
Abstract
Influenza A viruses (IAVs) remain a major health threat and a prime example of the significance of innate immunity. Our understanding of innate immunity to IAV has grown dramatically, yielding new concepts that change the way we view innate immunity as a whole. Examples include the role of p53, autophagy, microRNA, innate lymphocytes, endothelial cells and gut commensal bacteria in pulmonary innate immunity. Although the innate response is largely beneficial, it also contributes to major complications of IAV, including lung injury, bacterial super-infection and exacerbation of reactive airways disease. Research is beginning to dissect out which components of the innate response are helpful or harmful. IAV uses its limited genetic complement to maximum effect. Several viral proteins are dedicated to combating innate responses, while other viral structural or replication proteins multitask as host immune modulators. Many host innate immune proteins also multitask, having roles in cell cycle, signaling or normal lung biology. We summarize the plethora of new findings and attempt to integrate them into the larger picture of how humans have adapted to the threat posed by this remarkable virus. We explore how our expanded knowledge suggests ways to modulate helpful and harmful inflammatory responses, and develop novel treatments.
Introduction
Novel concepts regarding innate immunity to influenza.

IAVs are major pathogens that represent an ongoing threat to human and animal health principally through their ability to cause respiratory morbidity and mortality. Two major features of influenza biology result in continuous viral evolution and a critical role for the innate immune system in recovery: the presence of animal reservoirs of IAV and the propensity of the virus to undergo point mutations at high frequency. 2 The presence of animal reservoirs allows for exchange of whole gene segments of animal strains with human strains leading to pandemics (e.g. that of 2009) and propensity to point mutations results in yearly seasonal epidemics. In either case, the population is exposed to novel IAV strains that can spread rapidly in the absence of specific adaptive responses to the new strain. Adaptive immune T-cell effectors first reach the lung approximately 5 d after infection. 3 Hence, for the first 5 d or so after infection with novel IAV strains we depend on our innate immune system to control viral replication without resulting in undue damage to the delicate respiratory epithelium. In most cases, IAV infection is self-limited; however, IAV can lead to major morbidity and mortality. While pandemics of IAV are the most frightening in that they are associated with severe outcomes in otherwise healthy subjects, seasonal IAV epidemics cause substantial morbidity and mortality in vulnerable populations.
Understanding the innate immune response to IAV has begun to elucidate why some viral strains cause more severe outcomes and why some people are more vulnerable. It has become increasingly clear that there are important differences in the innate immune response to seasonal and pandemic viruses, as well as to milder and more severe IAV infection. Two major complicating features of seasonal or pandemic IAV are the risk of bacterial super-infection (principally pneumonia) and exacerbation of reactive airways disease.4,5 Recent findings have revealed that these complications can be accounted for by aspects of the innate immune response to IAV (see below).
Innate immunity refers to immune responses encoded in the genome that do not require prior exposure to a certain infectious agent to be effective. This distinguishes innate from adaptive immune responses. Innate responses are generally, and appropriately, viewed as the first line of host defense against novel pathogens. Innate immunity relies on recognition of pathogen-related patterns that broadly distinguish pathogens from healthy mammalian cells or commensal organisms. In addition to providing a first line of host defense, innate immunity paves the way for effective adaptive immune responses.
6
Adaptive immune responses can also modulate innate responses. We will not focus extensively on interactions of the innate and adaptive responses to IAV, although recent reviews can be consulted.
6
We attempt to be fairly comprehensive in this review but the topic is so large and expanding so fast that we refer to recent reviews in certain areas. Like the adaptive immune system, the innate system has soluble and cellular components, and we review both aspects, highlighting major new discoveries. Figure 1 summarizes the various aspects of innate defense to IAV.
The complex innate immune response to IAV infection.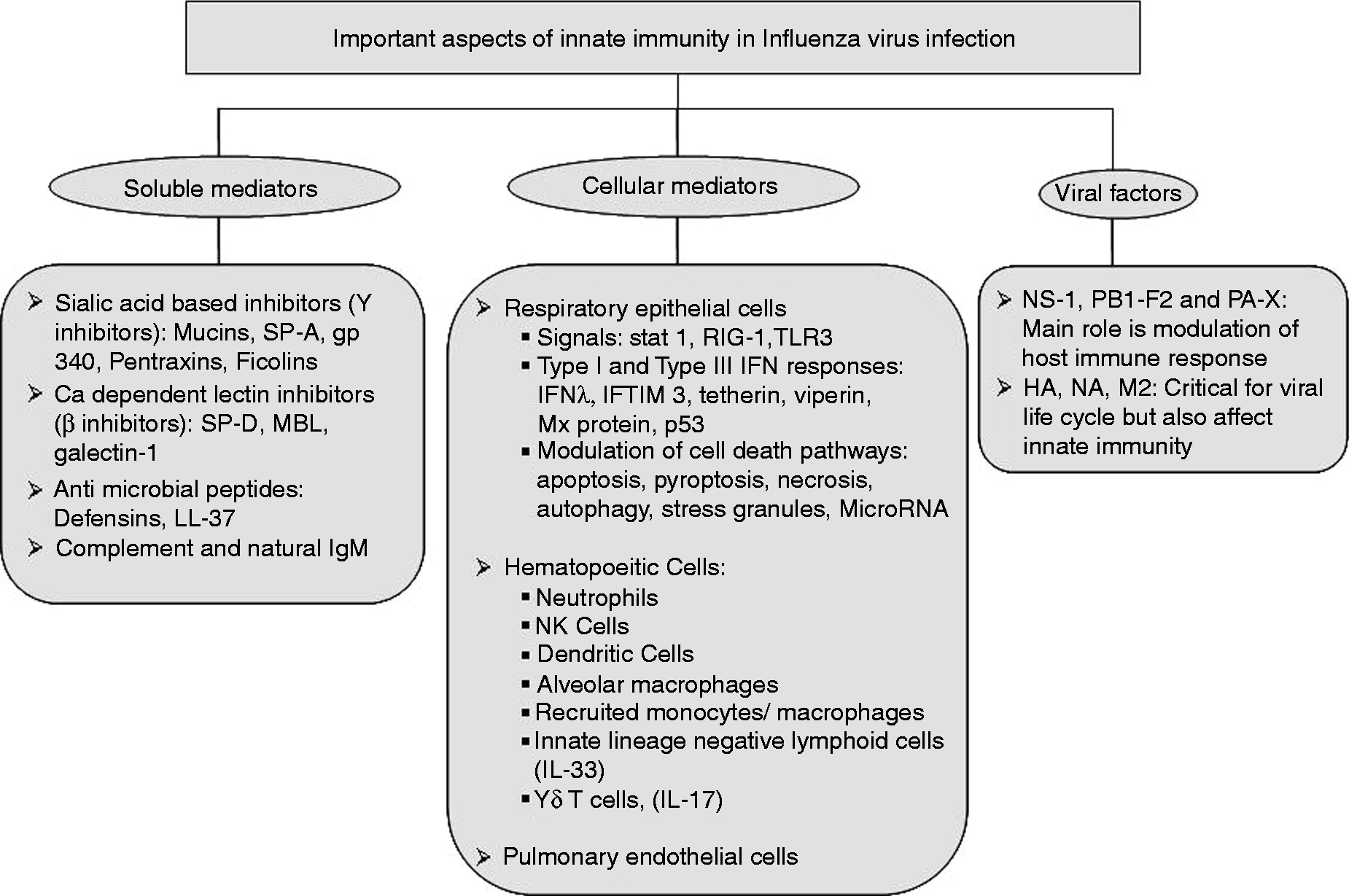
Soluble innate immune mediators
Soluble innate immune inhibitors of IAV play an important role both in blunting viral replication in the respiratory tract and in modulating the immune response to IAV (see Figure 1). The portals of entry of IAV are mainly the nasal and oral cavities, although the conjunctiva is also a potential route of infection.7–9 Of interest, several innate inhibitors, including β-defensins, surfactant protein D (SP-D) and LL-37, are produced by conjunctival cells and present in tear fluid where they could play a role in inhibiting IAV infection of this site.10–12 The nasal and oral cavities also contain many soluble inhibitors of IAV that can interact with each other. 13 We performed a detailed analysis of components of saliva that inhibit IAV, and these included the mucin MUC5B, salivary agglutinin [also known as scavenger receptor cysteine rich glycoprotein 340 (gp-340)], human neutrophil peptides (HNPs) and histatins. 14
Sialic acid-based inhibitors of IAV: γ Inhibitors
If the virus gains a foothold in the nasopharynx then further infection of the upper respiratory tract or the lung is possible. Fortunately, viral pneumonia from IAV is relatively uncommon, perhaps owing to a complex array of inhibitors present in respiratory lining fluids. These inhibitors act via different mechanisms. Several inhibit IAV by presenting sialic acid ligands for the viral hemagglutinin (HA), which impede viral binding to epithelial cells. These have been termed γ inhibitors. 15 The effectiveness of γ inhibitors varies depending on the degree to which the sialic acids resist cleavage by the viral neuraminidase (NA). 16 For example mucins in the respiratory tract or oral cavity can inhibit IAV, but their activity is greatly potentiated by addition of the NA inhibitor oseltamivir, indicating that the viral NA counteracts the action of the mucins.14,17,18 Some of the γ inhibitors are much more resistant to the effects of NA for reasons that are not fully understood. Such inhibitors include the lung surfactant protein A (SP-A),19–21 a member of the collectin family, and lung gp-340. 22 More recently, two other groups of γ inhibitors, the pentraxins and ficolins (see below), have been shown to have inhibitory activity for IAV strains.16,23
Calcium-dependent lectin inhibitors of IAV: The collectins SP-D and Man-binding lectin
Another group of inhibitors, termed β inhibitors, acts through calcium-dependent binding to carbohydrates present on viral proteins. In humans, β inhibitors found in airway secretions include the collectins, SP-D and Man-binding lectin (MBL), H-ficolin and galectin. SP-D and MBL will be discussed together based on their similar mechanisms of binding to IAV, although their impact on inflammatory responses during pandemic IAV infection may differ. SP-D is constitutively present in respiratory secretions, and the levels increase in response to inflammatory stimuli, including IAV infection.19,24 SP-D constitutes the most important innate factor in human bronchoalveolar lavage (BAL) fluid for seasonal IAV strains. 25 Inhibition of IAV strains by SP-D or MBL depends on the presence of Man-rich glycans on the viral HA and strains that lack glycosylation on the HA, including most mouse-adapted strains (e.g. PR-8 and WSN) are resistant. An important recent finding is that IAV isolates carrying the HA of human pandemic strains (including H1N1 of 1918 and 2009, H3N2 of 1968 and H2N2 of 1957) and also H5N1 strains, have few glycan attachments on the HA and are resistant to inhibition by SP-D or MBL.26–29 This may, in part, account for the increased pathogenicity of pandemic IAV strains as such strains bypass the inhibitory effects of SP-D and show increased pathogenicity in mice. Seasonal IAV strains acquire additional glycans on the HA and become increasingly sensitive to inhibition by SP-D and less pathogenic in mice. In contrast, removal of specific glycans from the HA of seasonal IAV strains makes them more pathogenic in mice. 28 Certainly, the increased pathogenicity of pandemic strains reflects the effects of multiple genes in addition to the HA (notably the polymerase genes and NS1; see below); however, the relative importance of the HA is illustrated by the fact that replacement of the HA of a relatively low pathogenicity seasonal strain with those of pandemic strains is sufficient to cause a marked increase in pathogenicity.26,30
A recent study showed decreased levels of SP-D in fatal cases of H5N1 infection in humans, 31 suggesting that in some settings failure to generate or increase SP-D expression (perhaps owing to widespread epithelial damage) can contribute to development of acute lung injury and viral pneumonia. Deficiencies in SP-D levels or function may also account for the susceptibility of specific vulnerable groups to severe outcomes with seasonal IAV, including diabetics, smokers, and patients with cystic fibrosis or chronic obstructive pulmonary disease.32–36 There are polymorphisms of SP-D that are associated with reduced serum levels of the protein and decreased in vitro activity against IAV,37,38 but the role of these polymorphisms in susceptibility to IAV infection in vivo has not yet been studied. As in the case of many other innate immune mediators, SP-D has important modulatory effects on inflammation and other aspects of innate and adaptive immunity, including promotion of viral uptake by phagocytes possibly through causing viral aggregation and down-regulation of chemokine and cytokine generation and lung pathology in vivo during IAV infection.24,39,40 SP-D has also been shown to modulate lymphocyte activation and dendritic cell (DC) function,41,42 although the relevance of these findings to IAV is not yet known. Although SP-D overall appears to play an anti-inflammatory role in the context of IAV infection, it can have pro-inflammatory effects in some settings, including when complexed with certain ligands or modified through nitrosylation.43,44 Whether such pro-inflammatory effects occur in some types of IAV infection has not been studied.
MBL is not expressed in the lung under resting conditions, but it is present in lung lavage fluid during IAV infection and it has been recently shown to play a role in clearance of seasonal IAV infection in mice.45,46 In contrast, MBL may play an adverse role in defense against highly pathogenic IAV strains. 47 This finding is consistent with potential pro-inflammatory effects of MBL that may relate, in part, to its ability to fix complement in the absence of Abs.45,48
Porcine SP-D: A combined γ and β inhibitor
Porcine SP-D and SP-A are of interest given the importance of pigs in development and transmission of new human pandemic strains. Porcine SP-D, in particular, has the unique feature among SP-Ds of various species of having a highly sialated N-linked sugar on its carbohydrate recognition domain. Human, rodent and porcine SP-A have a similar N-linked sugar on the carbohydrate recognition domain and it is this glycan that mediates the anti-influenza activity of SP-A. 49 Porcine SP-D inhibits IAV by a dual mechanism in which it binds to HA-associated carbohydrates through its calcium-dependent lectin activity and it presents the sialylated glycan to which the HA binds.50–52 As a result, porcine SP-D inhibits a broader spectrum of viral strains than human SP-D and has greater inhibitory activity against strains inhibited by human SP-D. This may, in part, account for the ability of pigs to be infected with IAV strains without obvious illness, facilitating transmission or simultaneous infection with more than one IAV strain possibly promoting reassortment.
A calcium-dependent lectin related to the collectins that functions as a γ inhibitor: H-Ficolin
The ficolins are a group of three distinct proteins in humans that resemble MBL in terms of the ability to fix complement and interact with similar receptors on immune cells.16,53 The ficolins may be more relevant to IAV than MBL. H-Ficolin, in particular, is present at higher levels in serum, and is also expressed by respiratory epithelium and is present in BAL of healthy donors. 16 Two studies recently demonstrated that ficolins inhibit IAV at relatively low concentrations and also fix complement in presence of IAV.16,54 The ficolins have lectin activity distinct from the collectins in that they bind mainly acetylated glycans or proteins. Pan et al. 54 first demonstrated that ficolins bind to and inhibit IAV in a calcium-dependent manner, suggesting that their lectin activity is important to inhibition. We found, however, that inhibitory activity of H-ficolin was not calcium-dependent and was that of a γ inhibitor. 16 Consistent with this, the inhibitory activity of H-ficolin was potentiated by the NA inhibitor oseltamivir and was lost after NA treatment of the protein. Of interest, deficiency states of both MBL and H-ficolin have been identified and associated with increased risk of respiratory and other infections.55,56
Galactose-binding lectins and IAV: Galectin 1
The galectins are a group of Gal-binding lectins that play pivotal roles in the immune response. A recent article demonstrated that galectin-1 inhibits infectivity of IAV, that it is up-regulated in the lung after IAV infection, and that mice lacking galectin-1 are more susceptible to IAV infection. 57 This is of interest, as complex glycans on IAV terminate in Gal due to the action of the NA, and the collectins have low affinity for Gal. Hence, galectins and collectins may complement each other as IAV inhibitors. Further studies on the role of galectins in IAV infection will be of interest.
Hydrophobic proteins and lipids as inhibitors of IAV
Other components of respiratory lining fluid appear to contribute to antiviral defense, including the hydrophobic surfactant protein SP-C 58 and surfactant lipids, 59 and further studies of these components will be of interest.
Anti-microbial peptides as antiviral proteins and immune modulators: Defensins and LL-37
Antimicrobial peptides represent another important, major group of soluble innate inhibitors of IAV infection (see recent reviews by Tecle et al. 60 and Doss et al. 61 ).
α- and β-defensins and retrocyclins
The defensins are one major category of antimicrobial peptides found in lung fluids. The human α-defensins present in the lung are the HNPs, which are delivered to the lung by neutrophils in inflammatory states. These have strong neutralizing activity for many IAV strains.62–64 The mechanism of antiviral activity of HNPs has not been fully elucidated. We have found that HNPs induce viral aggregation and inhibit infectivity mainly through direct interactions with the virus.62,64–66 Salvatore et al. 63 have found that HNPs also inhibit IAV through binding to epithelial cells and inhibition of protein kinase C. Unlike the collectins and other proteins that bind the viral HA, HNPs do not inhibit HA activity of IAV. 64 Mice do not have neutrophil α-defensins, but have other antimicrobial peptides that may play a similar role.
Another class of defensins, the β-defensins, is produced by respiratory epithelial cells either constitutively or in response to inflammatory stimuli and also inhibit IAV.62,67,68 The β-defensins are less potent as direct inhibitors of IAV than the HNPs; however, they may have important immunomodulatory roles during IAV infection as well. Ryan et al. 68 have demonstrated that mice lacking β-defensin 1 have more severe lung inflammation when infected with IAV, although viral titers were not different compared with control mice. This article also demonstrated that IAV infection increases production of β-defensin 1 by plasmacytoid DC. Further studies of the in vivo contributions of this and other β-defensins during IAV infection will be of great interest.
Of interest, there is a third class of defensins called theta defensins or retrocylins (because of their cyclic nature) that occur in primates, but not humans, and have very strong anti-IAV activity.62,69 The retrocyclins, like HNPs, can induce aggregation of IAV and they appear to have stronger intrinsic antiviral activity than HNPs. 62
LL-37: An antiviral and immunomodulatory peptide in IAV infection
A distinct group of antimicrobial peptides are called the cathelicidins, and the one representative of this class in humans is LL-37. Recent reviews have discussed the extraordinary range of activities of LL-37, which include direct antimicrobial and antiviral activities, chemotactic activities for various immune cells and modulation of macrophage responses to inflammatory stimuli, and modulation of DC responses.60,61 LL-37 is released from neutrophil granules, but is also produced by epithelial cells, including those in the respiratory epithelium. Leukotriene B4 has been shown to promote defense against IAV probably through its ability to stimulate release of LL-37 and β-defensins from respiratory epithelial cells. 70 Two studies have recently established a role of LL-37 during IAV infection. Barlow et al. 71 first demonstrated that LL-37 has direct antiviral activity against IAV and contributes to host defense against the virus in vivo both by limiting viral replication and virus-induced inflammation Our laboratory then reported on the mechanism of antiviral activity of LL-37, which is distinct from that of collectins or defensins. 72 Unlike these proteins, LL-37 did not cause viral aggregation or alter viral uptake by epithelial cells. LL-37 did, however, cause disruption of viral membranes on electron microscopy. It is unclear whether the antiviral or immune modulatory effects of LL-37 are more important in vivo, but further studies in mice (including mice in which the homologue of human LL-37 is knocked out) will, hopefully, clarify this.
Complement, natural IgM and IAV
Recent findings have linked genetic variations in proteins related to the complement system with outcome of IAV infection73,74 and showed that complement component C3 specifically has a beneficial role in response to IAV. 75 In contrast, a recent report from Sun et al. 76 showed that inhibition of complement with a C3 receptor antagonist or an anti-C5a Ab reduced acute lung injury in mice infected with highly pathogenic H5N1 The complement system can be activated during the innate response by MBL or H-ficolin. Another way in which complement can be activated prior to the development of an effective Ab response is through natural IgM, which has been shown to play a role in IAV infection. 77
High mobility group box 1
The high mobility group box (HMGB) family of proteins was first identified as nuclear proteins that bind damaged DNA, and are involved in transcription and DNA repair.78,79 More recent findings show that these proteins are also secreted or released from necrotic cells, and HMGB1 has been shown to act as an ‘alarmin’, signaling through various receptors on DC, monocytes and other cells, resulting in release of pro-inflammatory cytokines and causing chemotaxis. 80 These findings have led to interest in targeting HMGB1 to reduce inflammatory injury. 81 HMGB1 levels in serum were elevated in humans with severe H1N1 2009 infection 82 and those suffering bacterial infection after IAV infection. 83 Elevated levels of HMGB1 were also found in a mouse model of severe IAV infection, but levels were not correlated with survival. 84 Most recently, HMGB1 was found to bind to viral nucleoprotein (NP) in infected cell nuclei and to promote viral replication. 85 These findings illustrate the remarkable multi-functionality of some innate defense-related proteins and, specifically, how some innate immune proteins also often have roles in normal cellular physiologic processes.
Cellular innate immune mediators
Respiratory epithelial cells
The respiratory epithelium itself is considered to be an important component of the innate immune response system to lung infection. The importance of the epithelium per se (as opposed to bone marrow-derived cells) in initiation of the immune response was recently confirmed in a study by Shornick et al. 86 In this study, mice that lacked stat 1 in the epithelium, but had bone marrow containing stat 1, had a strongly impaired response to respiratory viral infection (Sendai virus), while the inverse was not true. Using similar techniques Unkel et al. 87 recently showed a critical role for production of granulocyte macrophage colony-stimulating factor (GM-CSF) by IAV-infected alveolar epithelial cells in causing recruitment of DC and reduction in virus-induced lung injury in mice. 87 Mice lacking the GM-CSF gene in the epithelial cells (but not in hematopoetic cells) had defective DC recruitment and increased mortality from infection. Mice are not a natural host for IAV, so it has also been an important goal to determine the response of human epithelial cells. Primary human alveolar type II cells have a robust innate immune response to IAV infection in vitro88–90 that differs from the response of primary human alveolar macrophages (AM). The most salient differences include the finding that type II cells support productive viral infection, while macrophages generally do not, that type II cells produce abundant amounts of IFN-λ (also called IL-29, IL-28 A and B, or type III IFN in other studies) and little TNF-α (TNF), whereas macrophages produced abundant TNF and minimal IFN-λ. Adding exogenous IFN-λ was also shown to down-regulate viral replication, increase IFN-responsive genes and reduce cytokine production by the type II cells. Other studies have confirmed that IFN-λ is an important part of the initial immune response of respiratory epithelium to IAV in mouse models, 91 and that it is the predominant IFN produced during intranasal IAV infection. 92 The evolving picture is that IFN-λ is the predominant IFN involved in the initial antiviral response at the epithelial surface, while type I IFNs are produced to a greater extent by DCs (see below).
Of course, the major target of IAV infection in most cases is the upper airway (e.g. nasal, tracheal or bronchial) epithelium, rather the alveolar epithelium as the latter would only become infected in the relatively uncommon cases of viral pneumonia. Studies using primary human bronchial epithelial cells have begun to demonstrate distinct responses of these cells to IAV infection as compared with the response of cell lines like A549 or Calu-3 cells.93,94 Importantly, the study by Hsu et al. 94 demonstrates that human bronchial epithelial cells grown at the air–liquid interface are able to release preformed IFN-β during IAV infection despite the ability of IAV to inhibit retinoic acid inducible gene 1 (RIG-1) signaling. This constitutively produced IFN-β was found to play a key role in restriction of viral replication in these cells.
IAV recognition pathways in epithelial cells
It is now clear that there are at least three pattern recognition pathways through which cells recognize and respond to IAV infection: (i) the cytoplasmic RNA recognition protein, RIG-1; (ii) TLR3 and TLR7, which recognize viral RNA at the cell surface or in endosomal compartments; and (iii) Nucleotide oligomerization domain (NOD)-like receptors (NLR) that trigger the inflammasome pathway leading to caspase 1 activation and IL-1β and IL-18 production. RIG-1 and TLR3 are very important for epithelial cell responses,95–98 while TLR7 is particularly important for responses of DC,99,100 and NLR for macrophages and DC (see below). Figure 2 illustrates the various pathways of cellular response to IAV infection.
Overview of innate immune recognition pathways of IAV in lung epithelial cells. (A) RIG-I-like receptor (RLR) pathway:
101
RIG-I is a cytoplasmic protein that detects 5’triphosphate single-stranded RNA (ssRNA). It then activates signaling pathways via MAVS or Fas-associated death domain containing protein (FADD) leading to production of type 1 IFN or pro-inflammatory cytokines, respectively. The RIG-I pathway can be regulated by TRIM 25 (activation), RNF125 (inhibition) or DUBA (inhibition). (B) TLR pathway: TLR3 and TLR7 recognize viral RNA at the cell surface or in endosomal compartments. TLR7 recognizes ssRNA and activates downstream signaling via myeloid differentiation factor 88 (MyD88), while TLR3 recognizes dsRNA and activates downstream signaling via TIR containing adapter inducing IFN-β (TRIF). (C) NLR pathway:
175
The cytoplasmic inflammasome complex which consists of NLRP3 (one of the best characterized NLR) and ASC recruits, binds and activates caspase-1 resulting in production of pro-IL-1β and IL-18. RNF: ring finger protein; TRAF3: TNF receptor associated factor 3; DUBA: deubiquitinating enzyme A; TBK1: TANK binding kinase1; IKK-I: inducible IκB kinase; IRAK4: IL-1R associated kinase 4.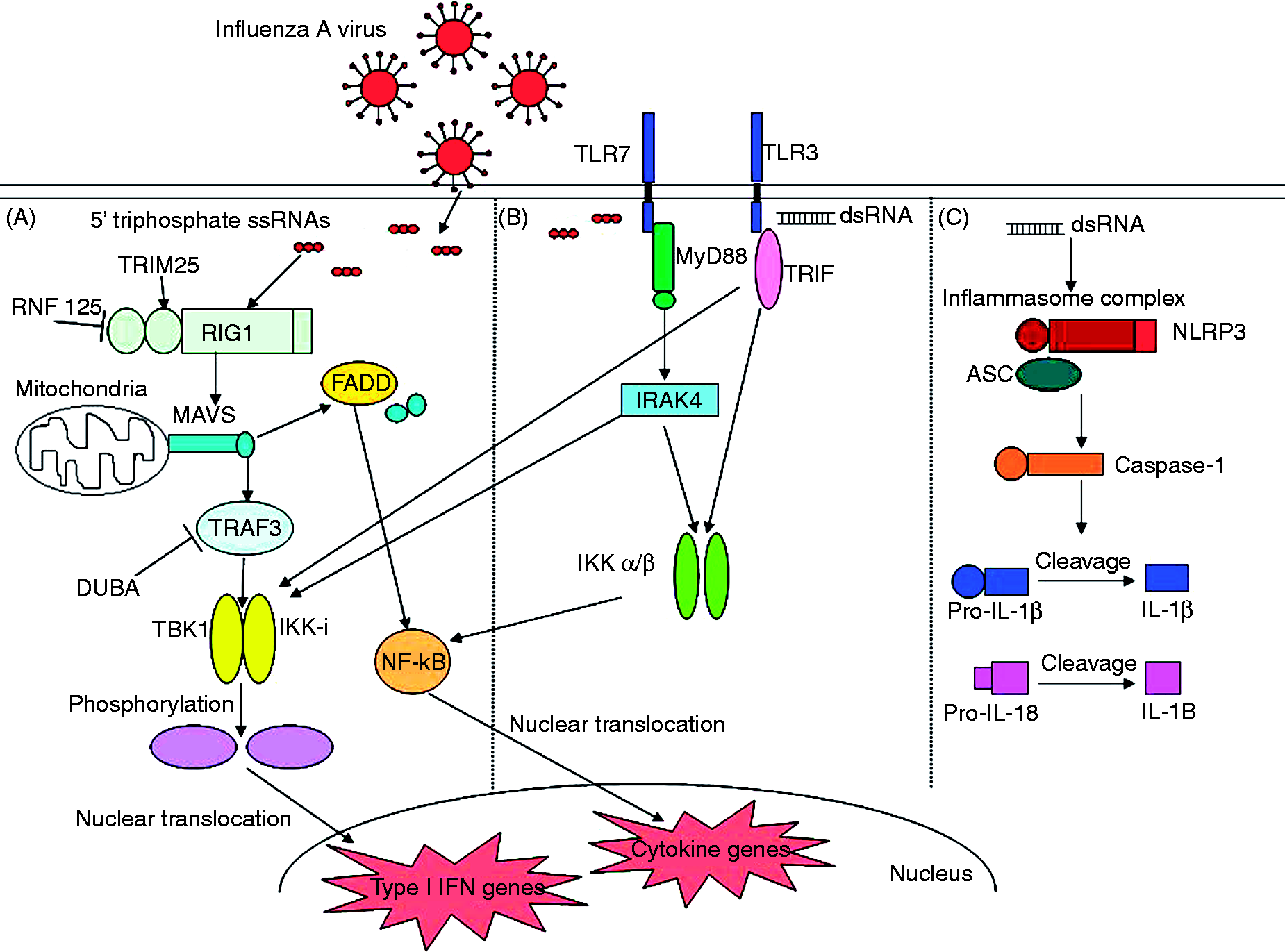
Extensive data now show that RIG-1, which is an RNA helicase, is a critical mediator of response to RNA viruses, including IAV, through its ability to recognize 5’ capped single-stranded RNA in the infected cell cytoplasm. 95 RIG-1 then signals through mitochondrial antiviral signaling protein (MAVS), and then NF-κB and IFN regulatory factor 3 (IRF3), leading to pro-inflammatory cytokine and type I IFN generation (see Takeuchi and Akira 101 for an excellent review). Of interest, the related RNA helicase, melanoma differentiation-associated gene 5, which is involved in poly I:C response, and response to picornaviruses and other viruses, is not involved in the response to IAV. 96 Given the importance of RIG-1 in host defense against IAV it is not surprising that IAV has evolved to inhibit RIG-1 activation through its NS1 protein (see below).
TLR3 has been shown to be involved in the response of respiratory epithelial cells to IAV and it is up-regulated by IAV infection. 97 Of note, activation of TLR3 appears to play mostly an adverse role in IAV infection by increasing immunopathology, in that TLR3−/− mice had improved survival and reduced inflammatory responses despite an increase in viral titers. 97 Although TLR4 was not found to be directly activated by IAV components, it has been shown to contribute to acute lung injury with highly pathogenic H5N1 infection. 102 Activation of TLR4 in this setting was dependent on formation of oxidized phospholipids and reduced in mice lacking a component of the NADPH oxidase. This may be a common pathway of acute lung injury, as other infectious agents and acid aspiration had similar effects.
A fascinating, recently developed, concept is that the coagulation system participates in innate host defense against infections through bacterial entrapment in fibrin clots and stimulation of signaling via protease activity.103,104 This system appears to be an ancient mechanism of early host defense. Thrombin or other proteases are known to mediate signaling in cells via activation of protease activated receptors 1 and 2 (PAR-1 and PAR-2). PAR-1 and PAR-2 expression in airway epithelium is increased during IAV infection 105 and activation of these receptors can modulate signaling through TLRs. 104 It is not yet clear in what settings PAR-1 or PAR-2 play beneficial or harmful roles in IAV infection. A compelling study showed that blockade of PAR-1 signaling or deletion of the PAR-1 gene in mice improved outcome of infection with pandemic H1N1 or H5N1 infection. 106 These results led to the suggestion that PAR-1 inhibition might be a therapeutic strategy during severe IAV infection. In contrast, another compelling study found that PAR-1−/− mice had improved outcome with PR-8 (mouse-adapted H1N1) infection, 107 suggesting that further study is necessary before attempting therapeutic blockade of PAR-1. Both studies do indicate that PAR-1 does mediate important signaling events for recruitment of immune cells. It is too soon to try to explain the different outcomes of these studies, but differences in the viral strains used could have been important. PAR-2 appears to have different, but also important, activities in IAV infection, including stimulation of IFN-γ 108 (beneficial) or activation of TLR4 104 (adverse).
The central role of type I and type III IFN response systems
The type I IFN system has long been known to have a key role in containment of IAV infection, but the diverse mechanisms through which type I IFNs mediate this effect, and how the virus counteracts these effects, are still being elucidated. 109 Intracellular events triggered by type I IFNs include oligoadenylate synthetase, protein kinase R (PKR) and a host of other IFN-responsive genes. 110 Recently, extracellular receptors whose expression is regulated by IFNs have also been described.
Major role of IFN-inducible transmembrane protein 3, tetherin and viperin in host defense against IAV
A functional genomic screen of factors involved in restriction of IAV replication in osteosarcoma cells in vitro identified (among other proteins) the IFN-inducible transmembrane (IFITM) proteins 1, 2 and 3 as important antiviral factors. 111 IFITM3 was particularly important in mediating the antiviral activity of IFN type I. The IFITMs were found to inhibit early replication of IAV, but also of West Nile and Dengue virus, and deletion of IFITM3 in mouse cells resulted in increased IAV replication. This was followed by another pivotal study showing a major impact of deletion of IFITM3 in mice on the course of infection with various IAV strains. 112 Mice lacking IFITM3 infected with otherwise low pathogenicity IAV had markedly increased mass loss, mortality, viral loads in the lung, respiratory epithelial apoptosis, infection in the lung (as opposed to upper airways for wild type mice), neutrophil infiltration and inflammatory cytokine production. Overall, the lack of IFTIM3 converted a low pathogenicity viral infection into an infection with all the features of high pathogenicity viruses (e.g. H5N1 or 1918 H1N1). In addition, the authors found increased incidence of an uncommon allelic variant of IFTIM3 in patients hospitalized during the 2009 H1N1 pandemic. This variant was found to have a truncation of the N-terminal domain of the protein and to have reduced antiviral activity in vitro. Another recently described cell surface, type I IFN-induced antiviral protein, tetherin, has been found to inhibit IAV (and other viruses, including HIV) by tethering newly produced viral particles at the cell surface. 113 The IAV NS1 protein counteracts tetherin expression and the viral neuraminidase counteracts tetherin activity. Viperin, another recently identified IFN-inducible protein, was found to restrict budding and release of IAV particles by impeding formation of lipid raft domains in cellular membranes. 114 Despite confirming this in vitro effect of viperin, a further study by Tan et al. 115 found that viperin knockout mice did not have a different course of IAV infection in vivo than wild type mice.
Role of the Mx protein in IFN-mediated host defense against IAV
The Mx protein is an IFN-inducible protein that has long been known to mediate antiviral effects in IAV-infected cells. The mouse and human Mx proteins are referred to as Mx1 and MxA, respectively. Both confer protection, but Mx1 acts in a nuclear location, whereas MxA acts within the cytoplasm. Common laboratory mouse strains (e.g. Balb/c) lack a functional Mx1 protein, and knock in of this gene in Balb/c mice protects against lethal IAV infection. 116 Recently, important new insights have emerged regarding the molecular mechanisms of Mx activity. Viral strains vary in sensitivity to Mx proteins and sensitivity segregates with the NP. Avian viral strains (e.g. H5N1) are highly sensitive to Mx mediated inhibition, whereas the human pandemic H1N1 strains are not. 117 Zimmerman et al. 118 found that exchange of NP of pH1N1 with that of H5N1 resulted in loss of Mx protein sensitivity and increased lung pathology, mass loss and lethality in mice, despite the fact that the modified H5N1 strain replicated to a lower degree in vivo and in vitro than the wild type H5N1 strain. These findings strongly suggest that Mx proteins act through interacting with NP and that Mx1 is capable of down-regulating adverse pro-inflammatory responses occurring with IAV infection. Wisskirchen et al. 119 showed that MxA also binds to two RNA helicases (UAP56 and URH49) that are required for replication of IAV. These RNA helicases bind NP and viral ribonucleoprotein and are involved in nuclear export of viral mRNAs and prevent the accumulation of double-stranded RNA (dsRNA), thus reducing cellular type I IFN responses. The findings that Mx proteins interact with RNA helicases and viral NP indicate that interference with viral RNA replication is central to their antiviral activity. Cilloniz et al. 116 used global transcription profiling in lungs of wild type or Mx1+/+ Balb/c mice infected with the highly pathogenic 1918 H1N1 IAV strains to demonstrate the molecular events associated with protection conferred by the Mx1 protein. The Mx1 protein alone improved survival from 0 to 50%, and treatment of Mx1-expressing mice with type I IFN improved survival further to 100%. This marked protective effect of IFN was only seen in the Mx1 expressing mice and was associated with specific down-regulation of pro-inflammatory cytokines (e.g. IL-1, IL-6, TNF) and chemokines (e.g. CCL5, CXCL-10). Hence, at least in the context of highly pathogenic IAV infection, the combination of Mx1 and type I IFN is able to down-regulate damaging pro-inflammatory responses. Mx proteins therefore appear to improve the outcome of IAV infection by both antiviral and anti-inflammatory effects. They may also play a role in restricting interspecies transmission of IAV strains (e.g. avian H5N1 is highly sensitive to inhibition by MxA). 117
p53 as an antiviral protein
p53 has mainly been thought of as a regulator of cellular apoptosis in cells that have undergone DNA damage and as an anti-oncogene; however, it is now clear that p53 has a pivotal role in regulating antiviral responses as well. Turpin et al. 120 demonstrated that levels of p53, phosphorylation of p53 and p53 accumulation in the nucleus in A549 cells are all increased during IAV infection. These changes occur late in the infection cycle (e.g. 8–24 h post-infection). Deletion or silencing of p53 resulted in marked reduction of apoptosis induced by IAV in respiratory epithelial cells (human or mouse). Recent studies have confirmed this increase in p53 expression and activation, and shown that IAV inhibits ubiquitination of p53 and modulates expression of p53 isoforms (which regulate expression of full length p53) resulting in the p53 increase.121,122 An additional striking finding in the Turpin et al. 120 study was that deletion of p53 resulted in elevation of viral titers and reduction of type I IFN transcription. These findings were recently expanded upon by Munoz-Fontela et al., 123 who demonstrated that infection of p53−/− mice results in reduced type I IFN, monocyte chemoattractant protein-1 (MCP-1), and macrophage inflammatory protein-1α and β in the lung accompanied by increased viral replication at d 3, more mass loss and higher mortality in the mice. DC responses and CD8 cell recruitment were also impaired. Hence, lack of p53 impaired both innate and adaptive immunity to IAV. To determine if these effects result from loss of p53 in non-hematopoietic cells or hematopoietic cells chimeric mice were generated (as in the study by Shornick et al. 86 ) in which the bone marrow expressed p53, but the other cells did not. These mice still had a defect in DC migration. This finding suggests that loss of p53 in the respiratory epithelium or other non-hematopoietic cells indirectly affects DC migration in this model. It has long been established that p53 plays numerous important roles that limit cancer development and progression (e.g. recognition of damaged DNA, promotion of apoptosis, inhibition of angiogenesis). It is remarkable to find that this extraordinary protein also has wide ranging effects in antiviral defense.
p53 directly modulates expression of the genes for MCP-1, IRF9, PKR and ISG15 through transcriptional activation and hence is central in initiation of antiviral responses through mechanisms separate from induction of apoptosis. 123 Of interest, despite the ability of IAV to cause increased late accumulation and activation of p53, the viral NS1 protein has been found to associate with p53 and inhibit its transcriptional activity and pro-apoptotic effects. 124 This finding is consistent with the various ways in which the NS1 protein inhibits the development of the IFN-mediated antiviral state in cells. IAV appears, therefore, to inhibit p53 activity early in the course of infection of epithelial cells, but to increase its activity later. This apparent dichotomy may make sense when one considers further evidence of how IAV modulates cell death pathways.
Modulation of cell death pathways during IAV infection: Apoptosis, pyroptosis, necrosis and autophagy
It is now considered that there are at least three distinct pathways of cell death, all of which involve specific signals and have distinct effects on inflammatory responses. Lamkanfi and Dixit 125 provide an excellent recent review of these pathways in relation to microbial infection. It has become increasingly clear that pathogens (including IAV) actively modulate cell death pathways and that the host can regulate cell death pathways as a mechanism of host defense.125,126 Although it has long been recognized that IAV induces cellular apoptosis, it has recently been shown that the virus inhibits apoptosis early in the course of infection. One important mechanism involves up-regulation of anti-apoptotic signaling through the PI3kinase/Akt pathway mediated by the viral NS1 protein127,128–131 (see Herold et al. for review 132 ). This activation of the PI3kinase pathway is observed with viral infection or recombinant expression of NS1 alone 127 and is reduced when infecting with NS1-deleted virus. This effect was not mediated by NS1’s inhibition of type I IFN as it occurred in IFN-deficient VERO cells as well. Another mechanism through which IAV counteracts apoptosis in epithelial cells involves interaction of the viral NA with CEACAM6 leading to stimulation of pro-survival pathways, including activation of Akt. 133
In the study by Zhirnov and Klenk, 127 the activation of PI3kinase peaked at 7.5 h post-infection, whereas p53 activation and caspase 3 activation occurred later. This led to the proposal that IAV may inhibit apoptosis at earlier time points after infection to facilitate maximal viral RNA and protein production, and accelerate apoptosis at later points to facilitate later phases of the viral life cycle. It has been shown that caspase 3 activation is necessary for viral propagation132,134 through promoting release of viral ribonucleoproteins from the nucleus of the cell. Blocking caspase 3 or NF-κB activation causes viral ribonucleoprotein retention in the nucleus and inhibits viral production.134,135 The apoptotic phase of IAV infection involves the extrinsic apoptotic pathway mediators TRAIL and Fas ligand. 136 The PB1-F2 protein is encoded in some viral strains through an additional open reading frame within the PB1 polymerase gene. PB1-F2 can mediate apoptosis in monocytes through the intrinsic pathway by permeabilizing mitochondrial membranes. 137
There is an extensive literature from murine studies indicating that IAV infection stimulates oxidant generation in the lung, and that this is injurious.88,138–140 Recent studies with isolated primary alveolar epithelial cells have demonstrated that the nuclear factor-erythroid 2 related factor 2 (Nrf2) is increased after IAV infection and protects against IAV induced apoptosis through an IFN-independent mechanism. 88 Nrf2 was also shown to reduce IAV-induced pulmonary pathology in mice. 141 These findings are of interest as Nrf2 expression appears to increase ability of respiratory epithelial cells to restrict IAV replication as well. 142 Nrf2 expression is depressed by cigarette smoking and in chronic obstructive pulmonary disease, and this correlates with increased susceptibility to bacterial infection, which can be ameliorated by treatment with phytochemicals that increase Nrf2 expression.142,143 Nrf2 was found to increase macrophage expression of phagocytic receptors and bacterial phagocytosis. 143
Apoptosis is generally felt to be immunologically silent and not pro-inflammatory, whereas pyroptosis and necrosis are pro-inflammatory modes of cell death. Pyroptosis is the end result of inflammasome activation, but, thus far, this mode of cell death has not been reported in context of IAV infection, despite clear evidence of inflammasome activation. 144 Yatim and Albert 126 present the very interesting hypothesis that there is a integral relationship of cell death pathways and immunity, and that viruses and other microbes have driven the evolution of eukaryotic cell death pathways. Hence, we should perhaps not look at the eukaryotic cell as standing in isolation through evolution and fending off pathogen invaders, but that it has evolved through time in interaction with pathogens.
Control of cell death pathways is a fundamental to how the host deals with viral infection or infection with other intracellular pathogens. 126 Autophagy is a constitutive process conserved from yeast to mammals that is important for recycling of cellular proteins and protein complexes, and for allowing cellular survival under stress conditions (e.g. nutrient deprivation). This process is also intimately involved in the response of cells to infection with all types of pathogens and in triggering both innate and adaptive immune responses (for excellent reviews see Dumit and Dengjel145, and Kuballa et al. 146 ). As a well researched clinical example, defects in proteins needed for autophagy have been linked to Crohn’s disease and increased inflammatory responses triggered by LPS or muramyldipeptide. We will be using the term autophagy here, although a more precise term is macro-autophagy to differentiate the process from micro-autophagy and chaperone-mediated autophagy, which occur at the level of lysosomes and do not involve the formation of the larger double-membraned autophagosome structure. In macro-autophagy this structure is formed through cooperative interaction of numerous proteins, it engulfs additional cytoplasmic proteins or ‘cargo’ and, ultimately, fuses with lysosomes to result in degradation and recycling of the cargo proteins. Viruses, in particular, have evolved ways of interfering with autophagy in different ways, often through interacting with a key autophagy-related protein called beclin-1. Beclin-1 is involved in formation of the autophagosome and also in the fusion of the autophagosome with lysosomes.
DNA viruses (e.g. herpes viruses) can inhibit autophagy formation by blocking this action of beclin-1, whereas RNA viruses (including IAV) inhibit the ability of beclin-1 to promote fusion with lysosomes. 147 During IAV infection of A549 cells an accumulation of autophagosomes is observed owing to direct interaction of the M2 viral matrix protein with beclin-1. 148 This results in cell death via apoptosis. This may result in blunting of innate responses to the IAV as Law et al. 149 have shown that IAV-induced CXCL10 and IFN-α generation are promoted by autophagy. In addition, autophagy has been shown to be involved in degradation of viral proteins for Ag presentation via class II MHC molecules, although it appears that proteosomal processing is more important for presentation to CD4 cells in the case of IAV. 150 Clearly, we have presented a simplified picture of this complex and rapidly evolving topic, as, in some instances, viruses promote autophagy (e.g. to result in death of uninfected CD4 cells mediated by the HIV envelope protein), 146 and the role of autophagy in viral replication and immune responses appear to vary based on cell type and virus strains. For instance, H5N1, but not H1N1, IAV was found to promote autophagy in epithelial cells and inhibition of autophagy increased survival of mice infected with H5N1.151,152
Autophagy is a distinct process from proteosomal degradation of ubiquitinated proteins; however, there is crosstalk between these two systems. 145 Inhibition of proteosome activation reduces IAV replication by blocking viral RNA synthesis, but also by causing retention of viral NP in the cytoplasm after viral entry. 153 In addition, in the setting of bacterial infection autophagy can limit IL-1β generation by inflammasomes providing another site of interaction between autophagy and innate immunity. 146 Clearly, much has yet to be learned about the role of autophagy in response to specific pathogens, including IAV.
Cellular stress granules and IAV infection
Another mode through which cells deal with stress is the formation of stress granules. These granules form when cellular protein translation is impaired and contain non-translating mRNAs and translation initiation components. Stress granules are felt to play a role in the ‘decision’ of whether a stressed cell should undergo apoptosis or not, in part as they sequester some apoptotic regulatory proteins. 154 Viruses have been shown to induce formation of granules resembling stress granules.155–158 Some viruses utilize stress granules to facilitate their replication.157,159,160 This is noteworthy as stress granules can contain RIG-1 and PKR and to function in innate antiviral immunity. 161 Consistent with this, IAV inhibits the formation of stress granules and induction of type 1 IFN generation through actions of the NS1 protein. 161 NS1 was recently shown to mediate this effect through interacting with the stress granule protein, RNA-associated protein 55. 162
microRNA changes triggered by IAV and the innate immune response
There are microRNAs (miRNAs) present in a variety of epithelial cells that can directly reduce IAV replication through acting on the PB1 gene (viral polymerase). These miRNAs (323, 491 and 654) are not perfect matches for the viral target but, nonetheless, cause degradation of viral RNA. 163 RNA silencing is also implicated as an important antiviral mechanism for IAV 164 as the NS1 protein of IAV inhibits RNA silencing in mammalian cells. IAV infection also leads to a rapid increase in miR29 in A549 cells infected with IAV in vitro or in monocytes of patients infected with IAV, and this miRNA causes up-regulation of expression of COX-2, which, in turn, leads to increased expression of IFN-λ through suppression of DNA methyltransferases. 165 Clearly, our understanding of how IAV infection modulates innate immunity through induction of miRNAs is far from complete, as illustrated by a recent article by Buggele et al. 166 in which miRNA microarrays were used to identify the spectrum of miRNAs induced in human lung cells by IAV strains. This study showed that at least seven miRNAs are consistently induced by IAV and that the degree of induction is greater than that induced by antiviral mediators alone. The up-regulated miRNAs are known to modify a wide array of signaling pathways. Specifically, MAPK3 and IRAK1 were shown to be targeted for mRNA degradation through the combined effect of three of the IAV-induced miRNAs. Hence, future studies will not only have to address the role of numerous miRNAs, but also their potential combinatorial effects to understand how they affect innate immunity.
Pulmonary endothelial cells
A major paper by Teijaro et al. 167 demonstrated that pulmonary endothelial cells are major producers of pro-inflammatory cytokines and chemokines (e.g. IFN, CCL5, IL-1, IL-6 and TNF) during infection of mice with WSN or pandemic H1N1 of 2009, and that they also mediate recruitment of activated monocyte/macrophages and NK cells to the lung 168 These effects were independent of adaptive immunity and could be suppressed by activation of the SIP1 receptors present on endothelial cells. Others have shown that stimulation of aryl hydrocarbon receptors during IAV infection results in increased mortality, 169 in part through stimulation of inducible nitric oxide synthase (iNOS) in lung endothelial cells. 170
Innate immune cells
Many innate immune cells participate in the response to IAV, including cells that are resident in the lung and involved in the initial recognition of, and response to, infection like AMs and resident DCs, and cells that are recruited in response to infection (e.g. monocyte, macrophages, DCs and neutrophils). There are major new findings regarding how macrophages and DCs recognize and respond to IAV, and the role of NK cells and neutrophils in IAV infection. Another important discovery is that there are innate lymphocytes that are integrally involved in the early response to IAV infection prior to development of adaptive lymphocyte responses.
AMs and recruited monocytes and macrophages
There have been numerous studies demonstrating that AMs play a key role in the initial response to IAV infection.171,172 These cells are resident in the lung and part of the first line of defense. There is recent evidence that AM-mediated resistance to IAV can be potentiated by GM-CSF treatment. GM-CSF treatment has been found to improve survival in IAV infection, in part through acting on macrophages.173,174 GM-CSF treated mice had increased numbers of alveolar macrophages, and the macrophages showed increased resistance to apoptotic effects of IAV. Removal of macrophages eliminated the beneficial effects of GM-CSF. As noted above alveolar epithelial cells appear to be a key source of GM-CSF in vivo and this GM-CSF also plays a critical role in recruitment of DC during IAV infection. 87
NLR and inflammasome activation
Inflammasomes are multiprotein complexes that lead to the activation of caspase-1 and generation of active IL-1β and IL-18. These two pro-inflammatory cytokines have been shown to have independent and important roles in promoting innate and adaptive responses to IAV and many other pathogens. 175 Inflammasome activation is mediated by NLR, which recognizes viral and microbial patterns, although the exact mechanism of recognition of IAV by NLRs is unknown. There are many types of inflammasomes, but thus far IAV has been shown to principally activate the NLRP3 inflammasome.176,177 Key components of the NLRP3 inflammasome include the NLRP3 protein itself, the adaptor protein apoptosis-associated speck like protein (ASC) and caspase-1. Mice lacking NLRP3, ASC or caspase-1 were found to have increased mortality after PR-8 infection in three studies.176–178 Of note, lack of MyD88 (adapter protein for TLR7, TLR8 and TLR9 signaling) or an alternative inflammasome molecule, NLRC4, did not affect mortality when tested in parallel studies. 177 Of interest, in mice lacking the inflammasome components, cytokine, chemokine, monocyte and neutrophil responses were reduced, but lung injury was, nonetheless, increased, as evidenced by epithelial cell necrosis and collagen deposition. 176 These results are similar to those reported by Schmitz et al. 179 with respect to deletion of IL-1 receptors in which inflammatory responses were reduced, but survival was impaired. Viral loads were similar in NLRP3−/− mice at d 3 after infection, but were higher by d 7, indicating a delay in viral clearance.177,179 These results indicate that the inflammatory response was protective in this setting and that healing may be impaired in the absence of NLRP3 activation. The role of NLRP3 activation in control of adaptive responses to IAV is controversial and under active study.177,178
Inflammasome activation by IAV mainly occurs in macrophages and DCs, and in contrast to the results obtained with IFITM, hematopoietic, and not stromal, NLRP3 components were critical. 178 The mechanism through which IAV triggers the inflammasomes in macrophages and DC involves viral RNA in part; however, in an elegant study by Ichinohe et al., 180 an additional signal for NLRP3 activation was provided by the viral M2 ion channel. In general, inflammasomes require two signals for activation, the first causing generation of pro-forms of IL-1β, IL-18 and IL-33 and the second activating caspase-1 to cleave the pro-forms of these cytokines to the active forms. In the case of IAV, viral RNA acting through TLR7 (but not RIG-1) mediates step 1, but does not result in caspase-1 activation. The second signal involves viral replication and specifically the M2 ion channel through a mechanism that involves ion fluxes in the Golgi. Inflammasome activation also has the potential to be damaging to the host. In this regard a recent article by Lupfer et al. 181 demonstrated that receptor interacting protein kinase 2 (RIPK2) down-modulates inflammasome activation during IAV infection, and RIPK2 knockout mice suffer increased illness and immunopathology. 181 Of interest, a recent article by Stout-Delgado et al. 182 showed that elderly mice have impaired NLRP3 function in response to IAV and that this can be restored by treating the mice with nigericin, which provides another source of signal 2 for inflammasome activation through a similar mechanism as the M2 protein. These results are of great interest with regard to the impaired immune response and more adverse outcome of IAV infection in the elderly.
Spooky effects at a distance: Gut commensal bacteria modulate innate immune responses to IAV infection in the lung
Two recent studies indicate that loss of normal commensal bacteria impairs innate and adaptive immune response to IAV. Ichinohe et al. 183 found that oral treatment of mice with neomycin alters specific subpopulations of gut flora and results in delayed viral clearance and impaired adaptive responses to IAV infection, including impaired migration of DCs to draining lymph nodes and reduced T-cell and Ab responses. This finding is of particular interest as neomycin was not absorbed through the gut lumen and did not alter airway flora. They further determined that the impaired immune responses to IAV infection resulted from diminished pro-ILβ, pro-IL18 and NLRP3 expression in the lung, and the immune deficit was seen with IAV, but not with two other lung infections that do not involve inflammasome activation (herpes simplex virus and Legionella). Hence, they concluded that the neomycin-sensitive gut bacteria provide signal 1 for NLRP3 activation at a basal level prior to IAV infection. Rectal administration of TLR agonists was able to restore IAV responses in antibiotic-treated mice. Abt et al. 184 demonstrated that antibiotic treatment of mice actually results in increased mortality from IAV infection associated with increased respiratory epithelial necrosis. There was similar recruitment of neutrophils, macrophages and DCs to the lungs of antibiotic treated mice; however, macrophages isolated from these mice had defective response to type I and type II IFNs. These responses could be restored and outcome of infection in vivo improved by treating the antibiotic fed treated mice with Poly I:C. Overall, these remarkable results suggest that the dependence of immune responses to IAV on inflammasomes accounts, in part, for the ability of gut flora to modulate the course of IAV infection.
Protective role of viral uptake or phagocytosis
Phagocytosis by macrophages and neutrophils has been shown to play a protective role during IAV infection in mice.185,186 This could, in part, reflect direct ingestion of viral particles by these cells, as demonstrated in vitro using human cells.25,66,187–192 Although free viral particles are internalized by endocytosis, treatment of IAV with collectins, defensins, ficolins or other innate inhibitors results in viral aggregation and ingestion of large viral aggregates by phagocytes through a process resembling phagocytosis.16,65,193 Human AMs have recently been shown to ingest apoptotic alveolar epithelial cells. 88 This may be the most important manner through which macrophage phagocytosis promotes recovery from IAV infection.
AMs have a variety of receptors capable of mediating viral uptake (see the excellent recent review by Londrigan et al. 194 ). The primary mode of IAV entry into macrophages is through binding of the HA to appropriate sialic acids on the macrophage surface; however, macrophages can also be infected in a sialic acid-independent manner through C-type lectins expressed on the macrophage surface, including macrophage Man receptor, macrophage Gal receptor and DC-Sign.194–196 Uptake through these receptors is calcium-dependent and dependent on glycosylation of the viral envelope proteins. 195 Optimal infection of macrophages involves both sialic acid-dependent and non-sialic acid-dependent uptake. 194 Scavenger receptors (SR-A and MARCO) on AMs play important roles in uptake of bacteria; however, mice lacking SR-A did not have altered infection with IAV, and MARCO−/− mice had improved survival with IAV infection.173,197 Of interest, the protective effects of treatment of mice with GM-CSF correlated with increased expression of the SP-A receptor SP-R210 on macrophages, and was diminished by increased expression of MARCO. 173 In vivo, this could reflect increased SP-A-mediated clearance of IAV. 21 IAV infection of human AMs in vitro reduces expression of C type lectin and scavenger receptors correlating with reduced uptake of zymosan. 198
AMs are major source of chemokines and cytokines
AMs exhibit a potent chemokine and cytokine response upon IAV infection.89,198 AMs are generally not productively infected with IAV, although they express viral antigens. 198 Nonetheless, viable virus is required for stimulation of cytokine and chemokine production by AMs. 198 Of note, highly pathogenic H5N1 viral strains have been shown to productively infect AMs of mouse or human origin,199,200 possibly contributing to increased pathogenicity. Human AMs produce abundant amounts of IFN-α, pro-inflammatory cytokines and chemokines, and up-regulate expression of many innate defense genes in response to viable IAV infection.89,198 There is evidence that pathogenicity of H5N1 may result from profound stimulation of macrophage cytokine production,201,202 although blockade of this cytokine response was not found to be protective in mice. 203 Furthermore, elimination of IL-1β production in response to severe IAV infection led to reduced lung pathology, but increased mortality. 179 These findings suggest that simple blockade of IL-1β or TNF production in response to IAV is unlikely to benefit patients with severe IAV infection and may even worsen outcome.
Macrophage defects and bacterial superinfection
IAV infection impairs expression of phagocytic receptors and phagocytosis of bacteria or zymosan particles by AMs.198,204 This could contribute to co-infection of patients with IAV and bacteria, a potentially lethal combination. 4 There is also extensive evidence of depressed neutrophil function and accelerated neutrophil apoptosis caused by IAV infection,205–210 and this may contribute to bacterial superinfection.211–213
Depression of macrophage and DC functions by IAV can also persist after the peak of IAV infection and may relate to the observation of waves of bacterial pneumonia occurring in patients seemingly in recovery from IAV infection. Didierlaurent et al. 214 found that IAV infection in mice led to prolonged desensitization to bacterial TLR ligands, reduced chemokine generation and impaired recruitment of neutrophils after bacterial infection. Sun et al. 215 found that production of IFN-γ by T cells after IAV infection causes depression of alveolar macrophage uptake of bacteria. In a recent study, Ghoneim et al. 216 tracked the fate of alveolar macrophages after IAV infection and showed that the virus caused marked loss of these cells and that this loss correlated with susceptibility to lethal pneumoccal super-infection. This study showed protection with local GM-CSF treatment.
Recruited monocyte macrophages may have distinct roles compared with AMs
Monocytes and macrophages recruited to the lung during infection appear to play a more complex role than AMs in that there is evidence that they are protective, but also that they can be harmful.217, 218 Recently, Gong et al. 219 showed that mice lacking SerpinB1 show increased lung inflammation and mortality after infection with seasonal IAV. 219 There was no difference in viral clearance in this setting; however, infiltrating monocytes producing excessive TNF and IL-6 and elevated numbers of pulmonary γδ T-cells (see below) were noted in the SerpinB1 knockout mice. These results indicate that serpinB1 acts to restrain overly vigorous monocyte and γδ T-cell responses.
DC
We will not be able to do justice in this review to the extensive and fascinating role of DC in host defense against IAV and refer to reviews that deal with this topic.1,220 Myeloid DC clearly play a key role in early detection of viral infection and in transfer of viral Antigens to draining lymph nodes to initiate an antiviral response, and plasmacytoid DC are major producers of type I IFN during IAV infection in vivo.221,222 Of interest, myeloid and plasmacytoid DC produce distinct waves of cytokines and chemokines over time after IAV infection regulating recruitment of various immune cell types to the lung. 223 Of interest, the NS1 protein of IAV inhibits DC maturation and activation resulting in lowering of the ability of DC to induce T-cell responses. 224 Furthermore, different viral strains have distinct interactions with different DC subsets. 225 As with AM, DC are generally not productively infected or killed by IAV; however, H5N1 can productively infect monocyte-derived DC leading to cell death. 226 This may contribute to the lymphocyte depletion and severe pathogenicity of this viral subtype. 227 Another recent finding of interest is that recruited monocytes can rapidly differentiate into myeloid DC in vivo, and constitute a major source of type I IFNs. 218 A number of soluble innate mediators have been found to modulate DC responses to infection, including SP-D, defensins and LL-37.228–231
Neutrophils
Neutrophils are the first wave of recruited immune cells responding to IAV infection. Given their presence in the lung of subjects dying of severe IAV infection, there was early speculation that neutrophils might be mediators of lung injury and mortality. Neutrophil oxidants may be injurious, 232 and neutrophil-derived proteases233,234 and defensins 64 can impair the functions of SP-D. IAV has recently been found to provoke formation of neutrophil extracellular traps (Tripathi S, White MR and Hartshorn KL, unpublished data) and these have been implicated in lung injury indirectly. 235 In fact, the bulk of the evidence now supports the concept that neutrophils are protective in IAV infection,185,236–238 even as part of the profound influx of myeloid cells caused by highly pathogenic 1918 viral strains. 30 Inhibition of neutrophil extracellular trap formation by deletion of the PAD4 gene was not found to alter survival of IAV infection. 239 Neutrophils can take up IAV and various innate immune proteins, including collectins, ficolins and defensins, markedly increase neutrophil uptake of the virus. Neutrophils are also a source of defensins and LL-37. Unanticipated roles of neutrophils in facilitating the adaptive immune response to IAV have now been reported. For instance, neutrophils were shown to serve as APC 3 and to promote T-cell responses to IAV infection. 240 Given the abundance of these cells in the first wave of response to IAV infection in the airway, these roles in adaptive immunity could be very important.
As noted above potentially adverse effects of neutrophils include production of oxidants as mice lacking functional NADPH oxidase have improved outcome with IAV, 232 and many studies now show that anti-oxidants are protective in the context of IAV infection. IAV directly stimulates oxidant production by neutrophils, and this response is linked to neutrophil apoptosis and impaired responses of neutrophils to subsequent bacterial challenges. Of interest, pre-incubation of IAV with collectins protects the cells against this IAV induced impairment.25,205
NK cells
Once again, we do not propose to do justice to the major and complex role of NK cells in influenza viral infection, but we refer to a recent review. 241 Recent studies have demonstrated that mice lacking NK cells have worse outcome with IAV. 242 NK cells contribute to control of viral replication and produce cytokines that modulate the immune response to IAV. Production of IL-22 by NK cells and NKT cells has an important role in repair of epithelial damage caused by IAV.243,244 IL-15 contributes to host defense against IAV by recruiting NK cells to the lung. 245 NK cells have also been shown to contribute to lung pathology in models of IAV infection.246,247 Of interest, NKT cells have been found to suppress immuno-pathology caused by IAV, in part through suppressing accumulation of inflammatory monocytes.248,249 Impaired function of NK cells resulting from IAV infection is also reported,250,251 and this may also contribute to bacterial super-infection. 252 Notably, the response of NK cells to IAV infection is mediated through binding of the viral HA to sialic acids present on the NK receptor NKp46.253,254 This appears to be another example of what, at first, appears to be a non-specific, carbohydrate-dependent binding event, leading to a directed innate immune response. As with many other topics we have reviewed, NK cells also play important roles in bridging the innate and adaptive responses to IAV. 255
Discovery of innate lymphocyte cells and their role in IAV infection
It was previously assumed that lymphocytes (other than NK cells) do not participate in the initial innate response to infection with a novel IAV strain, but that they come into play during the adaptive immune response. It is now clear, however, that there are several populations of innate lymphoid cells that exist and that some of these participate in the initial response to IAV infection. Neill et al. 256 used mice engineered to express GFP under the IL-13 promoter and identified a novel subset of IL-13 producing lymphoid cells, which they termed ‘nuocytes’ (as nu is the thirteenth letter of the Greek alphabet). These cells are rare in the resting state, but expand greatly in response to IL-25 and IL-33. They could not be assigned to any known hematopoetic cell lineage and they were shown to be critically important in mediating protective type-2 immunity to helminth infection. 256 IAV infection induces IL-33 generation, 257 raising the question of whether innate lymphoid cells are generated during IAV infection. Chang et al. 258 demonstrated that a H3N1 strain of IAV induces rapid development of airway hyper-reactivity (AHR) in mice, even in the absence of adaptive immunity (i.e. in Rag2-/-, mice which lack T- and B-cells) or in NKT cells (i.e. in CD1d-deficient mice). This AHR response was shown to depend on the presence of a subset of cells the authors referred to as ‘natural helper cells’, which are not present in mice lacking the IL-33 receptor ST2. Of note, mice lacking ST2 could still develop AHR through the adaptive immune pathway triggered by ovalbumin sensitization. Hence, the AHR pathway triggered by the natural helper cells is a fully innate response and distinguishable from classic adaptive type-2 responses. These extraordinary findings may explain, in part, the ability to IAV to trigger asthma exacerbations which is a major source of morbidity. 259
An article by Monticelli et al. 260 demonstrated another, more beneficial, aspect of innate lymphoid cells in response to IAV. First, they demonstrated that a similar population of cells (lineage-negative, with similar markers and producing IL-13 and IL-5 in response to IL-33) exists in human lungs. Then, they showed in a mouse model using PR-8 H1N1 that these innate lymphoid cells accumulate in response to IAV infection and that they are a major source of amphiregulin, which mediates their ability to promote epithelial tissue repair after infection. Amphiregulin alone was found to promote tissue repair and improve oxygenation in mice depleted of the innate lymphoid cells. Again, this process occurred in Rag2−/− mice as well. The subject of innate lymphoid cells is rapidly evolving and an excellent recent review is provided by Spits and Cupedo. 261 It is clear that these cells, IL-33 and amphiregulin may play roles in both host recovery and tolerance of infection and immunopathogenesis of IAV and hopefully many new insights will emerge in the near future. A potential adverse effect of amphiregulin is that it is also involved in development of fibrotic responses in the lung; however, this aspect may not be important in the context of an acute, self-limited infection like IAV.
γδ T-cells and IL-17
A surprising, and important, recent finding is that IL-17 plays an important role in the immune pathology induced by severe IAV infection (in this case the PR-8 infection model in mice). 262 IL-17 is an important trigger of neutrophil recruitment that can be generated as part of an adaptive immune response; however, IL-17 is also produced by a subset of innate T-cells (γδ T-cells). These cells led to a rapid and sustained generation of IL-17 in PR-8 infected mice (2–7 d post-infection) that reached high levels prior to the recruitment of Ag-specific lymphocytes to the lung. Of note, deletion of the IL-17 response by use of mice lacking the IL-17 receptor resulted in improved survival and reduced mass loss in the mice without changing the viral load. This effect correlated with reduction in neutrophil influx and reduction in generation of oxidized phospholipids, which have been shown to mediate acute lung injury related to severe influenza infection (e.g. by H5N1) or acid aspiration through activation of TLR4 signaling.102,262 Of note, the adverse effect of IL-17 was not mediated through IL-6 or TNF. A common point between the IL-17 and TLR3 studies is that in both settings deletion of these innate responses improves outcome of infection without affecting viral load. It is important to make a distinction between the innate γδ T-cells and the protective subset of CD4 T-cells that produce IL-17 as part of the adaptive immune response (Th17 cells). For instance, McKinstry et al. 263 showed that IL-10 deficiency promotes a Th17 response leading to improved survival in mice challenged with high dose IAV infection.
Viral proteins that modulate innate immune responses
How can a virus with only eight RNA genome segments cause so much havoc in human and animal populations? IAV makes optimal use of its small genome by producing proteins with multiple sometimes seemingly unrelated functions and by use of alternative reading frames within its major genes.
Proteins whose main role appears to be modulation of innate responses: NS1, PB1-F2 and PAX
The NS1 protein has been mentioned frequently in this review, and it is extraordinary how many aspects of the innate antiviral response are inhibited by this multifunctional protein (see Hale et al. 264 for a review). The NS1 protein blocks generation of type 1 IFN responses during IAV infection through multiple mechanisms, and viruses lacking NS1 are greatly attenuated in vitro and in vivo. 265 NS1 inhibits activation of the RIG-1 RNA recognition system through direct effects and through sequestration of RNA. The NS1 proteins of different IAV strains are important in determining pathogenicity (e.g. that of the 1918 H1N1 strains partially accounts for their dramatic pathogenicity).266–268 We have highlighted some of the recently discovered roles of NS1, including activation of PI3k and interference with tetherin induction, p53 activation, RNA silencing, formation of stress granules and maturation of DCs. Very recent articles have revealed additional remarkable features of the NS1 protein, including the presence of a histone mimic in the NS1 protein of H3N2 viruses that allows it to target the human PAF1 transcription elongation complex resulting in suppression of antiviral responses regulated by this protein, 269 binding to tripartite motif (TRIM) 25 ubiquitin ligase resulting in suppression of RIG1 activation, 270 and binding to RNA helicase A, which facilitates the ability of this helicase to promote viral replication. 271 NS1 also inhibits caspase 1 activation in macrophages, resulting in reduction in cytokine release. 272 Thus, studies of the IAV NS1 protein have been a very effective tool in revealing multiple layers of innate antiviral response in the cell.
Recently, two previously unknown IAV proteins, PB1-F2 and PA-X, have been discovered. They are produced through use of alternative reading frames in the PB1 and PA viral genes, and are important determinants of pathogenicity in an IAV strain and host-dependent manner. 273 Initial studies demonstrated that PB1-F2 promotes apoptosis through a mitochondrial pathway. 137 In particular, PB1-F2 induces apoptosis of monocytes. 274 PB1-F2 has additional effects that promote viral pathogenicity and viral replication as well,273,275–277 especially with respect to highly pathogenic viruses like the 1918 H1N1 and H5N1 strains. Of note, a recent study showed that PB1-F2 is a mediator of increased susceptibility to bacterial superinfection. 278
The PA-X protein is the most recent addition to a small, but expanding, IAV protein family. This protein was discovered through a search for alternative reading frames in the viral genome.279–281 It is produced in small amounts in infected cells from a reading frame in the viral PA protein through a process called ribosomal frame shifting. It is nearly ubiquitous in IAV strains and deletion from a 1918 H1N1 strain resulted in increased weight loss and mortality at intermediate infectious doses of the virus in mice. PA-X reduces host gene transcription and appears to specifically reduce the intensity of the host inflammatory response. Hence, PA-X appears to down-regulate potentially injurious inflammatory responses. This may serve the virus as excessive illness and death of the host prior to viral transmission would be counterproductive in terms of long-term viral survival in the human reservoir.
Proteins that are critical for the viral life cycle per se, but which also modulate innate responses: HA, NA and M2 polymerase
While IAV has dedicated three proteins out of its small genome to modulate host responses, other structural and polymerase proteins play double roles as host response modulators as well. In this review, we have highlighted the role of the HA in triggering the profound inflammatory response of pandemic viruses and activating NK cells through binding to the NKp46 receptor. 254 The viral NA counteracts tetherin activity and inhibits apoptosis of epithelial cells through interacting with CEACAM6. 132 The viral M2 interacts with beclin-1 altering autophagy in IAV-infected cells 148 and provides the second signal for inflammasome activation through its ion channel activity. 180 Shapira et al. 282 used a non-biased approach involving yeast two-hybrid assays to assess interaction partners of viral proteins with cellular proteins. This study revealed, among other things, a role of the viral polymerase in modulating IFN-β production. 282 It seems likely that such studies will reveal additional unexpected ways in which viral proteins interact with the innate proteins.
Therapeutic considerations
Therapeutic strategies based on our understanding of innate immunity.

How to harness innate immunity for therapy of IAV
We have not explored in depth the ways in which innate immune responses initiate the adaptive immune response as this is a major topic in itself. Suffice it to say that nearly every innate response mediator we have discussed, including soluble mediators (e.g. collectins and anti-microbial peptides), RIG-1, TLR, inflammasomes, macrophages, DC, NK cells and even neutrophils contribute in some way to the elaboration of the adaptive immune response. Hence, stimulation of the innate response (e.g. through activation of TLRs) may be an important way to increase the effectiveness of vaccines. Of interest, seemingly non-specific stimulation of inflammation in the respiratory tract by use of an aerosolized bacterial extract had protective effect against subsequent IAV infection. 283 Subsequent studies by this group showed similar effects using a combination of TLR2/6 and TLR9 agonists. 284 Furthermore, dsRNA mimics given prior to IAV are also protective. 285 In contrast, prior infection of mice with Bordetella pertussis markedly worsened the course of subsequent IAV infection by a pertussis-toxin mediated suppression of innate immunity. 286 Similarly, activation of aryl hydrocarbon receptors during IAV infection (as could occur as a result of environmental hydrocarbon exposures) worsens outcome of infection. 169 It seems likely that more research will clarify which types of innate immune stimulation will be beneficial as vaccine adjuvants or even as prophylaxis for IAV infection.
Another approach for treating or prophylaxing against IAV could involve modeling of antiviral treatments based on properties of innate immune proteins. Collectins are attractive in this regard as they have direct antiviral activity, promote phagocytosis and down-modulate lung inflammation. We have used molecular modeling and X-ray crystallographic methods to generate truncated collectin carbohydrate-binding domains (CRDs) with increased ability to bind to IAV associated carbohydrates and inhibit IAV infection in vivo and in vitro.287–290 Some of theses modified collectins acquire the ability to inhibit pandemic IAV strains (Hartshorn KL, Seaton B and Crouch EC, unpublished data). Cross-linking of these CRD preparations causes a further increase in antiviral activity.193,291,292 Van Eijk et al.293,294 have created constructs based on pig SP-D containing N-linked sialylated glycans on the CRD, which have significantly increased antiviral activity. Full-length molecules containing the N-terminal structural domains of ficolins, or of SP-D, combined with the CRD or MBL also have increased antiviral activity.45,295 Synthetic retrocyclins have also increased antiviral and opsonizing activity.65,69,296 Another approach might include enrichment of surfactant with phosphatidyl glycerol. 59
As noted above, treatment with GM-CSF improved outcome in murine IAV infection through enhanced macrophage function.173,174 In addition, it was found to improve outcome of bacterial superinfection after IAV through increasing survival of macrophages. 216 Further study of the use of GM-CSF in severe IAV infection should be explored.
Modulating immuno-pathology induced by IAV
Following the compelling hypothesis advanced by Medzhitov et al., 297 there are two types of injury that can occur during infection: direct cellular injury by the pathogen itself (e.g. respiratory epithelial cell injury by IAV) or injury due to over-exuberant immune response. For some tissues (e.g. skin, bone marrow or liver) there may be fairly extensive ability to tolerate injury due to functional autonomy of the cells making up these organs and the ability to rapidly regenerate function. At the opposite extreme, brain or heart tissue cells are highly specialized and function cannot readily (or ever) be restored. The lung does not tolerate extensive injury owing to the requirement for gas exchange and has limited capacity for regeneration dependent on the degree of injury. There is evidence that mortality resulting from severe IAV infection can be either due to rampant infection per se or immunopathology. In humans, examples of severe influenza infection include infection with pandemic strains (most notably the 1918 strain which was highly pathogenic in humans and in other mammalian hosts, including mice, ferrets and primates), and H5N1 avian influenza which causes a >50% mortality in infected people. Some specific aspects of the innate response that have been associated with injury in mouse models include activation of TLR396,97 in respiratory epithelial cells, production of reactive oxygen and nitrogen intermediates, or activation of aryl hydrocarbon receptors.232,298 As noted above, viral triggering of IL-17 production appears to result in harmful pro-inflammatory responses. In some of these cases, deletion of the pro-inflammatory mediator resulted in both reduced inflammation and reduced viral titers 232 and in others improved outcome resulted from reduced inflammation without change in, or with actual increased, viral titers. 97 The fatal outcome of pandemic H1N1 2009 infection in pregnant mice was recently shown to result from increased immunopathology and lung repair, and not increased viral replication. 299
Such findings have led to the speculation that various methods of inhibiting the innate response during IAV infection could be beneficial in humans. At present, however, there is no clinical evidence to support this and our understanding of the complex innate response to the virus is not sufficient to select an appropriate strategy to be tried in humans. In mouse models, blockade of specific aspects of the innate response has, in some cases, been shown to increase mortality, despite reducing the extent of inflammation. Notable examples include blockade of the neutrophil and macrophage response in mice infected with the highly pathogenic 1918 strain 30 and genetic deletion of IL-1 in mice infected with PR-8. 179 In both cases lung inflammation was improved, but mortality was increased, suggesting that the robust inflammatory response was necessary for survival from infection. Salomon et al. 203 found that deletion of genes for IL-6, TNF and CCL2 did not protect mice against mortality from H5N1 infection, and cyclooxygenase inhibitors were not shown to be protective in another murine study. 300
A possible alternative strategy has recently been reported. Aldridge et al. 301 found that lethal influenza infection (PR-8 or H5N1) resulted in a marked increase in recruitment of a subpopulation of TNF and iNOS-positive DCs they called tipDCs into the lung compared with sublethal infection. 301 Abrogation of the tipDC recruitment through use of CCR2−/− mice did not improve outcome in the mice; however, partial reduction of the response by oral administration of the peroxisome proliferator activated receptor gamma (PPARγ) agonist pioglitazone improved survival and reduced inflammatory cytokine generation. Similarly, Cloutier et al. 302 showed that treatment of mice with prostanoid 15-deoxy-Δ12,14-prostaglandin-J2 reduced mass loss and increased survival after PR-8 infection by stimulating PPAR. Another study found that gemfibrozil was protective. 303 These studies suggest a possible route to using immune modulation in treatment of severe influenza that appears worthy of further investigation.
The limitations of inhibiting innate immune responses as a therapeutic strategy are also apparent when one considers the important phenomenon of bacterial superinfection. Such superinfections were responsible for many, if not most, of the deaths in the 1918 pandemic 304 and have been important causes of mortality (likely exceeding mortality caused by viral pneumonia per se) in other pandemics (including that of 2009). 4 The phenomenon of bacterial superinfection in influenza-infected people suggests that important host defense mechanisms against the virus may actually counteract effective defense against bacteria in the lung. The mechanisms behind bacterial superinfection are complex, but our understanding of the IAV-bacterial pneumonia linkage has increased dramatically in recent years through use of robust mouse models. Sublethal infection with IAV results in dramatically increased morbidity and mortality from subsequent infection with either Streptococcus pneumoniae, Staphylococcus aureus or Haemophilis influenzae.305–308 It appears that increased susceptibility of IAV-infected hosts to bacterial pneumonia are the result of innate immune responses to IAV. Important among these is the type I IFN response. Recently, Shahangian et al. 309 showed that IFNAR−/− mice infected with IAV followed by S. pneumoniae have improved neutrophil recruitment and survival compared with wild type controls. Of interest, two articles have now shown that this effect of type I IFN is the result of its ability to cause impairment of γδ T-cells activity and IL-17 generation, which is critical for neutrophil recruitment and clearance of bacteria including, Klebsiella pneumoniae, S. aureus and S. pneumoniae.308,310 Hence, inhibition of the IL-17 response to IAV might further increase susceptibility to bacterial superinfection.
Certainly, one recurring theme throughout the literature on IAV infection is that there are significant differences between the host response to seasonal and pandemic (or avian) IAV and hence the results of suppression of immuno-pathology may be quite different in these different settings.
Identifying vulnerable groups or individuals based on polymorphisms of innate response genes
The genes encoding many components of innate immunity exhibit single nucleotide polymorphisms (SNPs), which can modulate host susceptibility to infection. Juno et al. 311 recently reviewed various immonogenetic factors associated with disease severity caused by H1N1 and H5N1 IAVs, and emphasized the need for more studies in this area. In one study, the presence of the -238A SNP allele of TNF was associated with increased susceptibility to 2009 H1N1 virus and increased pulmonary complications such as viral pneumonia and acute lung injury/acute respiratory distress syndrome. 312 However, TNF polymorphisms were not found to correlate with IAV-associated death in another study. 313 The minor rs12252 C-allele of the IFITM3 gene has been shown to be associated with enhanced disease severity leading to hospitalization of patients infected with pH1N1. 112 Similarly, TLR3 mutations have been reported to be associated with severe influenza complications.314,315 The NLRP3 inflammosome protein complexes also exhibit multiple SNPs, which are associated with dysregulated inflammasome immune responses; however, their effect on susceptibility to IAV infection has not yet been studied. Similarly, SNPs of MBL, SP-A, SP-D and MxA genes have also been reported to be associated with other respiratory illness, but have not yet been examined in the context of IAV infection.316,317 Polymorphisms associated with low levels of MBL were associated with methicillin-resistant S. aureus superinfection of IAV-infected patients. 313
Increasing lung repair during IAV infection
We have highlighted possible mechanisms through which lung epithelial repair could possibly be increased during IAV infection, including treatment with IL-22 or amphiregulin. As noted, a potential adverse effect of amphiregulin is induction of lung fibrosis, and IL-22 has been found to have adverse effects in West Nile Virus infection, 318 and to induce colon tumor formation after long exposure; 319 hence, further study is needed before recommending these as therapies.
Seeking an integrated picture of innate defense against IAV
A major challenge for future research will be to integrate the various elements of the innate response to IAV into a unified picture and to understand the relative importance of the response components. Some studies have elucidated interactions of innate response components (e.g. we and others have shown both cooperative and antagonistic interactions among some soluble inhibitors).13,17,18,62,64,233 Non-biased approaches to the study of the global response of cells to IAV have been reported, and these have started to provide valuable insights.90,198,282 Shapira et al., 282 in particular, have used such an approach and revealed important elements to the cellular response to IAV infection that were not appreciated by other means, including a role for Wnt signaling and effects of the viral polymerase on IFN-β production. 282
Conclusions
There has been an extraordinary explosion in our understanding of the workings of the innate immune response to IAV infection in the last several years. Clearly, many of the findings summarized in this review have relevance to other infections and inflammatory diseases. Nonetheless, better understanding of the pathophysiology of IAV infection alone remains a very high priority given its on-going impact on human health. It is striking that a virus with just eight RNA segments can have so many effects on the human host. One explanation is that the virus encodes more genes than we previously knew. Another is that the virus parasitizes the cell’s machinery for RNA and protein synthesis, resulting in myriad perturbations of cell physiology. Hence, the study of IAV has been an effective means of revealing how cells or the lung manage to retain homeostasis under stress. An important insight is that we have co-evolved with viruses like IAV and that some of the most basic cellular processes developed as part of this co-evolution. One feature of innate immunity as distinct from adaptive immunity is that most innate immune mediators have other roles in normal cellular or lung physiology, and the question of whether immune or normal physiological functions came first is not clear or perhaps even relevant. It is of interest that so often the literature imputes intentionality to the virus, raising the question of where in this small group of genes a ‘brain’ or ‘motivation’ is hiding. The question of why even the simplest life forms seek to replicate themselves is a philosophical one beyond the scope of this article.
The key challenges facing the field of innate immunity to IAV include how to incorporate the profusion of new discoveries into a more unified picture, how to understand better the difference between milder and more severe IAV infection, how to use our knowledge of immunopathology during IAV to develop therapies that can reduce injury incurred by severe IAV infection without impairing host defense or causing compensatory increase in other harmful inflammatory responses, and how to use our knowledge of innate immune mechanisms to better inhibit viral replication. It is likely that new findings in this field will continue to emerge at a rapid pace and, hopefully, this review will provide the reader with a framework to understand these new discoveries.
Footnotes
Funding
This work was supported by NIH grants AI083222 and HL069031.