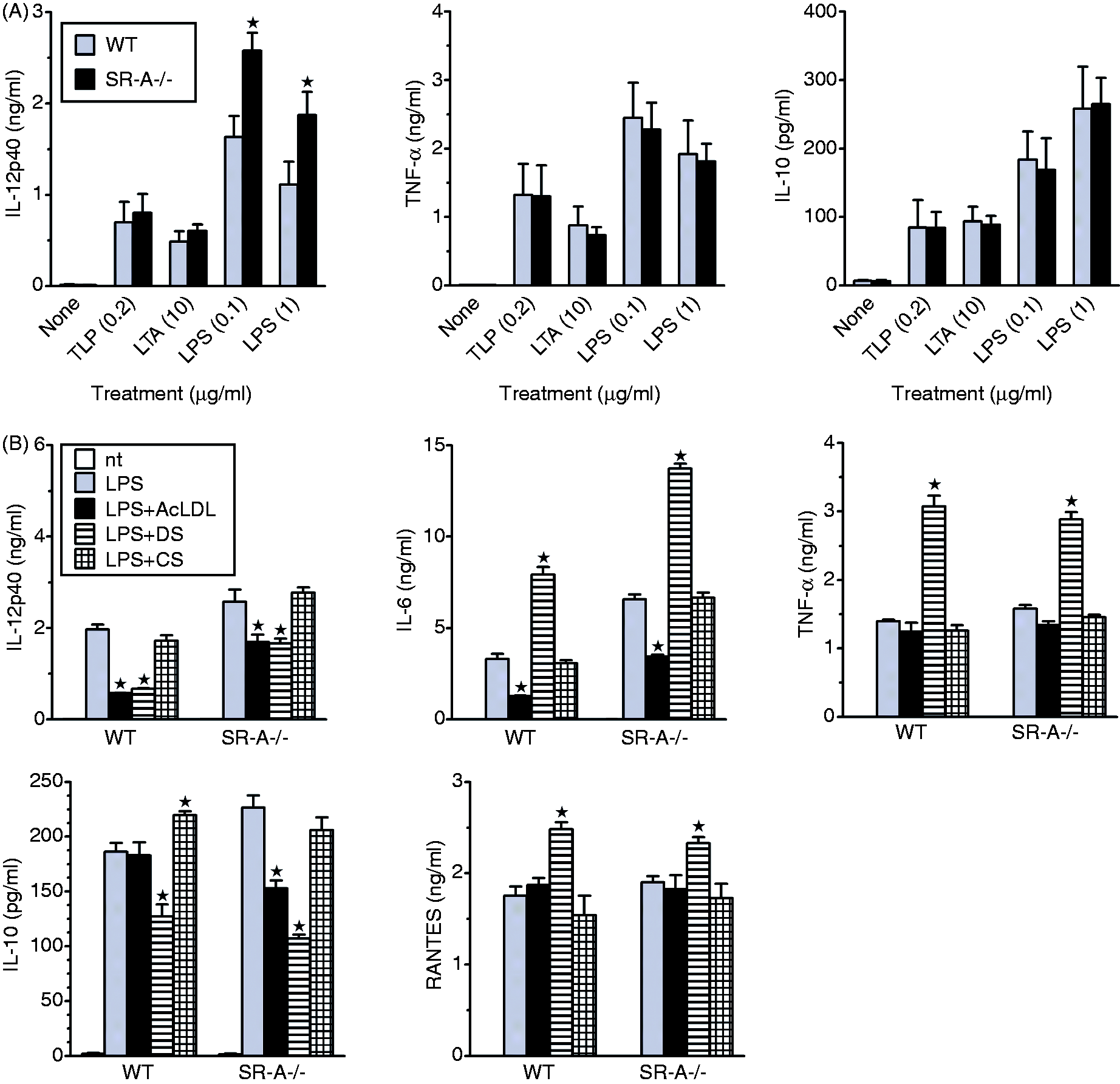
A frequently applied method in studies on SR-A signaling involves comparison of responses of WT and SR-A−/− cells to sulfated polysaccharides.18,28–30 We applied this approach to examine the role of SR-A in DS effects on cytokine production. We decided to use DS rather than the chemically similar sulfated polysaccharide, fucoidan, because of its superior purity in comparison to commercially available crude preparations of fucoidan. As a control we studied the effects of CS. The concomitant use of DS and CS allows to discriminate effects mediated by SR from those mediated by non-SR DS receptors because we found that several SR, including SR-A, CD36, SREC-I and LOX-1 bound DS but not CS, whereas non-SR receptors of DS, such as RAGE and the mannose receptor, bound both DS and CS (unpublished observations). We found that DS alone did not stimulate cytokine release (not shown), except of low IL-10 production, which was similar in WT and SR-A−/− PEM (∼ 45 pg/ml). In contrast, DS significantly increased LPS-stimulated TNF-α, RANTES and IL-6 production, but strongly inhibited LPS-stimulated production of IL-12/23p40 (Figure 1B). None of these DS effects was abolished by SR-A deficiency (Figure 1B). However, DS inhibited LPS-stimulated production of IL-12/23p40 less strongly in SR-A−/− than in WT PEM, which indicates that this DS effect is mediated, in part, by SR-A. Cytokine production was unaffected by CS (Figure 1B), consistent with SR involvement in DS effects. Similar effects on LPS-stimulated IL-12/23p40, TNF-α, IL-6 and RANTES production were exerted by DS in Balb/c PEM (Figure 2A and data not shown). In contrast, whereas DS enhanced LPS-stimulated IL-10 production in Balb/c PEM (Figure 2A), it inhibited IL-10 production in C57BL/6 PEM, less strongly in WT than in SR-A−/− cells (Figure 1B). These results indicate that at least two different SR, with different relative expression levels/activities in Balb/c and C57BL/6 PEM, mediate DS effects on IL-10 production: IL-10 production is enhanced by SR-A, but inhibited by another SR, which is more active in C57BL/6 PEM.
SR-A ligation with mAb inhibits LPS-stimulated IL-12/23p40 and IL-6 production, enhances production of IL-10, but does not modulate TNF-α release. PEM isolated from Balb/c (A), WT or SR-A-deficient (SR-A-/-) C57BL/6 mice (B) were treated with LPS or LPS plus DS and anti-SR-A (MαSR-A) or control mAb and cytokine concentrations were determined in culture supernatants by ELISA Graphs show results (means + SEM) of a representative experiment out of three such experiments performed. *Significant effect of MαSR-A relative to isotype-matched control rat IgG2b (P < 0.05 using Student’s t-test).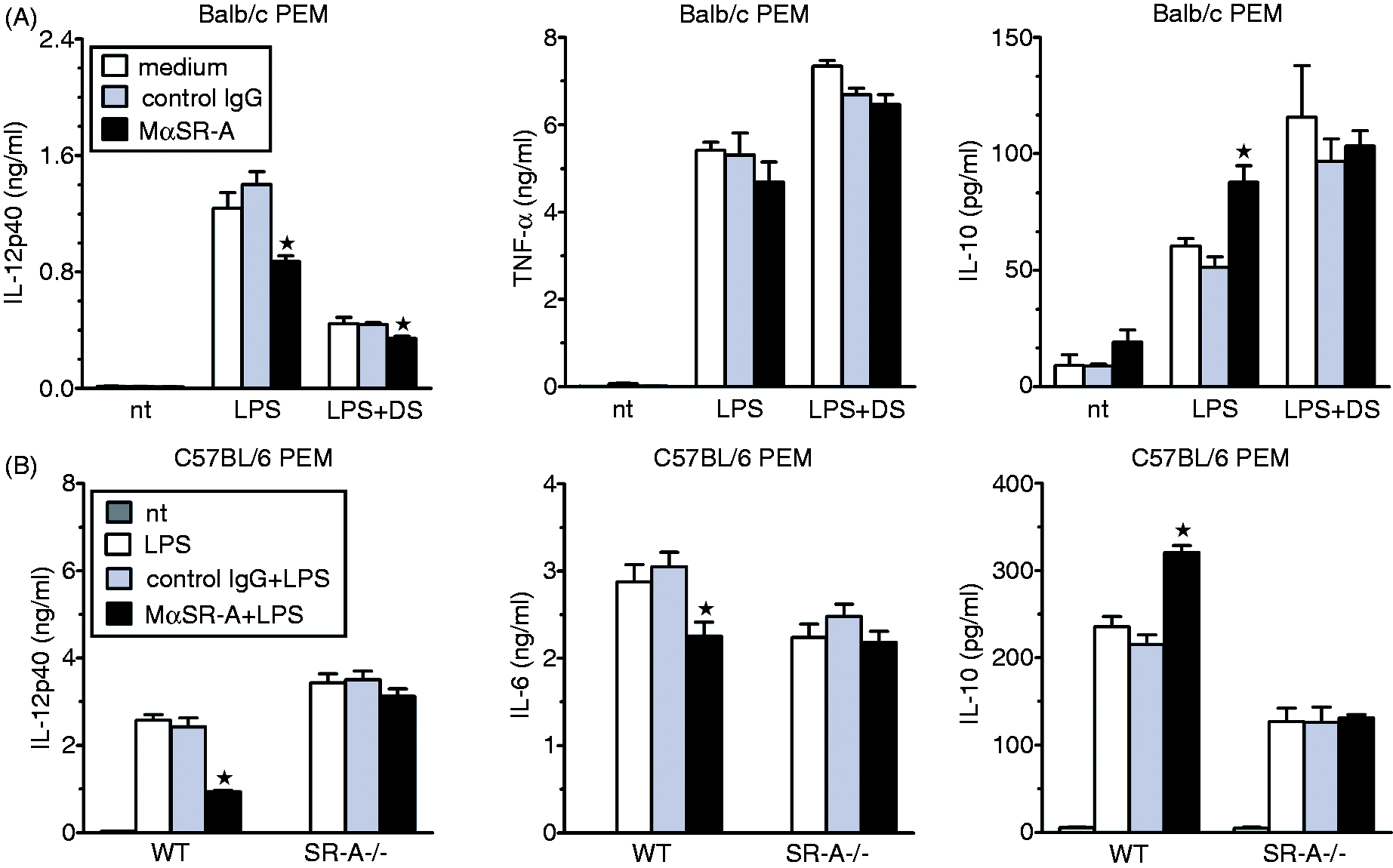
Among a variety of identified SR-A ligands, AcLDL seems to be among the most selective ones for this receptor,12,22,31 and it was used as a SR-A-selective agonist.30,32,33 We therefore examined the role of SR-A in AcLDL effects by comparing responses of WT and SR-A−/− PEM. By itself, AcLDL did not stimulate production of any cytokines (not shown), but inhibited LPS-stimulated production of IL-6 and IL-12/23p40, and had no effect on TNF-α and RANTES production (Figure 1B). However, AcLDL produced weaker suppression of IL-12/23p40 and IL-6 production also in SR-A−/− PEM. In contrast, AcLDL inhibited LPS-stimulated production of IL-10 in SR-A−/−, but not WT PEM (Figure 1B). These results indicate that also AcLDL effects in PEM are mediated by at least two different receptors: SR-A inhibits IL-12/23p40 and IL-6, but enhances IL-10 production, whereas an alternative receptor(s) inhibits production of all three cytokines. The observation that LPS-stimulated production of TNF-α and RANTES is enhanced by DS, but unaffected by AcLDL and CS, suggests involvement of at least one more SR, which is activated by DS, but not by AcLDL.
SR-A ligation with mAb enhances LPS-stimulated production of IL-10, but inhibits production of IL-6 and of IL-12/23p40 subunit
The results of the experiments comparing responses to AcLDL and DS of WT and SR-A−/− PEM suggest that, acting through SR-A, both ligands enhance LPS-stimulated IL-10, but inhibit IL-12/23p40 production. However, these ligands also stimulated qualitatively similar responses in SR-A−/− PEM, which makes them unsuitable for studies on SR-A signaling. In contrast, in our previous study we demonstrated that stimulation of H2O2 production by anti-SR-A 2F8 mAb (MαSR-A) in mouse AM was mediated exclusively by SR-A, as this response was absent in SR-A−/− AM. 21 We therefore assessed effects of specific SR-A ligation with MαSR-A on cytokine production. In Balb/c PEM MαSR-A alone had no effect, but inhibited LPS-stimulated production of IL-12/23p40 and enhanced IL-10 release (Figure 2A and data not shown). MαSR-A and DS had less than additive effects on the production of these cytokines (Figure 2A), consistent with the possibility that DS effects on IL-12/23p40 and IL-10 production are mediated, in part, by SR-A. In contrast, MαSR-A had no effect on LPS-stimulated TNF-α or RANTES production during both 4 h and 22 h treatment (Figure 2A and data not shown). In our previous study, MαSR-A did not modulate LPS plus IFN-γ-stimulated production of IL-6 in AM. 21 In contrast, in PEM the mAb significantly inhibited LPS-stimulated IL-6 production. However, on average from eight experiments, MαSR-A produced stronger suppression of IL-12/23p40 (49 ± 3.0%) than IL-6 (28 ± 4.5%) production (P = 0.008 in paired t-test). It should be noted that, in comparison with Balb/c PEM, in AM isolated from these mice MαSR-A produced much weaker, only ∼ 25% inhibition of IL-12, production, likely as the result of a much lower level of SR-A expression, 21 so an even weaker effect of MαSR-A on IL-6 production in AM might not reach statistical significance. Alternatively, SR-A may be able to regulate IL-6 production in response to stimulation by LPS, but not by LPS plus IFN-γ.
To verify SR-A-specificity of MαSR-A we compared its effects in WT and SR-A−/− C57BL/6 PEM. It has been reported that MαSR-A does not bind to an allelic isoform of SR-A expressed in C57BL/6 mice. 34 However, subsequently, several other groups observed labeling of cells derived from C57BL/6 mice by this mAb. 35 We therefore performed saturation binding experiments and found that MαSR-A bound to C57BL/6 and Balb/c PEM with similar affinities, with Kd values of 0.62 ± 0.094 and 0.87 ± 0.034 µg/ml, respectively (Supplementary Figure S1B). Values of maximal binding were also similar in both cases, which indicates that Balb/c and C57BL/6 PEM express similar levels of SR-A. However, MαSR-A also bound to SR-A−/− PEM, although with ∼ 10 times lower affinity (Kd = 5.7 ± 0.95 µg/ml) and with lower values of maximal binding. Previously, Becker et al. 36 reported that epitopes cross-reacting with MαSR-A are also present on the surface of SR-A-deficient DC. Nevertheless, MαSR-A modulated production of IL-10, IL-12/23p40 and IL-6 in WT, but not SR-A−/− PEM (Figure 2B), which confirms the exclusive role of SR-A in these effects.
We reported previously that MαSR-A stimulated H2O2 production and regulated IL-12p70 release in Balb/c, but not C57BL/6 AM. 21 Attempting to explain this discrepancy, we assessed SR-A expression on AM and found that SR-A was weakly expressed on the surface of Balb/c, but not C57BL/6 AM (Supplementary Figure S1C, D). Thus, the lack of MαSR-A effects in C57BL/6 AM seemed to result from the lack of SR-A expression rather than from the inability of MαSR-A to bind to an allelic isoform of SR-A expressed in C57BL/6 mice.
Effects of SR-A ligation are reversed by pertussis toxin and wortmannin
We reported previously that pertussis toxin, the selective inhibitor of heterotrimeric Gi/o proteins, inhibited SR-A-stimulated H2O2 production in AM,
21
whereas Post et al.
37
reported that pertussis toxin inhibited SR-A-mediated adhesion and AcLDL uptake in transfected cells. We found that pertussis toxin also abolished modulating effects of MαSR-A on cytokine production (Figure 3). One of the major modes of action of heterotrimeric Gi/o proteins is inhibition of adenyl cyclase by their Gα subunits. However, MANT-GppNHp—an inhibitor of adenyl cyclase—did not mimic effects of MαSR-A (Figure 3), which suggests that MαSR-A effects are not due to inhibition of adenyl cyclase. Moreover, in the presence of MANT-GppNHp MαSR-A preserved the ability to modulate cytokine release, which indicates that also activation of adenyl cyclase is not responsible for the MαSR-A effects (Figure 3).
Modulating effects of SR-A ligation with anti-SR-A mAb (MαSR-A) on LPS-stimulated cytokine production are mediated by pertussis toxin (PTX)-sensitive heterotrimeric Gi/o proteins and phosphatiylinositol 3-kinases (PI3K), but do not involve inhibition or stimulation of adenylate cyclase. Balb/c PEM were pre-incubated for 2 h with PTX or for 40 min with wortmannin (Wortman), MANT–GppNHp (MANT-G) or equivalent amount of solvent (DMSO), and then treated with LPS and MαSR-A or control rat IgG2b in the continuous presence of inhibitors. Following 22 h of treatment, cytokine concentrations in culture supernatants were determined by ELISA. Graphs show results (means + SEM) of a representative experiment out of three such experiments performed. *Significant effect of MαSR-A relative to control rat IgG2b.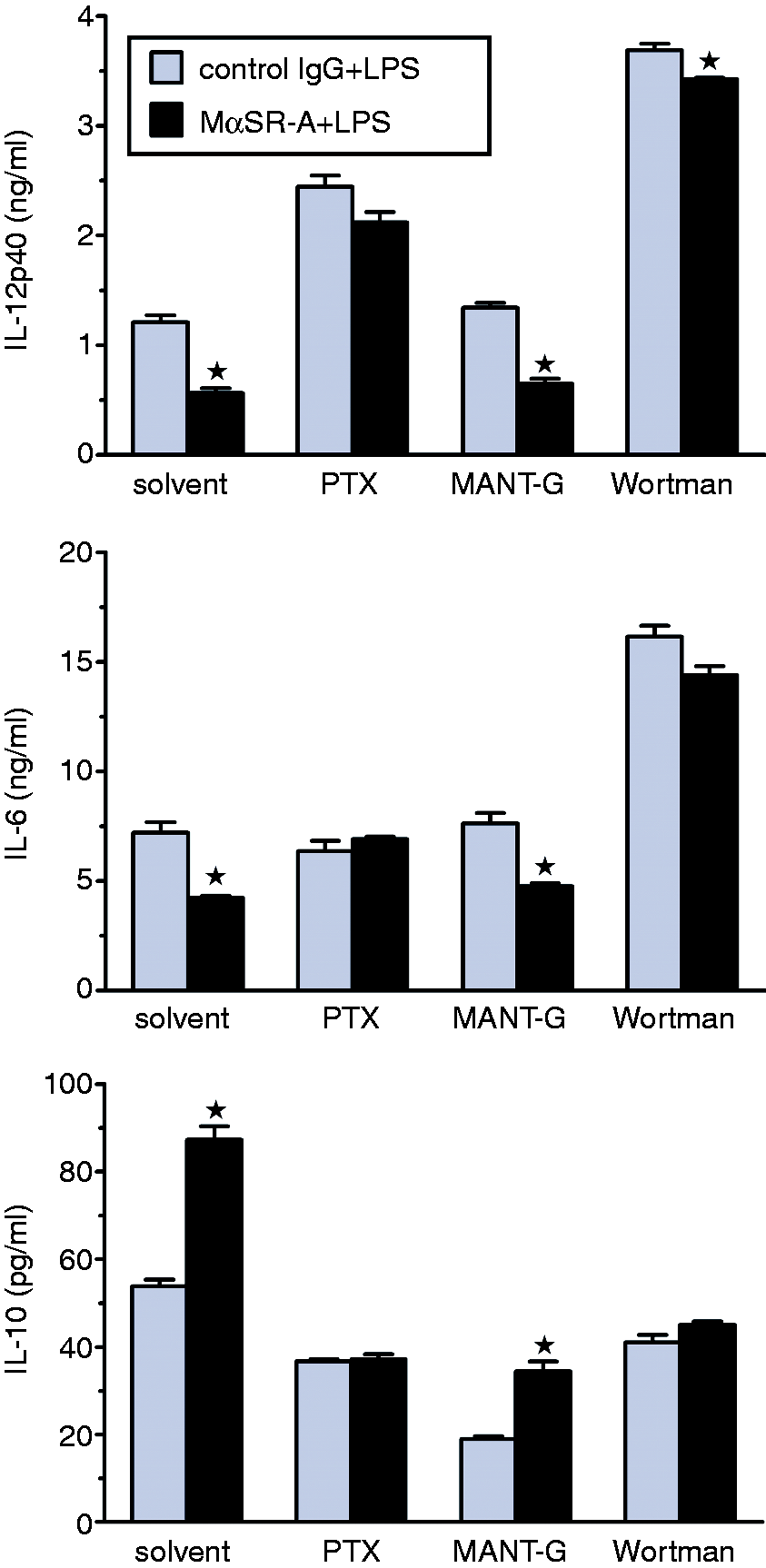
It has been reported that PI3K mediate the suppressive effect of heterotrimeric Gi proteins on IL-12 production in human monocytes. 38 We therefore assessed the effects of wortmannin—a selective inhibitor of these enzymes. In the presence of wortmannin, MαSR-A lost its ability to modulate cytokine release (Figure 3), which indicates that activation of PI3K is an element of SR-A signaling responsible for the modulation of cytokine release.
Responses of macrophages to CD36 ligation
We next examined the effects of CD36 ligation on the production of inflammatory cytokines by PEM. For this purpose we used function-blocking CRF D-2712 anti-CD36 mAb (MαCD36) which has been demonstrated recently to stimulate production of TNF-α and RANTES in PEM.
39
The CD36 specificity of the observed effects of MαCD36 in that study has been proven by their lack in CD36-deficient PEM. In striking contrast to ineffectiveness of MαSR-A in this regard, MαCD36 alone stimulated in PEM production of all cytokines tested (Figure 4). The most prominent feature of MαCD36 effects was stimulation of very high production of IL-10, which was several fold higher than that stimulated by TLR agonists (Figure 4C).
Effects of anti-CD36 mAb (MαCD36) alone and in combinations with TLP, LTA or LPS on the production of TNF-α (A), RANTES (B), IL-10 (C), IL-6 (D) and IL-12/23p40 (E) in Balb/c PEM, and the role of autocrine/paracrine IL-10 in these MαCD36 effects. PEM were co-treated for 4 or 22 h with MαCD36 or control IgA and LTA, TLP or LPS, and cytokine concentrations in culture supernatants were determined by ELISA. Where indicated, neutralizing anti-IL-10 or control mAb were additionally included in the medium. Graphs show results (means + SEM) of single experiments, each representative of 3–5 similar experiments performed. *Significant effect of MαCD36 relative to control mouse IgA; # significant effect of neutralizing anti-IL-10 mAb.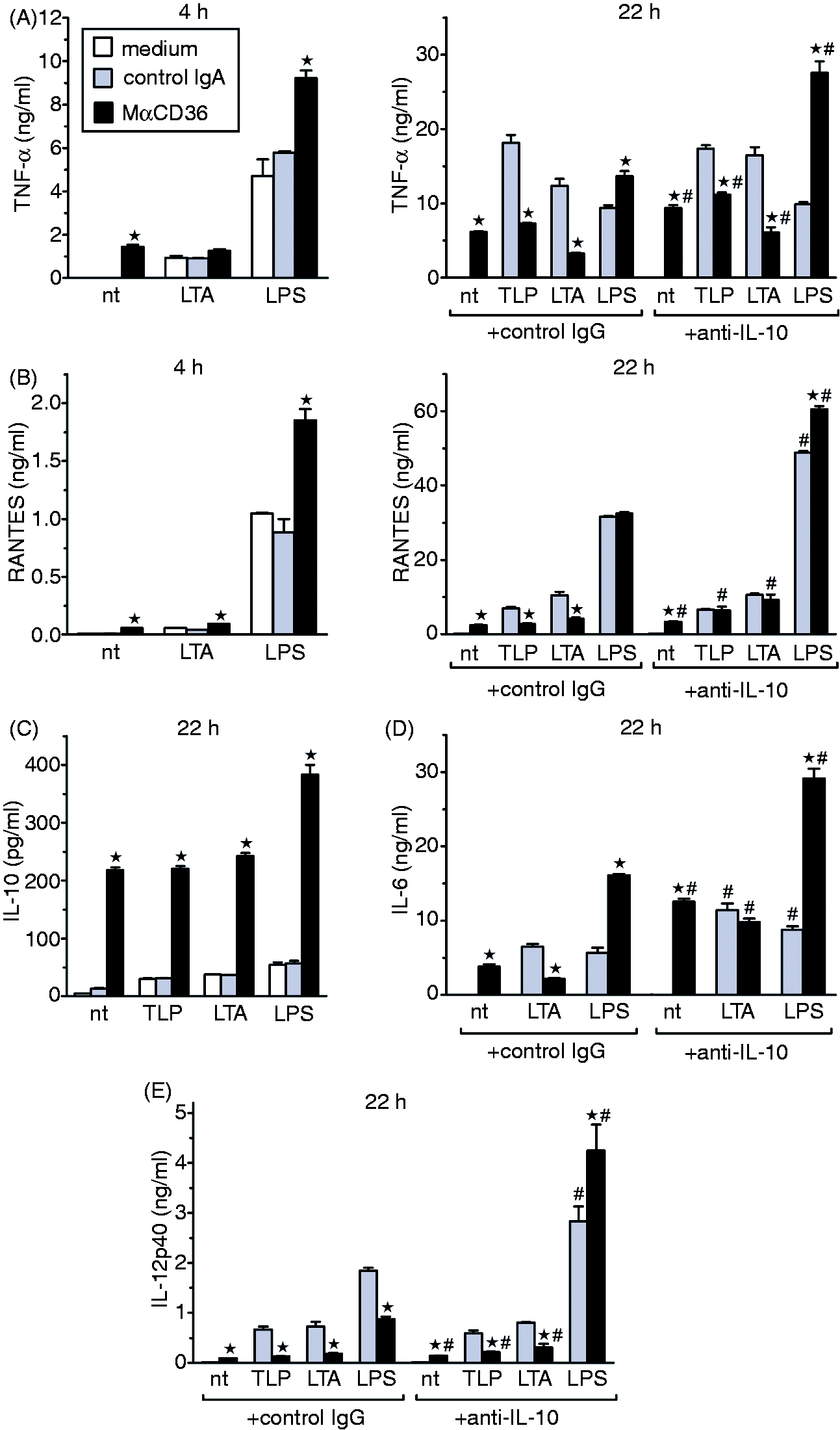
CD36 has been reported to cooperate with TLR2 in generating macrophage responses to some TLR2 ligands. 40 We therefore assessed effects of MαCD36 on macrophage responses to LTA, a ligand of both TLR2/TLR6 and CD36, to TLP, which is a ligand of TLR2/TLR1, but not of CD36, 27 as well as to LPS. MαCD36 and LTA stimulated similar production of TNF-α (Figure 4A) and RANTES (Figure 4B) in PEM during 4 h incubation, as well as of IL-6 during 22-h incubation (Figure 4D). Effects of the combination of MαCD36 and LTA or TLP on cytokine production were less than additive, whereas those of MαCD36 and LPS were additive or even synergistic (Figure 4A–D). In contrast, in PEM treated for 22 h with TLR2 agonists, co-treatment with MαCD36 inhibited production of pro-inflammatory cytokines (Figure 4A, B, D, E). The combined effect of 22-h co-treatment with LPS and MαCD36 was cytokine-specific: MαCD36 enhanced LPS-stimulated production of TNF-α (Figure 4A) and IL-6 (Figure 4D), had no effect on RANTES (Figure 4B) and inhibited IL-12/23p40 (Figure 4E) production.
Of note, MαCD36 and TLR2 agonists also share similar kinetics of cytokine induction, which was delayed relative to that induced by LPS (Figure 4A, B). Taken together, the lack of additivity in MαCD36 and TLR2 agonists effects on cytokine production, as well as similar magnitude and kinetics of production, are consistent with the possibility that effects of all three ligands are mediated by shared receptors, distinct from those mediating effects of LPS.
IL-10 mediates negative feedback regulation of MαCD36-stimulated cytokine production, but not effects of MαSR-A on IL-6 or IL-12/23p40 release
IL-10 is known as a potent negative regulator of pro-inflammatory cytokine production. As MαCD36 stimulated high IL-10 production with delayed kinetics (no IL-10 could be detected in cultures of PEM treated for 4 h) we hypothesized that negative feedback regulation by autocrine/paracrine IL-10 might be responsible for differences in MαCD36 effects in PEM treated for 4 h versus 22 h. We tested this possibility by the inclusion of neutralizing anti-IL-10 mAb in the culture medium. In the case of LPS and MαCD36 co-treatment, the inclusion of anti-IL-10 mAb potentiated the enhancing effect of MαCD36 on LPS-stimulated production of TNF-α (Figure 4A), RANTES (Figure 4B) and IL-6 (Figure 4D), and reversed the effect of MαCD36 on IL-12/23p40 production from ∼ 50% inhibition to ∼ 50% enhancement (Figure 4E). Importantly, IL-10 neutralization fully reversed the inhibitory effect of MαCD36 on TLP- or LTA-stimulated production of RANTES (Figure 4B) and IL-6 (Figure 4D), and partially on TNF-α (Figure 4A) and IL-12/23p40 (Figure 4E) production. Under these conditions the combination of MαCD36 and a TLR2 agonist stimulated similar cytokine production as MαCD36 alone, which may be explained by higher affinity of MαCD36 for the shared receptor system.
In contrast, although MαSR-A also enhances IL-10 production, neutralization of IL-10 did not reverse, to any extent, the inhibitory effect of MαSR-A on LPS-stimulated IL-12/23p40 or IL-6 production (Supplementary Figure S2), which suggests that these effects of MαSR-A are direct, rather than IL-10-mediated.
Inhibitors of MAPK and STK have similar effects on PEM responses to agonists of TLR and to MαCD36, but do not affect responses to MαSR-A
Effects of pharmacological inhibitors on 22-h cytokine production, stimulated in PEM by MαCD36, LPS or TLP.
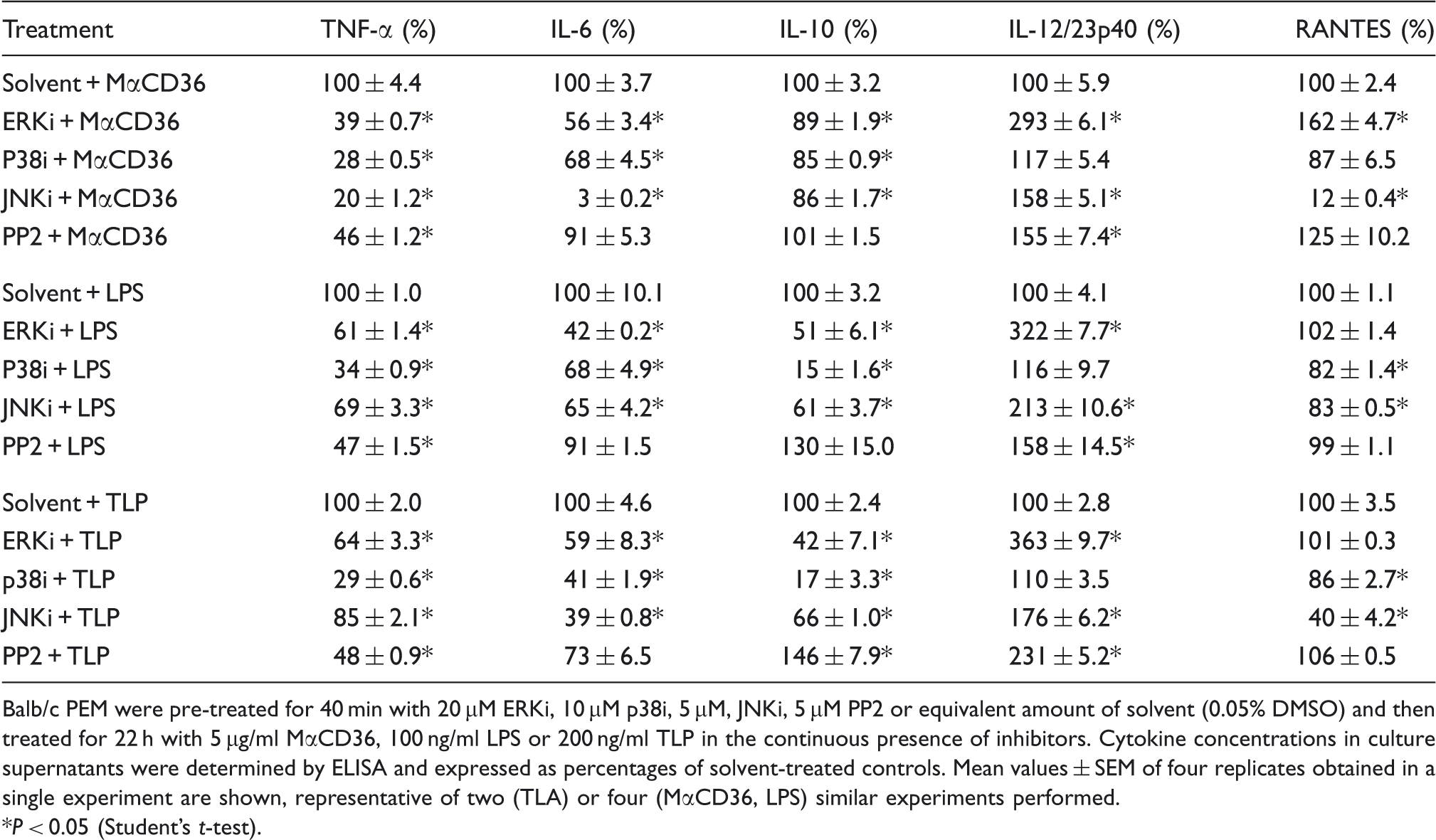
Balb/c PEM were pre-treated for 40 min with 20 μM ERKi, 10 μM p38i, 5 μM, JNKi, 5 μM PP2 or equivalent amount of solvent (0.05% DMSO) and then treated for 22 h with 5 µg/ml MαCD36, 100 ng/ml LPS or 200 ng/ml TLP in the continuous presence of inhibitors. Cytokine concentrations in culture supernatants were determined by ELISA and expressed as percentages of solvent-treated controls. Mean values ± SEM of four replicates obtained in a single experiment are shown, representative of two (TLA) or four (MαCD36, LPS) similar experiments performed.
P < 0.05 (Student’s t-test).
Interestingly, macrophage responses to MαCD36 and to TLR agonists shared similar susceptibilities to MAPK inhibitors (Table 1), being indicative of similarities in signal transduction pathways. There were, however, two major differences. MAPK inhibitors inhibited strongly IL-10 production stimulated by TLP or LPS, but had little effect on this macrophage response to MαCD36. Also, JNKi produced much stronger inhibition of cytokine production in response to MαCD36 than to TLR agonists.
In contrast to their modulating effects on CD36 signaling, none of MAPK inhibitors affected responses to SR-A ligation, also at two-times higher concentrations (not shown). MAPK inhibitors did not impair cell viability, as assessed by MTT reduction (not shown).
It has been reported that STK Lyn co-precipitated with SR-A and was phosphorylated in response to AcLDL. 42 STK were also shown to associate with CD36 and their activation was reported to be the crucial step in the initiation of intracellular signaling by the C-terminal cytoplasmic domain of CD36.8,43 We therefore assessed the role of STK in SR-A and CD36 signaling with the use of PP2, a selective inhibitor of these enzymes. Like MAPK inhibitors, PP2 also had similar effects on cytokine production stimulated by MαCD36 and by TLR agonists. It had little or no effect on IL-6, IL-10 or RANTES production, enhanced IL-12/23p40 release, but inhibited TNF-α production by ∼ 50% in response to all three stimuli (Table 1). Thus, effects of PP2 do not support the major role of STK in intracellular signaling linking CD36 ligation with cytokine production. Also, PP2 did not antagonize to any extent the effects of SR-A ligation on LPS-stimulated IL-10 and IL-12/23p40 production (data not shown).
Effects of MαCD36 are mediated by TLR2 and CD14, but not by TLR4
The results so far obtained provide several pieces of indirect evidence that effects of MαCD36 and TLR2 agonists are mediated by shared receptors. We therefore directly assessed the role of TLR2 in MαCD36-induced signaling with the use of blocking T2.5 mAb. In Balb/c PEM anti-TLR2 mAb inhibited not only responses to TLP and LTA, but also blocked TNF-α, IL-12/23p40 and RANTES production, and partially inhibited IL-10 production in response to MαCD36 (Figure 5A and data not shown). Anti-TLR2 mAb had no effect on TNF-α or IL-12/23p40 production stimulated by LPS, which confirms its specificity. These results indicate that the production of pro-inflammatory cytokines in response to MαCD36 is mediated entirely by TLR2, whereas CD36 signaling leading to IL-10 production involves mainly TLR2-independent pathways. These TLR2-independent pathways of CD36 signaling do not seem to involve activation of MAPK, PI3K or STK, as indicated by the lack of effects of selective inhibitors of these enzymes (Figure 5C).
Stimulation of pro-inflammatory cytokine production by anti-CD36 mAb (MαCD36) is mediated by TLR2 and CD14. Balb/c PEM were pre-incubated with indicated pharmacological inhibitors (C) anti-TLR2 (A, C), anti-CD14 (B) or isotype-matched control mAb, and then stimulated with MαCD36 or TLR agonists. Cytokine concentrations in culture supernatants were determined by ELISA. Graphs show results (means + SEM) of representative experiments out of three (A, B) or two (C) such experiments performed.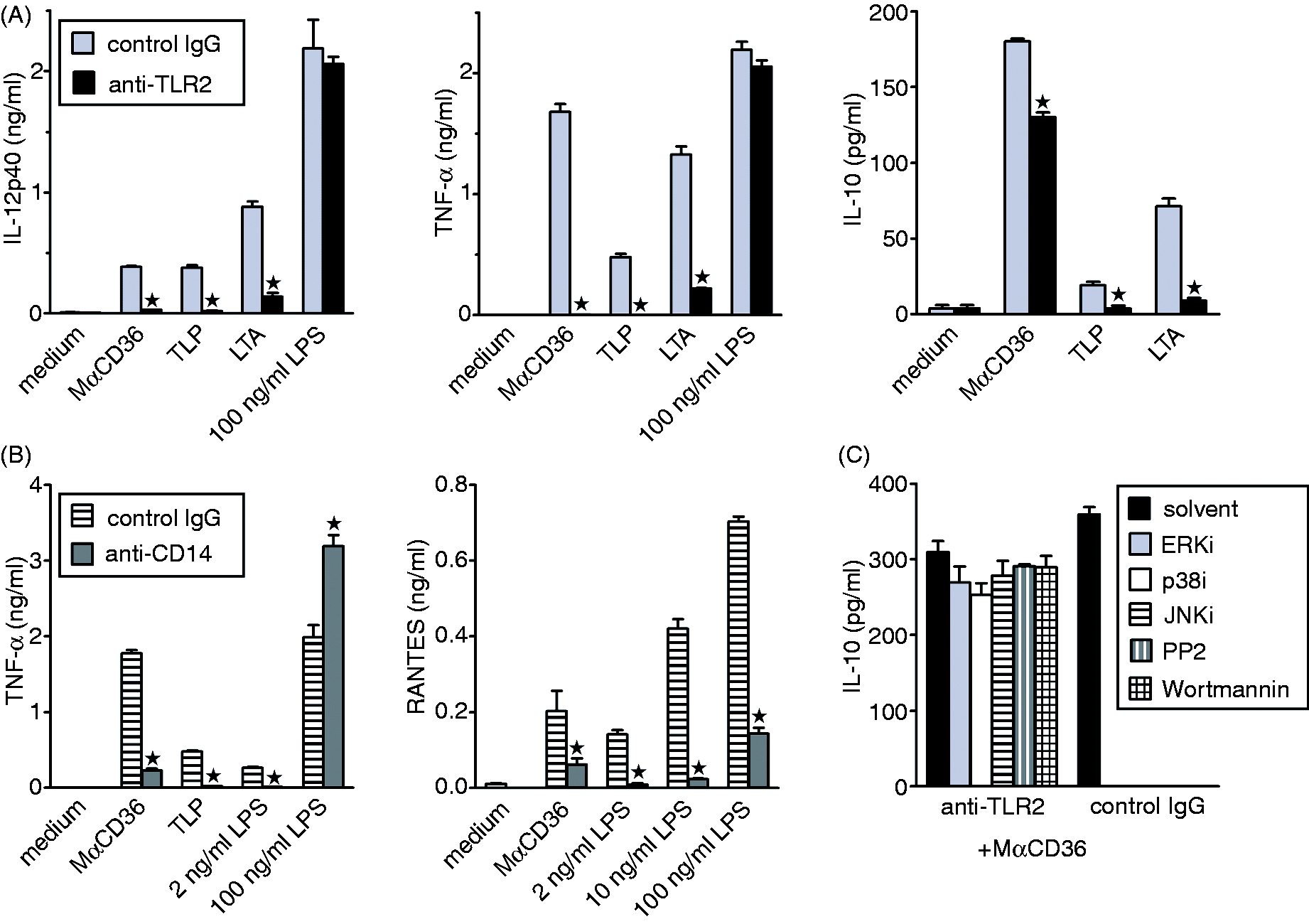
In addition to TLR2, CD14 has also been reported to participate in macrophage responses to LTA and to some lipopeptides, including TLP. 44 In Balb/c PEM, function-blocking anti-CD14 mAb inhibited TNF-α and RANTES production stimulated by both TLP and MαCD36 (Figure 5B), but had no effect on MαCD36-stimulated IL-10 production (data not shown). Anti-CD14 mAb also blocked TNF-α production in response to low (up to 10 ng/ml) LPS concentrations, but strongly enhanced TNF-α production stimulated by 100 ng/ml LPS. In contrast, the mAb inhibited production of RANTES stimulated by the whole range of LPS concentrations (Figure 5B). These effects of anti-CD14 mAb are consistent with the reported differential involvement of membrane CD14 in the activation of TRIF- versus MyD88-dependent pathways of TLR4 signaling, responsible for RANTES and TNF-α production, respectively. 45
The abovementioned results suggest that propagation of intracellular signaling triggered by MαCD36 depends on CD36 association with TLR2 and CD14. In contrast, Stewart et al. 46 reported that macrophage responses to other CD36 ligands—β-amyloid fibrils and oxidized LDL—were not mediated by TLR2, but by the newly described heterodimer of TLR4 and TLR6. We therefore examined the role of TLR4 in CD36 signaling with the use of PEM isolated from LPS non-responsive C3H/HeJ mice, which express TLR4 unable to trigger intracellular signaling owing to a point mutation in its cytoplasmic domain. As might be expected, C3H/HeJ PEM did not produce any cytokines in response to LPS, even at a concentration as high as 20 µg/ml (Supplementary Figure S3). In contrast, responses to LTA, TLP and MαCD36 were preserved in C3H/HeJ PEM, indicating that the TLR4/TLR6 heterodimer does not mediate responses to CD36 ligands LTA and MαCD36.
CD36, but not SR-A, mediates LPS clearance from the culture medium
It has been suggested in two recent reports that SR-A is devoid of signaling ability and that the observed enhancing effects in these studies of SR-A deficiency, AcLDL or MαSR-A on cytokine production, stimulated in macrophages by ligands of TLR4, are caused by impaired clearance of these ligands from the culture medium, leading to their higher concentrations in the proximity of TLR4.33,47 Our results are inconsistent which such a mechanism of MαSR-A effects. For instance, if MαSR-A acts by blocking LPS uptake and therefore increasing LPS concentration in the proximity of TLR4, it should affect production of all LPS-stimulated cytokines, but this was not the case. Nevertheless, we addressed the effect of MαSR-A on LPS clearance from the culture medium directly. MαSR-A had no effect on LPS concentration in the supernatant of PEM during 40 min or 2 h co-incubation (Figure 6A). Surprisingly, MαCD36 partially prevented LPS clearance by PEM, whereas the effect of anti-CD14 mAb did not reach statistical significance. The majority of LPS clearance occurred during the first 40 min of incubation, and increasing its duration to 2 h had only a small additional effect. Next, we examined the ability of SR-A to bind LPS with the use of CHO cells transfected with human SR-A. AF488-LPS turned out to be a very low-affinity ligand of SR-A, as concentrations of AF488-LPS as high as 10 µg/ml were required to observe increased binding to SR-A-transfected CHO cells (Figure 6B).
LPS uptake in PEM is mediated by CD14 cooperating with CD36, but not by SR-A. (A) Following pre-incubation with the indicated mAb, 100 ng/ml LPS was added for 40 min or 2 h incubation, and LPS concentrations in the culture medium were determined with the Endpoint Chromogenic LAL Assay. (B) CHO cells, transfected or not with human SR-A, were incubated for 1.5 h with indicated concentrations of Alexa Fluor 488-labeled LPS (AF488–LPS) and fluorescence of cell-associated AF488–LPS was measured with the use of a plate reader. (C) Following pre-incubation with the indicated mAb, PEM were incubated with 5 µg/ml BODIPY-labeled LPS for 40 min or 2 h, detached and geometric mean fluorescence intensities (MFI) of cell-associated BODIPY-LPS were determined by flow cytometry. (D) WT and SR-A-/- PEM were incubated for 1 h on ice with 0.1, 1 or 10 µg/ml biotinylated LPS (bLPS), washed and bLPS bound to surfaces of PEM was detected with streptavidin–HRP conjugate. (E) PEM were pre-incubated with the indicated mAb, and the binding of 100 ng/ml bLPS to PEM was determined as described above. Graphs show mean + SEM from either three independent experiments (B) or single experiments, each reproduced in another similar experiments (A, C–E). Asterisks indicate significant effect (P < 0.05) according to the Student’s t-test.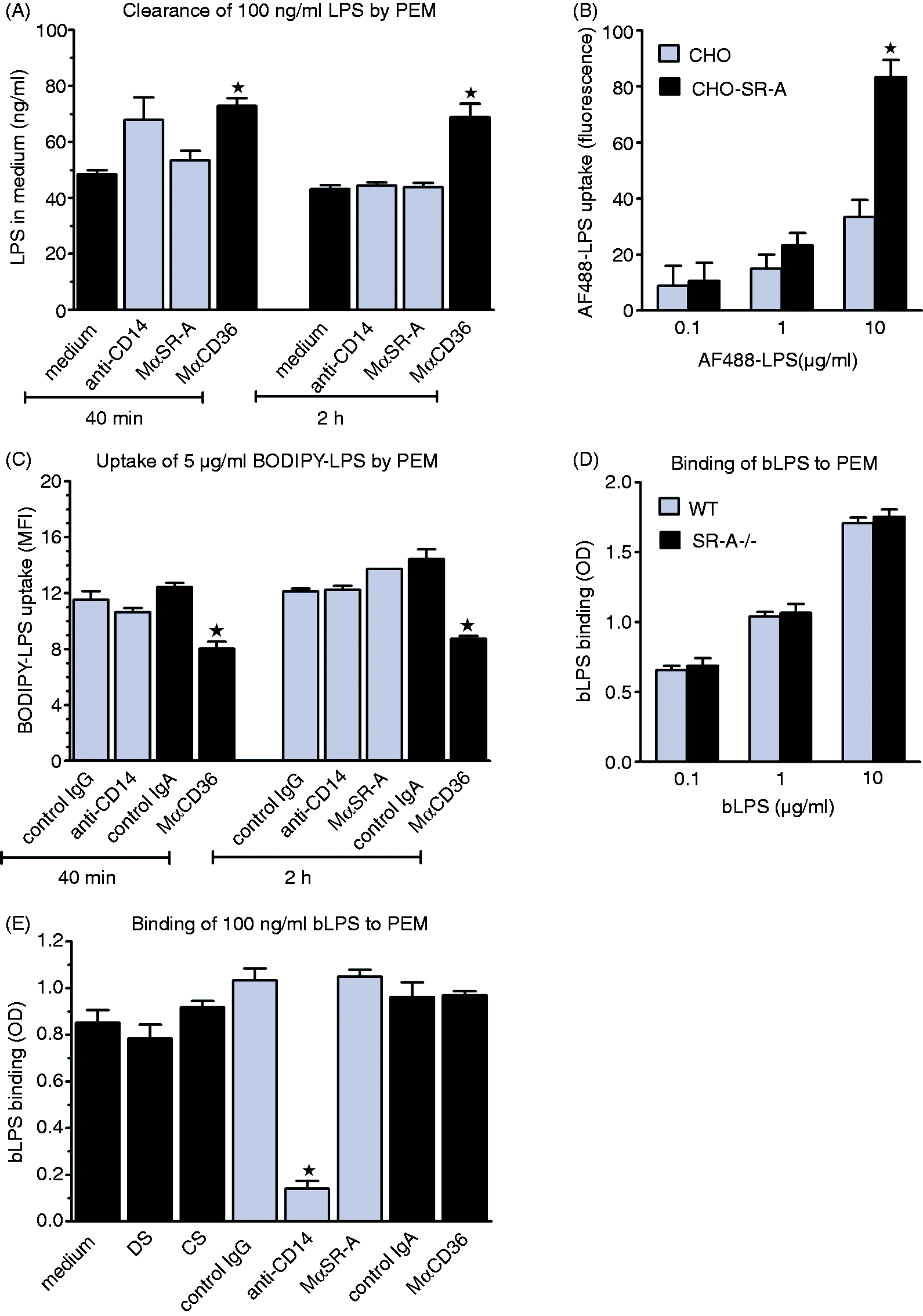
Effects of mAb on BODIPY-LPS uptake by PEM (Figure 6C) mirrored their effects on LPS clearance from the culture medium (Figure 6A). Also in this case, MαSR-A had no effect on 0.1, 1 or 5 µg/ml BODIPY-LPS uptake by PEM (Figure 6C and data not shown). However, MαSR-A also did not inhibit uptake by PEM of 10 µg/ml BODIPY-LPS (data not shown), the concentration at which LPS bound to human SR-A expressed in transfected CHO cells. Also, WT and SR-A−/− PEM exhibited indistinguishable binding of 10 µg/ml biotinylated LPS (Figure 6D). Thus, even the involvement of SR-A in binding to PEM of this high concentration of LPS seems overshadowed by other receptors. Alternatively, this discrepancy might result from inter-species differences in the ability to bind LPS between murine and human SR-A. 48 Like uptake of unlabeled LPS (Figure 6A), uptake of BODIPY-LPS by PEM was inhibited by MαCD36, but not by anti-CD14 mAb (Figure 6C).
The major difference between our study and that of Ohnishi et al., 33 who suggested (but did not examine) involvement of SR-A in LPS clearance, is the use of serum-free medium in the latter study. However, in the absence of serum, LPS clearance by PEM was impaired and also not affected by MαSR-A (Supplementary Figure S4). Taken together, the above results confirm that MαSR-A does not exert its effects on LPS-stimulated cytokine production by inhibiting LPS clearance.
Interestingly, whereas LPS uptake was inhibited by MαCD36, but not by anti-CD14 mAb, LPS binding to PEM was inhibited almost completely, by ∼ 86%, by anti-CD14 mAb, but unaffected by MαCD36, MαSR-A or DS, a ligand of both SR-A and CD36 (Figure 6E). We suspect that this discrepancy between effects of anti-CD14 mAb on LPS binding (blockade) versus uptake (no effect) may be related to the fact that incubation at 37℃ in the uptake assay, but not on ice in the binding assay, allows recycling of CD14 between the plasma membrane and intracellular compartments. This process may lead to the appearance on the macrophage surface of CD14 molecules not occupied by anti-CD14 mAb and therefore capable of LPS binding as the result of dissociation of anti-CD14 mAb from CD14 in the acidic environment of early endosomes 49 and redistribution of intracellular CD14 onto the cell surface.50,51 Consistent with this interpretation, when the LPS binding assay was performed at 37℃ instead of on ice, anti-CD14 mAb inhibited binding of 100 ng/ml biotinylated LPS by ∼ 17% only (data not shown).
MαCD36-stimulated production of pro-inflammatory cytokines, but not of IL-10, requires dynamin-dependent internalization of receptors
It has been reported that propagation of intracellular signaling from CD36 depends on endocytosis. Stuart et al.
3
reported that LTA-induced, CD36-dependent activation of NF-κB was abrogated by cytochalasin D, an inhibitor of several different endocytic pathways.
52
In contrast, Stewart et al.
46
reported that initiation of TLR4/TLR6-dependent CD36 signaling required dynamin-mediated endocytosis and was blocked by dynasore. In our study, pretreatment of PEM with dynasore blocked LPS-stimulated RANTES, but had no effect on TNF-α production (Figure 7), which confirmed the results of Kagan et al.,
53
who demonstrated that the TRIF-dependent RANTES production, but not MyD88-dependent TNF-α production, requires dynamin-mediated internalization of TLR4. Dynasore also inhibited LPS-stimulated production of IL-10 and IL-12/23p40, confirming its dependence on the TRIF pathway (Figure 7). In contrast, dynasore inhibited production of all pro-inflammatory mediators, including TNF-α, in response to MαCD36 and TLR2 agonists, but had no effect on IL-10 production stimulated by these ligands (Figure 7). Cytochalasin D had little or no effect on TNF-α, RANTES or IL-12/23p40 production when stimulated by MαCD36 or TLR2 ligands, inhibited to a different extent production of these mediators in response to LPS and strongly enhanced IL-10 production in response to all factors tested (Figure 7). Thus, initiation of the TLR2-dependent CD36 signaling, responsible for the production of pro-inflammatory cytokines, seems to require dynamin-dependent internalization of receptors, whereas the TLR2-independent signaling, responsible for high IL-10 production, may be activated by cell surface-localized CD36 and suppressed by processes involving actin polymerization.
Effects of endocytosis inhibitors, cytochalasin D and dynasore, on MαCD36-, LTA-, TLP- or LPS-stimulated cytokine production. Graphs show results (mean + SEM) obtained in a single experiment, repeated three times with similar results. *Significant effect versus the solvent control.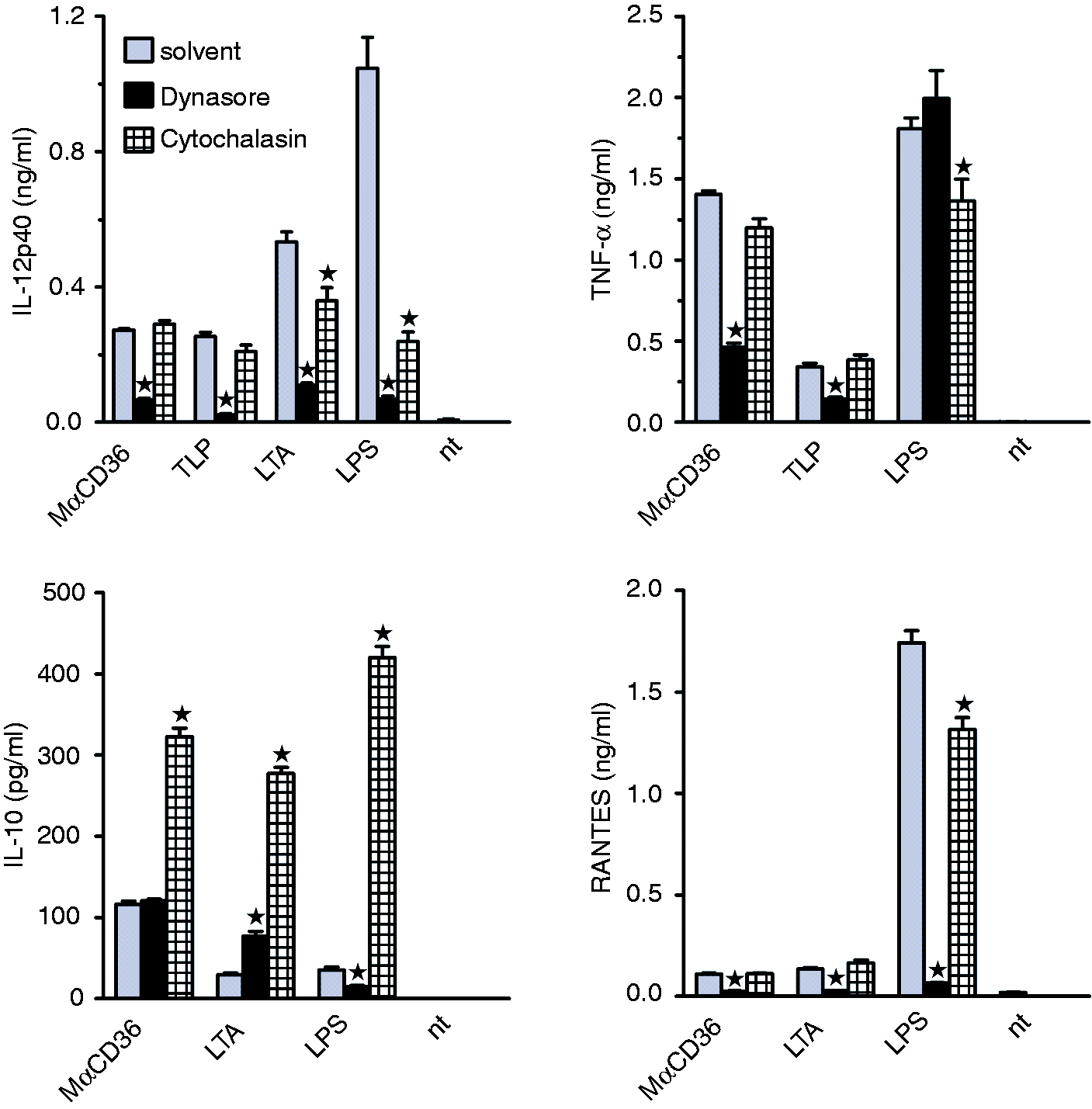
CD36 or SR-A ligation on PEM regulates differentiation of naïve Th cells
Of note, both MαCD36 and MαSR-A regulated production of cytokines known to play crucial roles in regulating differentiation of Th lymphocytes: p40 subunit of IL-12 and IL-23, IL-6 and IL-10. We therefore assessed effects of CD36 or SR-A ligation with mAb on the ability of PEM to drive differentiation of Th lymphocytes during Ag presentation in the mix culture of PEM and CD4+ lymphocytes isolated from spleens of OT-II mice. Lymphocytes of OT-II mice express transgenic TCR, which recognizes one of the OVA epitopes (OVA323-339) in the context of MHC class II (I-Ab) molecules. It is well-documented that Ag taken up through CD36 or SR-A are processed and effectively presented to lymphocytes.54,55 As we were interested in immunoregulatory effects of SR-A- or CD36-induced signaling, but not in the role of these receptors in Ag uptake and processing, we directly loaded MHC class II molecules on the surface of PEM by incubation with OVA323-339 and studied the effects of receptor ligation on the ability of PEM to regulate differentiation of CD4+ lymphocytes. We found that even OVA323-339 alone stimulated high-level IFN-γ production during 2 days of co-culture of PEM with naïve CD4+ lymphocytes (Figure 8A). Pre-incubation of PEM with MαCD36, but not with isotype-matched control IgA, produced ∼ 2.3-fold enhancement of IFN-γ production, comparable to that induced by LPS. In contrast, MαSR-A alone had no effect, but inhibited by ∼ 35% the stimulatory effect of LPS on IFN-γ production. Both MαCD36 and LPS also enhanced IL-10 production, and the effect of LPS on IL-10 production was increased significantly by MαSR-A (Figure 8A).
Effects of CD36 or SR-A ligation on IFN-γ and IL-10 production in the co-culture of naïve OT-II CD4+ lymphocytes and PEM presenting OVA323-339 peptide. (A) Following 2 h pre-incubation with mAb, OVA323-339 and LPS, PEM were washed and OT-II CD4+ splenocytes were added for 2 d co-culture. IFN-γ and IL-10 concentrations in culture supernatants were determined by ELISA. (B–D) The cells were cultured as described in (A) except that mAb, OVA323-339 and LPS were not removed after pre-incubation, but were present during the entire period of co-culture. In (D) neutralizing anti-IL-10 or control mAb were additionally included in the culture medium. Graphs show averages of indicated numbers (N) of independent experiments, each performed in four replicates (A, B) or results of single experiments, both representative of three similar experiments performed (C, D). *Significant effect of MαSR-A or MαCD36 relative to isotype-matched control mAb (P < 0.05 using Student’s t-test).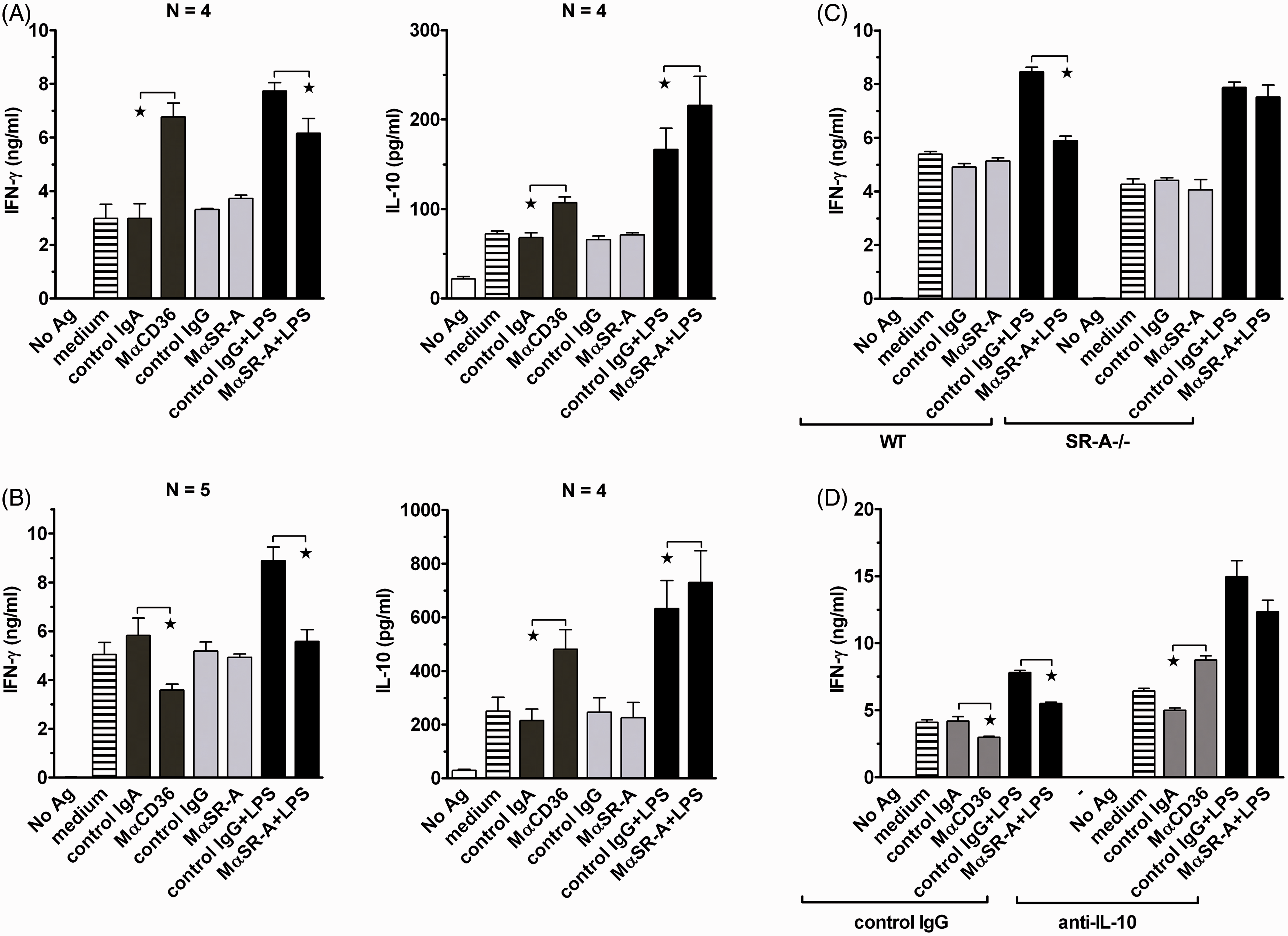
We speculated that a rather weak effect of MαSR-A on IFN-γ production might be related to the fact that the mAb was present during the 2 h pre-incubation only. Indeed, when MαSR-A was not removed following pre-incubation, but was present during the entire period of co-culture, it produced the complete reversal of the LPS effect on IFN-γ production (Figure 8B). This effect of MαSR-A was SR-A-specific, as it was not observed in the co-culture with SR-A−/− PEM (Figure 8C). Interestingly, under conditions of co-incubation, MαCD36 inhibited IFN-γ production (Figure 8B). We suspected that the opposite effects exerted by MαCD36 in experiments with pre-incubation versus co-incubation may be caused by differences in levels of IL-10 production under these conditions. In fact, MαCD36 stimulated ∼ 4.5-fold higher IL-10 production during co-incubation (Figure 8B) as compared to its presence during pre-incubation only (Figure 8A). Inclusion of neutralizing anti-IL-10 mAb in the culture medium under conditions of continuous presence of mAb caused the reversal of the effect of MαCD36 on IFN-γ production from inhibition to stimulation (Figure 8D), which confirms our hypothesis. Neutralization of IL-10 also enhanced the stimulatory effect of LPS on IFN-γ production and partially reversed the suppressive effect of MαSR-A on this production (Figure 8D).
No production of IL-4 higher than few pg/ml could be detected in the co-culture of PEM with CD4+ lymphocytes under any conditions, consistent with known Th1 skewing of immune responses in the C57BL/6 strain of mice.
CD36, but not SR-A, stimulation regulates expression of MHC class II and co-stimulatory molecules on the surface of PEM
In addition to regulating cytokine production, it was also possible that ligation of CD36 or SR-A might affect differentiation of Th lymphocytes by regulating expression of MHC class II or co-stimulatory molecules on the surface of PEM. As might be expected, 1-day co-incubation of PEM with LPS up-regulated strongly expression of CD40, CD86 and MHC class II molecules on these cells (Figure 9). MαCD36 produced increases of MHC class II and CD40 levels about twice smaller than LPS, but did not induce expression of CD86. Surprisingly, effects of MαCD36 on MHC class II and co-stimulatory molecules expression were not prone to the negative feedback regulation by autocrine/paracrine IL-10, as indicated by the lack of effects of neutralizing anti-IL-10 mAb. In contrast, when MαCD36 was present during the 2 h pre-incubation only, it up-regulated expression of CD40, but not of CD86 or MHC class II. MαSR-A alone had no effect on CD40, CD86 or MHC class II expression (not shown), as well as, it did not significantly modulate the LPS-induced effects (Figure 9).
Anti-CD36 mAb and LPS induce up-regulation of MHC class II and co-stimulatory molecule expression on the surface of PEM, but anti-SR-A mAb has no effect. Binding of PE-conjugated reagents to CD40, CD86 and MHC class II molecules on the surface of PEM was measured with the use of a fluorescence plate reader. Specific binding was calculated by subtracting binding of control mAb. Graphs show results of a single experiment, representative of three such experiments performed. *Significant effect (P < 0.05 using Student’s t-test) of a treatment, relative to a control group treated with isotype-matched non-immune mAb.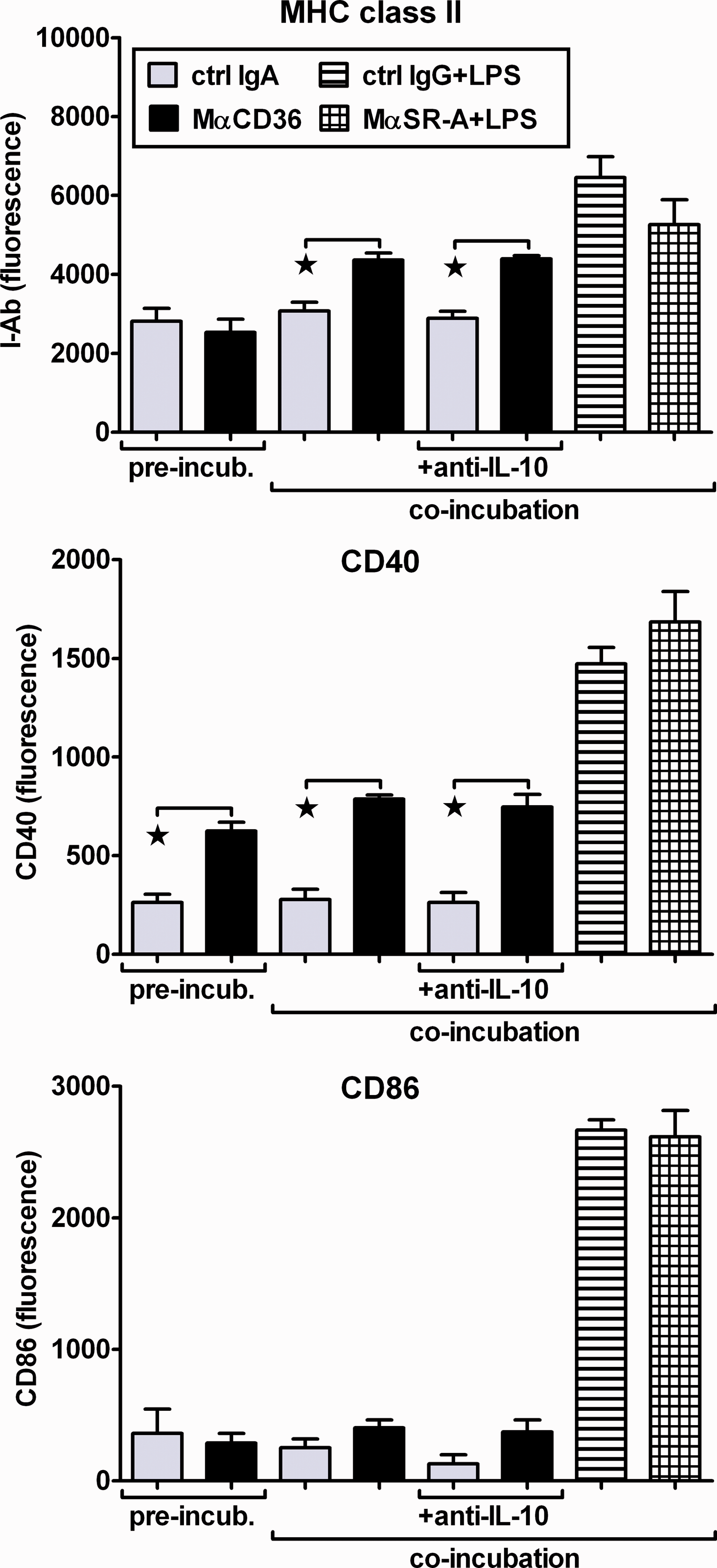
Discussion
Two major types of receptors mediate interactions of innate immune cells with unopsonized pathogens. Pattern recognition receptors (PRR), such as TLR, are responsible for triggering acute inflammation and affect the type of acquired immune responses that subsequently develop, but are unable to mediate phagocytosis. Phagocytosis of pathogens, followed by their intracellular destruction and presentation of pathogen-derived Ag to specific T lymphocytes, seems to be the major function in the immune system of the second group of receptors, endocytic receptors, represented by SR. Whereas PRR seem highly selective for specific chemical compounds, which expression invariably characterizes all related pathogens, 56 SR exhibit so-called broad-selectivity for numerous ligands produced by both several different, unrelated pathogens and by host cells. The lack of selective ligands was the major obstacle hampering studies on signaling abilities of individual SR. Consequently, mechanisms of signal transduction from SR remain poorly characterized, and even the ability of these receptors to trigger any intracellular signaling, other than that inherent to endocytosis, is still questioned by some authors.27,33,47 In our previous study, using specific mAb to selectively ligate receptors, we were able to demonstrate that SR-A mediated stimulation of H2O2 production and inhibition of LPS plus IFN-γ-stimulated IL-12p70 production, but did not affect NO release in AM or PEM. 21 Herein, we extended these observations by showing that in PEM specific SR-A ligation with 2F8 mAb is ineffective alone, but inhibits LPS-stimulated production of IL-6 and of the IL-12/23p40 subunit, enhances IL-10 release, and has no effect on the production of TNF-α or RANTES.
Effects of another SR-A ligand, AcLDL, on cytokine production were very similar to those of MαSR-A, but they were only partially SR-A-dependent as in SR-A−/− PEM effects of AcLDL were diminished, but not abolished. Also all DS-induced responses were preserved in SR-A−/− PEM, which questions applicability of both ligands in studies on SR-A signaling. We showed previously that DS also inhibited strongly LPS plus IFN-γ-stimulated NO production, and that this effect of DS was fully preserved in SR-A−/− macrophages. 24 In contrast, a few articles have been published recently in which it has been reported that stimulation of cytokine release in macrophages and induction of DC maturation by fucoidan alone are mediated solely by SR-A, as these responses were absent in SR-A−/− cells.28,29 These findings are surprising because of notorious non-selectivity of sulfated polysaccharides. Moreover, Kim et al. 18 reported that in WT and SR-A−/− PEM fucoidan stimulated the same TNF-α production and tyrosine phosphorylation of PI3K. In our current study, neither AcLDL nor DS alone stimulated production of pro-inflammatory cytokines, confirming that they were free of bioactive microbial contaminations. In contrast, both ligands exerted differential modulation of LPS-stimulated cytokine production. At least three different SR seemed involved in these AcLDL and DS effects. The lack of additivity with MαSR-A, as well as weaker responses of SR-A−/− PEM, indicated that acting through SR-A both AcLDL and DS inhibited LPS-stimulated IL-12/23p40, but enhanced IL-10 production. In addition, SR other than SR-A mediated inhibitory effects of DS and AcLDL on the production of both cytokines. Yet another SR, activated by DS, but not by AcLDL, was responsible for the enhancement of IL-6, TNF-α and RANTES production.
Pre-incubation with pertussis toxin, a selective inhibitor of heterotrimeric Gi/o proteins, fully abolished modulating effects exerted by MαSR-A on cytokine production. The lack in the short cytoplasmic domain of SR-A of known signaling motifs has suggested that signaling from SR-A may be mediated by other receptors. Consistent with this possibility, SR-A-mediated adhesion, which was also inhibited by pertussis toxin, 37 was not inhibited by the deletion of almost entire cytoplasmic domain of SR-A. 57 Taken together, these results indicate that signaling from SR-A may be propagated by interaction of the extracellular portion of SR-A with a seven transmembrane spanning receptor(s)—the only known receptor type that couples to heterotrimeric Gi/o proteins. The reversal of MαSR-A-induced effects by wortmannin indicates that further steps of signal transduction pathways downstream of Gi/o proteins involve activation of PI3K. PI3K from the IB family, namely PI3Kγ, is known to be activated upon binding to βγ subunits of heterotrimeric G proteins, so we suspect that PI3Kγ may be involved in SR-A signaling. In contrast, class IA PI3K, known to act downstream of TLR, 58 seem to negatively regulate production of all pro-inflammatory cytokines tested, as wortmannin strongly enhanced their production when stimulated by TLR agonists or MαCD36 (Figure 3 and data not shown). Further elements of signaling pathways linking SR-A-induced activation of PI3K to the reciprocal regulation of IL-10 and pro-inflammatory cytokine production remain to be identified. However, PI3K-mediated signaling pathways activated by other receptors, but producing similar effects have been already elucidated in details and their involvement in SR-A signaling is currently being verified in our laboratory. These pathways involve activation of Akt kinase downstream of PI3K and Akt-mediated phosphorylation-induced inactivation of glycogen synthase kinase-3 (GSK-3) and activation of the mammalian target of rapamycin complex 1 (mTORC1), together regulating the relative amounts of active transcription factors, CREB and the p65 subunit of NF-κB that compete for the common co-activator-CREB-binding protein (CBP). As inhibition of GSK-3 upon phosphorylation by Akt or mTORC1 results in augmented binding of CREB to CBP and competitive suppression of the binding of NF-κB p65 to the co-activator, activation of these pathways ultimately leads to the enhancement of the CREB-dependent production of IL-10 and suppression of NF-κB-dependent production of pro-inflammatory cytokines.59,60
We demonstrated that SR-A binds to LPS with very low affinity and is not involved in LPS clearance from the culture medium by PEM. The role of SR-A as a receptor for LPS has been generally accepted, although the evidence supporting this view was weak. This assumption was based on two early reports in which binding of high concentrations of LPS precursors or partial structures to SR-A was demonstrated.25,48 Our results showing that natural LPS is a very low-affinity SR-A ligand are consistent with results of studies undermining the role of LPS as the specific ligand for SR-A on the surface of Gram-negative bacteria. 7 As SR-A does not serve as LPS receptor, increased production of IL-12/23p40 in SR-A−/− cells exposed to LPS is unlikely to be caused by the loss of a suppressive effect exerted by SR-A upon its ligation by LPS. Among possible reasons of this selective effect of SR-A deficiency is higher expression of another class A SR−MARCO in SR-A−/− PEM (Supplementary Figure S1A), 22 as we showed previously that co-stimulation provided by MARCO is required for the LPS-stimulated production of IL-12, but not of NO in PEM. 22
The lack of effects of SR-A deficiency on the production of pro-inflammatory cytokines in response to TLR ligands in macrophages observed in our study is consistent with previous reports.7,61 In contrast, TLR agonists have been reported to stimulate much higher production of pro-inflammatory cytokines in SR-A-deficient DC, as well as upon stimulation with TLR agonists these cells exhibited increased potency in priming adaptive immune responses.20,35,36 Also in our study, LPS stimulated strongly increased IL-12p70 and IFN-γ production in the co-culture of Th lymphocytes with SR-A-deficient DC (unpublished observations), but not with SR-A-deficient PEM, despite much higher SR-A expression on WT PEM than WT DC (Supplementary Figure S1C). A recent report by Yu et al. 20 revealed that these properties of SR-A-deficient DC may be largely ascribed to the removal of tonic inhibitory effects exerted by intracellular SR-A on signaling from receptors belonging to the TLR/IL-1R family, rather than by the loss of receptor functions of SR-A. Thus, SR-A seems engaged in two different mechanisms regulating pro-inflammatory and immunoregulatory functions of immunocytes, dependent or not dependent on ligand binding to the receptor, and the relative role of these mechanisms seems to be cell type-specific.
Surprisingly, we found that LPS clearance from the culture medium by PEM is mediated, in part, by CD36. As CD14, but not CD36, was able to bind LPS directly, CD36 might be involved in the internalization of CD14-LPS complexes. Indeed, we found that LPS stimulated specific association of rCD14 with both rCD36 and endogenous CD36 on the surface of PEM, which was not accompanied by LPS transfer (unpublished observations). However, CD36 does not seem able to mediate pro-inflammatory activation of macrophages by LPS, as concentrations of LPS even as high as 20 µg/ml did not stimulate cytokine production in C3H/HeJ PEM, although these cells responded normally to CD36 ligands. This conclusion is in line with several reports demonstrating unimpaired responses to LPS in CD36-deficient cells3,9,40 and contrast those of Baranova et al.4,62
We have also shown that macrophage responses to the CD36 ligands, LTA and MαCD36, depend on CD36 cooperation with other receptors, namely TLR2 and CD14. Cooperation between TLR2 and CD36 in sensing infections caused by P. falciparum or by Gram-positive bacteria is already well documented,3,9,27,40 but its mechanism has remained unclear. In particular, there is a controversy over whether CD36 is a signaling receptor for microbial ligands4,41,43 or plays only a passive role, limited to ligand loading on TLR, in a manner analogous to CD14.3,27,40 Our results situate themselves between these possibilities. The effects of function-blocking mAb, as well as several pieces of indirect evidence, indicate that, with the exception of IL-10 production (which is largely TLR2/CD14-independent), the stimulation of cytokine production by MαCD36 is mediated by CD14 and TLR2. Moreover, responses to MαCD36 were not inhibited by an inhibitor of SFK, which were suggested to play a crucial role in signal transduction initiated by intracellular domains of CD36. However, the observation that anti-TLR2 and anti-CD14 mAb blocked effects of CD36-specific mAb indicates that cooperation of CD36 with TLR2 and CD14 is not based on a passive ligand transfer from CD36 to TLR2 through CD14, as suggested by Jimenez-Dalmaroni et al. 27 Rather, ligand binding induces physical association of CD36 with CD14 and TLR2, resulting in the activation of TLR2-mediated intracellular signaling. It has been demonstrated previously, with the use of fluorescent imaging techniques, that LTA or diacylated lipopeptides induce association of TLR2/TLR6 heterodimers with lipid raft-resident CD36, whereas TLP induces association of TLR2/TLR1 heterodimers with CD14, but not with CD36.63,64 We found that LTA induced strong, specific binding of rCD14 to endogenous CD36 on the surface of PEM (unpublished observations). However, although responses to LTA were inhibited by anti-TLR2 mAb, LTA-induced rCD14 binding to PEM was blocked by MαCD36, but unaffected by anti-TLR2 mAb (unpublished observations), suggesting that CD36 alone is sufficient to bind CD14 and only later the complex of CD14 with CD36 may associate with TLR2. The observation that MαCD36- or LTA-induced signaling is inhibited strongly by dynasore, but not by cytochalasin D, suggests that, like the TRIF-dependent pathway of TLR4 signaling, triggering of intracellular signaling by the CD36/CD14/TLR2 receptor complex requires dynamin-dependent internalization of receptors. Association of CD14/CD36 with TLR2 may occur before internalization of the whole receptor complex because addition of anti-TLR2 mAb to the extracellular medium was effective in blocking MαCD36-induced responses. This mechanism provides explanation for the apparent paradox that TLR2/TLR1 and TLR2/TLR6 heterodimers, which seem highly selective for, respectively, triacylated and diacylated lipopeptides or lipoglycans, as confirmed by crystallographic data, 65 mediates responses to ligands as diverse as lipopeptides, proteins, glycoproteins and even polysaccharides; 56 binding of these ligands may be mediated by multispecific receptors, such as CD36, which then associate with TLR2 and exploit its signaling abilities.
Interestingly, cytokine production stimulated by MαCD36 was much more susceptible to inhibition of JNK than that stimulated by the TLR2 agonist, TLP. It has been demonstrated that JNK is a major transducer of intracellular signaling initiated by the C-terminal cytoplasmic domain of CD36.4,41 Thus, although TLR2 plays an obligatory role in mediating effects of CD36 ligation on pro-inflammatory cytokine production, a co-stimulation provided by autonomous CD36 signaling might be also required.
Both MαSR-A and MαCD36, exhibiting agonistic properties in our study, are function-blocking mAbs. MαCD36 cross-competed in binding to rCD36 with diverse ligands of this receptor, including DS, LTA and glycolaldehyde-modified albumin (not shown), consistent with the possibility that the epitope of this mAb is localized in the ligand-binding domain of the receptor. Although MαSR-A seems to bind to the domain of the receptor (the coiled-coil domain) 34 different from that involved in ligand binding (the collagenous domain), it also effectively blocked binding to the receptor of all so-far tested ligands. Thus, we suppose that both mAb are able to trigger intracellular signaling because they induce in receptors conformational changes similar to those induced by binding of other receptor agonists.
It has been a puzzling observation that CD36 mediates both pro-inflammatory macrophage activation in response to microbial products 40 and immunosuppressive effects of apoptotic cells or P. falciparum-infected erythrocytes.66,67 We have demonstrated that CD36 is capable of triggering both pro-inflammatory and immunosuppressive signaling. We suppose that an important factor determining the choice of pathways activated in response to a given ligand may be the degree of CD36 cross-linking this ligand produces. According to this scenario, ligands of CD36 that do not produce aggregation of the receptor, such as LTA or GPI anchors from P. falciparum, would signal mainly through TLR2, following dynamin-dependent internalization of receptors. In turn, ligands producing moderate cross-linking of CD36, such as bivalent MαCD36, in addition to the TLR2/CD14-dependent pathway, also activate the TLR2-independent pathway leading to high IL-10 production. This TLR2-independent pathway is likely to be initiated from the cell surface as inhibitors of endocytosis, dynasore and cytochalasin D did not inhibit MαCD36-stimulated IL-10 production, and even this production was enhanced strongly by cytochalasin D. Extensive cross-linking of CD36 produced by large, multivalent ligands, such as apoptotic cells or P. falciparum-infected erythrocytes would prevent the interaction of CD36 with TLR2 and triggering of the TLR2-dependent pathway. Under these conditions, only the TLR2-independent, anti-inflammatory pathway would be initiated. Consistent with the model outlined above, when CD36-bound MαCD36 was cross-linked with secondary Ab or when macrophages phagocytosed erythrocytes coated with MαCD36 no induction of IL-6 and TNF-α production was observed.39,68 Elements of these TLR2-independent signal transduction pathways, as well as whether cooperation of CD36 with other receptor(s) is required for their initiation, remain to be determined. Effects of MαCD36 on LPS-stimulated cytokine production observed in our experiments resemble previously reported effects of other CD36-specific mAb, apoptotic cells or P. falciparum-infected erythrocytes on LPS-stimulated cytokine production in human monocytes 66 and monocyte-derived DC. 67
In our study, we also assessed the effects of receptor ligation with mAb on the ability of PEM to drive differentiation of Th lymphocytes. Confirming our expectations, based on the pattern of cytokine modulation, MαSR-A reversed the Th1-polarizing effect of LPS in the mixed culture of naïve Th lymphocytes and PEM presenting specific peptides. Both stimulation of IL-10 production and inhibition of IL-12 secretion seem to play a role in this effect of MαSR-A. As SR-A has been reported to serve as a receptor for parasite-derived Ag, 69 intracellular signaling accompanying SR-A-mediated endocytosis of these Ag might contribute to the suppression of Th1-type immune responses, characteristic of infections caused by parasitic worms. SR-A has been also demonstrated to participate in non-opsonic phagocytosis of Gram-negative bacteria. 7 However, in this case, ligands of TLR4 and TLR9, synthesized by these bacteria, are able to up-regulate expression of MARCO, which, as we have demonstrated previously, 22 takes over the role in non-opsonic phagocytosis from SR-A. Intracellular signaling triggered during MARCO-mediated phagocytosis of bacteria may provide the necessary co-stimulation for TLR-mediated production of IL-12p70,12,22 therefore promoting differentiation of Th1 lymphocytes.
Interestingly, pre-incubation with MαCD36 had the opposite to MαSR-A effect, namely it strongly stimulated IFN-γ production. This observation was somewhat unexpected as MαCD36 did not stimulate detectable production of IL-12p70 (not shown) under conditions at which it stimulated IFN-γ production in the co-culture. However, unlike effects of MαSR-A, effects of MαCD36 on the polarization of Th lymphocytes may involve MαCD36-induced up-regulation of MHC class II and co-stimulatory molecules expression on the surface of PEM. In particular, pre-incubation with MαCD36 up-regulated expression of CD40 on PEM, which was shown required for differentiation of Th1 lymphocytes. 70
In contrast, when MαCD36 was not removed following pre-incubation and was present during the entire period of co-culture, it inhibited IFN-γ production, as a result of IL-10 accumulation. Taken together, our results suggest that immunoregulatory effects of CD36 ligation in vivo may depend on several factors, such as the phase of immune response, type and density of APC, or degree of CD36 cross-linking. Thus, CD36 ligation on macrophages by pathogens or their products may initially promote the Th1 polarization of immune responses. However, in later phases this Th1 polarizing effect may be countered by the delayed production of IL-10, constituting a built-in autoregulatory mechanism preventing pathology known to be caused by excessive Th1 responses. The same CD36-induced, IL-10-based immunosuppressive mechanism seems to also be triggered in macrophages upon cross-linking of their CD36 receptors by apoptotic cells and to contribute to the resolution of inflammation. As these immunosuppressive effects require the accumulation of IL-10 in extracellular fluids, they may be more effective in sites with poor blood/lymph supply than in well-perfused organs.
Both SR-A and CD36 have also been demonstrated to be major receptors mediating endocytosis of oxidized LDL, amyloid-β fibrils and proteins modified with advanced glycation end products, which has implicated involvement of both receptors in the pathogenesis of atherosclerosis, Alzheimer’s disease and diabetes-associated pathologies. In addition to mediating endocytosis, CD36 has been also suggested responsible for the initiation and sustaining of chronic inflammation, characteristic of these diseases.43,46 Stewart et al. 46 reported recently that upon binding to β-amyloid fibrils or oxidized LDL, CD36 signals through SFK to induce a newly described heterodimer of TLR4 and TLR6 from which signal is propagated by both MyD88-dependent and TRIF-dependent signaling pathways. This apparent paradox of PRR activation by endogenous ligands has been resolved in the recent study by Kannan et al. 71 who demonstrated that microbial contaminants are fully responsible for the ability of oxidized LDL to stimulate pro-inflammatory macrophage activation. When oxidized LDL was prepared under conditions restricting this contamination, it not only did not stimulate production of any pro-inflammatory cytokines in human monocytes and monocyte-derived macrophages, but even inhibited LPS- or TLP-stimulated production of these cytokines in monocytes. These results, as well as the effects of receptor ligation with mAb or low-endotoxin AcLDL observed in our study, are consistent with the possibility that both SR-A- and CD36-induced signaling, triggered upon binding to self host ligand, such as oxidized LDL or apoptotic cells, might play an atheroprotective role, by limiting inflammation and by inhibiting development of pro-atherosclerotic Th1-type responses. 72 However, the CD36/CD14/TLR2 receptor complex might participate in mediating pro-atherogenic effects of bacteria-derived ligands. 73
Footnotes
Funding
The study was supported by the National Science Centre of Poland [grant number 2012/05/B/NZ6/00813]; and by Jagiellonian University Medical Collage [grant number K/ZDS/002326].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.