
Abstract
The mechanisms underlying pathophysiological states such as metabolic syndrome and obesity include endoplasmic reticulum (ER) stress and aberrant inflammatory responses. ER stress results from the accumulation of misfolded proteins during stress conditions. However, the precise mechanisms by which ER stress modulates inflammation remain incompletely understood. In this study, we hypothesized that ER stress alone could represent a sufficient signal for the modulation of inflammasome-dependent cytokine responses. We found that several ER stress-inducing chemicals and the free fatty acid palmitate can trigger IL-1β secretion in various cell types, including monocytic leukemia cells, primary macrophages and differentiated adipocytes. We show that ER stress primes cells for the expression of pro-IL-1β via NF-κB activation and promotes IL-1β secretion. Enhanced IL-1β secretion depended on the activation of the NLRP3 inflammasome through a mechanism involving reactive oxygen species formation and activation of thioredoxin-interacting protein. Chemical chaperone treatment and the pharmacological application of carbon monoxide inhibited IL-1β secretion in response to ER stress. Our results provide a mechanistic link between ER stress and the regulation of inflammation, and suggest that modulation of ER stress may provide a therapeutic opportunity to block progression of low grade chronic inflammation to metabolic syndrome.
Introduction
IL-1β is a multifunctional pro-inflammatory cytokine that is associated with acute, local and chronic inflammation.1,2 In contrast to most other cytokines, IL-1β secretion requires two steps of priming and activation.3,4 The priming step of IL-1β maturation involves the increased synthesis of immature pro-IL-1β, which is regulated by the NF-κB and MAPK signaling pathways. These responses are initiated by ligand-receptor interactions involving TLR, NOD-containing proteins or cytokine receptors (i.e. TNF-α receptor, IL-1 receptor). 5 The activation step of IL-1β secretion requires the caspase-1-dependent proteolytic cleavage of pro-IL-1β (35 ku) to generate the 17-ku mature form.6,7
The inflammasome, a newly discovered multi-protein signaling platform, regulates the maturation and secretion of select pro-inflammatory cytokines (i.e. IL-1β and IL-18). Inflammasomes have a basic structure consisting of a nucleating intracellular PRR and an adaptor protein (e.g. apoptosis-associated speck-like protein containing a caspase-1 activator domain), which recruit and activate pro-caspase-1. In turn, the activation of caspase-1 by the inflammasome complex processes pro-IL-1β and pro-IL-18 into their bioactive and secreted forms. 8
A number of PRR proteins are known to form inflammasome complexes, including nucleotide-binding-domain, leucine-rich repeat (LRR) domain containing proteins (NLRP)-1, NLRP-3 and NLRP-6, NLRC4/IPAF and AIM2.3,4,8 NLRPs contain a nucleotide-binding and oligomerization domain (NACHT), carboxy-terminal LRR, and may contain a pyrin domain (PYD) or caspase activation and recruitment domain (CARD), or both. 8
The NLRP3 inflammasome is activated in response to a wide range of exogenous and endogenous activators, such as pathogens (i.e. bacteria, fungi and viruses), extracellular ATP, crystalline substances (i.e. monosodium urate crystals and cholesterol crystals), and peptide aggregates (i.e. amyloid beta and islet amyloid polypeptide). 8 Recently, the involvement of reactive oxygen species (ROS) in the activation of the NLRP3 inflammasome has been demonstrated.8–14 In macrophages, most NLRP3 activators trigger the generation of ROS, and this common pathway engages the NLRP3 inflammasome. The thioredoxin (TRX)-interacting protein (TXNIP), also known as vitamin D3 up-regulated protein-1 or TRX binding protein-2, is a ROS-sensitive NLRP3 ligand that directs ROS-dependent NLRP3 inflammasome activation. 14 TXNIP is an endogenous inhibitor of TRX.15–17 During oxidative stress, TXNIP dissociates from TRX. 17 Free TXNIP interacts with the LRR and NACHT domains of NLRP3, but not with other intracellular PRRs, which results in the assembly of the NLRP3 inflammasome and maturation of pro-IL-1β. 8,14
The endoplasmic reticulum (ER) is a specialized cellular organelle that functions as the site for folding of proteins destined for several cellular compartments or the extracellular milieu. 18 The ER also serves as a site for the biosynthesis for steroids, cholesterol and other lipids, and for the storage and cytosolic release of Ca2+. 18 In addition, the ER can activate several signaling pathways collectively called the unfolded protein response (UPR) when it sustains stress that challenges its function. 19 In eukaryotic cells, three ER-resident transmembrane proteins are crucial for sensing ER stress and transducing signals during the UPR: inositol-requiring enzyme 1 (IRE1), double-stranded RNA-activated protein kinase-(PKR)-like eukaryotic initiation factor 2α kinase (PERK) and the activating transcription factor-6 (ATF6).20–22 During transient and mild ER stress, the UPR restores ER homeostasis by adaptive mechanisms, including the expansion of ER size, enhanced protein folding capacity, suppression of protein synthesis through transcriptional and translational controls, and degradation of unfolded or misfolded proteins.23,24 However, if the stress is persistent and strong, the UPR activates mitochondria-dependent or mitochondria-independent apoptotic pathways.23,25 Several reports suggest that UPR signaling pathways are involved in the activation of NF-κB, the major transcription factor regulating inflammatory processes.26–29 Furthermore, ER stress was recently shown to promote inflammasome activation in macrophages primed with pro-inflammatory stimuli such as LPS or phorbol esters. 30 On the basis of these findings, we hypothesized that ER stress alone may constitute a sufficient signal to stimulate cells to produce IL-1β. Here we show that ER stress can increase pro-IL-1β mRNA synthesis via NF-κB activation. Furthermore, we show that ER stress-induced ROS production activates the NLRP3 inflammasome through binding of a ROS-sensitive NLRP3 ligand TXNIP, resulting in IL-1β cleavage and secretion. These results suggest that ER stress can serve as a sufficient primer and activator for the production of IL-1β.
Materials and methods
Reagents
Tunicamycin (TM), thapsigargin (TG), homocysteine (Hcys) and 4-phenylbutyric acid (4-PBA) were from Sigma-Aldrich (St Louis, MO, USA). The Nuclear/Cytosol Fractionation Kit (K266-25) was from BioVision Research Products (Mountain View, CA, USA). Abs against NF-κB p50 and p65, lamin A/C, caspase-1, TXNIP and β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Abs against α-tubulin and cleaved IL-1β were from Cell Signaling (Danvers, MA, USA). The polyclonal Ab to NLRP3 was from IMGENEX (San Diego, CA, USA). All other chemicals were from Sigma-Aldrich.
Animals
Six-week-old male C57BL/6 mice were purchased from ORIENT (Busan, Korea). The NLRP3-/- mice were provided by Augustine M. K. Choi (Brigham and Women Louis, MO, MOMOSt Louis, MOuis, MOSt Louis, MO, USA). The mice were maintained under specific pathogen-free conditions at 22 ℃ and given access to food and water ad libitum. TM and Hcys were freshly dissolved 0.5% v/v DMSO/saline solution before each experiment. Mice were injected with TM (3 mg/kg, i.p.) or with Hcys (0.5 mg/kg, i.p.). The control mice received identical amounts of vehicle (0.5% DMSO/saline solution). Six h post-injection with TM, or 1 h post-injection with Hcys, mice were sacrificed by cervical dislocation, and then the serum was obtained for experiments.
Cell culture
Cell cultures were maintained at 37℃ in humidified incubators containing an atmosphere of 95% air, 5% CO2. U937 cells, and THP-1 cells [Korean Cell Line Bank (KCLB40202)] were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin/ streptomycin (100 U/100 µg) solution (Gibco, Life Technologies, Grand Island, NY, USA). Murine 3T3-L1 pre-adipocytes (Dr Rena Yu, University of Ulsan) and Raw264.7 cells were grown and maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin solution (Gibco, Life Technologies). Two d after plating, the cells were initiated to differentiate by adding adipogenic agents (1 µM dexamethasone, 10 µg/ml insulin and 0.5 mM 3-isobutyl-1-methylxanthine) in DMEM supplemented with 10% FBS, and 1% penicillin/streptomycin solution. The medium was changed every 48 h until the initiation of the experiments.
Primary macrophage cells
Six-week-old male C57BL/6 mice were purchased from ORIENT (Busan, Korea). Murine peritoneal macrophage cells were isolated from the peritoneal lavage of mice 3 d after injection with 4% sterile thioglycollate. Bone marrow-derived macrophages (BMDM) were isolated from the femurs of mice. Femurs were flushed out with PBS using a sterile needle. The BMDM were cultivated in DMEM medium (Gibco, Life Technologies), containing 10% FBS and 10 ng/ml macrophage colony stimulating factor. After 4–5 d, fully differentiated BMDM had formed on the bottom of the culture plates. The isolated macrophage cells were then incubated at 37 ℃ in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin solution. Macrophages isolated from TXNIP-/- mice were supplied by Dr In-Pyo Choi (Korea Research Institute of Bioscience and Biotechnology, Taejeon, Korea).
Transfection
Small interfering RNA (siRNA) against human TXNIP was purchased from Santa Cruz Biotechnology. U937 cells were transfected using the metafectene method according to the manufacturer Biontex (Planegg, Germany). The interference of TXNIP expression was confirmed by Western blotting using anti-TXNIP Abs. Corresponding scrambled siRNAs were used as transfection controls.
RT-PCR
Total RNA was isolated with the Trizol Reagent (Invitrogen, Carlsbad, CA, USA) from U937 cells or mouse peritoneal macrophages. For RT-PCR, first-strand cDNA was synthesized using 2 µg of total RNA using Moloney murine leukemia virus (M-MLV) reverse-transcriptase and oligo (dT) 15 primer (Promega, Madison, WI, USA). The samples were incubated at 42°.
Semi-quantitative PCR was performed using 2 µl cDNA. Specific primer sequences were as follows: sense strand human pro-IL-1β primer, 5′-CTC TCT CAC CTC TCC TAC TCA C-3′; antisense strand human pro-IL-1β primer, 5′-ACA CTG CTA CTT GCC CC-3′; sense strand human NLRP3 primer, 5′-CTG TGT GTG GGA CTG AAG CAC-3′; antisense strand human NLRP3 primer, 5′-GCA GCC CTG CTG TTT CAG CAC-3′; sense strand human C/EBP homologous protein (CHOP) primer, 5′-CTC CCA GAG CCC TCA CTC TC-3′; antisense strand human CHOP primer, 5′-CAC TTC TGG GAA AGG TGG GT-3′; sense strand human XBP-1(s) primer, 5′-GGG TCC AAG TTG TCC AGA ATG C-3′; antisense strand human XBP-1(s) primer, 5′-TTA CGA GAG AAA ACT CAT GGC-3′; sense strand human GAPDH primer, 5′-CCA CCC ATG GCA AAT TCC ATG GCA-3′; antisense strand human GAPDH primer, 5′-TCT AGA CGG CAG GTC AGG TCC ACC-3′; sense strand mouse NLRP3 primer, 5′-ATG GTA TGC CAG GAG GAC AG-3′; antisense strand mouse NLRP3 primer, 5′-ATG CTC CTT GAC CAG TTG GA-3′; sense strand mouse CHOP primer, 5′-CCC AGG AAA CGA AGA GGA AG-3′; antisense strand mouse CHOP primer, 5′-AGT GCA GTG CAG GGT CAC AT-3′; sense strand mouse XBP-1(s) primer, 5′-GAA CCA GGA GTT AAG AAC ACG-3′; antisense strand mouse XBP-1(s) primer, 5′-AGG CAA CAG TGT CAG AGT CC-3′; sense strand mouse 18S primer, 5′-CAG TGA AAC TGC GAA TGG CT-3′; antisense strand mouse 18S primer, 5′-TGC CTT CCT TGG ATG TGG TA-3′; sense strand mouse GAPDH primer, 5′-AGG CCG GTG CTG AGT ATG TC-3′; antisense strand mouse GAPDH primer, 5′-TGC CTG CTT CAC CTT CT-3′. Primers for mouse IL-1β was supplied by the Bioneer Coporation (N_4009) (Daejeon). The conditions for amplification were as follows: human pro-IL-1β, human NLRP3, human XBP-1(s), mouse NLRP3, mouse XBP-1(s)—denaturation at 94℃ for 30 s, annealing at 56℃ for 30 s, extension at 72℃ for 1 min 30 cycles and a final extension at 72℃ for 10 min; human CHOP, human GAPDH, mouse CHOP, mouse 18S and mouse GAPDH—denaturation at 94℃ for 30 s, annealing at 56℃ for 30 s, extension at 72℃ for 1 min 25 cycles and a final extension at 72℃ for 10 min. The PCR products were detected on 2% agarose gels. Gene expression data from RT-PCR was quantified relative to GAPDH or 18s RNA.
Real-time quantitative RT-PCR
Real-time quantitative RT-PCR (qRT-PCR) was performed using SYBR Green qPCR Master Mix (2×; USB products; Affymetrix, Santa Clara, CA, USA) on an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA). PCR primer pairs were as follows: IL-1β: 5′- TCGCTCAGGGTCACAAGAAA -3′, 5′- ATCAGAGGCAAGGAGGAAACAC -3′; TNF-α: 5′- AGACCCTCACACTCAGATCATC
TTC -3′, 5′- TTGCTACGACGTGGGCTACA -3′; NLRP3: 5′- ATGGTATGCCAGGAGGA GAG -3′, 5′- ATGCTCCTTGACCAGTTGGA -3′; CHOP: 5′- CTGCCTTTCACCTTGGAG AC -3′, 5′- CGTTTCCTGGGGATGAGATA -3′; splicing mXBP-1: 5′- GAGTCCGCAGCAG GTG -3′, 5′- AGG CTTGGTGTATACATGG -3′; GAPDH: 5′GGGAAGCCCATCACCATC T-3′, 5′- CGGCCTCACCCCATTTG-3′.
Treatment with fatty acids
Fatty acids (50 mM palmitate and 50 mM oleate) were dissolved the sodium salt in BSA (0.5:3 fatty acid/albumin) solution. Fatty acid mixtures (1:1, palmitate:oleate) were prepared as previously described. 31 The stock solutions were employed for the experiments in a dilution to a final concentration of 50–500 µM for FFAs.
Immunoblot analysis
Following experimental treatments, cells were harvested and washed twice with ice-cold PBS. Cells were lysed with 1× RIPA buffer containing protease and phosphatase inhibitors. Equal amounts of cell lysates were measured with the BCA protein assay reagent (Pierce Biotechnology, Rockford, IL, USA). The samples were diluted with 2× sample buffer containing β-mercaptoethanol, and then an equal amount of protein was separated on SDS-PAGE gels followed by transfer to polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat milk in PBS containing 0.1% Tween 20 (PBS-T) for 20 min and then incubated overnight (18 h) with Abs in PBS-T containing 1% nonfat milk. The blots were developed with a peroxidase-conjugated secondary Ab and reacted proteins were visualized using the ECL Plus Western Blotting Detection System (GE Healthcare Life Sciences, Buckinghamshire, UK). The relative signal intensity of the bands was determined and standardized using Scion Image Software (Scion Corporation, Frederick, MD, USA).
Immunoprecipitation
For immunoprecipitation studies, U937 cells were collected and gently re-suspended in 1×RIPA buffer containing protease and phosphatase inhibitors. Total protein was determined using a standard BCA protein assay and then anti-TXNIP Ab (sc-271237) was added to equal amounts of each sample. The samples were incubated overnight with rotation in a cold room (4℃). After overnight incubation, Dynabeads (100.03D; Invitrogen) were added to each sample and rotated again in the cold room for 1 h. A magnet was applied, and then the supernatant was removed and the beads were washed three times with 1× PBS. The bound protein was eluted off the beads by boiling in 2× sample buffer containing β-mercaptoethanol for 5 min. Immunoprecipitated proteins were assayed by Western immunoblotting.
Measurement of intracellular ROS
Intracellular ROS production was measured using the oxidant-sensitive fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (C6827; Invitrogen). Cells were exposed to TM (10 µg/ml) for the indicated intervals (3–9 h) and then treated with 5 µM DCF-DA for 10 min. 2′,7′-dichlorofluorescein (DCF) fluorescence (excitation, 488 nm; emission, 520 nm) was imaged on a laser confocal scanning microscope (Olympus Flouview-FV300 Laser Scanning Confocal system; Olympus, Tokyo, Japan). For FACS analyses, cells were harvested and washed twice with 1% BSA solution and fluorescence intensity was measured by flow cytometry (excitation at 488 nm; emission at 600 nm) (FACScanto II flow cytometer; BD Biosciences, San Jose, CA, USA).
ELISA analyses
Cell culture supernatants were assayed for cytokine production by ELISA using a commercial cytokine ELISA duo set kit for human IL-1β (DY201; R&D Systems, Minneapolis, MN, USA), mouse IL-1β (DY401, R&D Systems), mouse TNF-α (DY410; R&D Systems) and human TNF-α (2637 KI; BD Bioscience).
High-fat diet in mice
C57BL/6 mice were fed a high fat diet (D12492; Research Diet, New Brunswick, NJ, USA), or normal diet for 16 wk. Thirty mice were divided into three groups: normal diet group (ND; n = 10); high-fat diet group (HFD; n = 10); carbon monoxide-treated high-fat diet group (HFD + CO or H + CO; n = 10); and carbon monoxide-treated normal diet group (N + CO). Animals in the HFD + CO and N + CO groups were treated with CO (250 ppm) daily for 2 h for 10 wk in specialized chambers.
Streptozotocin-induced diabetes
C57BL/6 mice were given a daily i.p. injection with streptozotocin (STZ) (40 µg/g body mass in 0.1 µM citrate buffer) for 5 d to induce diabetes. Control mice were injected with buffer alone. Blood Glc levels were examined 3 d after the final STZ injection by obtaining blood from the lateral saphenous vein and measuring Glc concentrations with a glucoanalyzer (SD BIOSENSOR Instruments, CodeFree, Suwon-si, Korea). Mice with blood Glc levels > 300 mg/dl were considered diabetic. Thirty mice were divided into three groups: control group (CON; n = 6); diabetic group (STZ; n = 12); and carbon monoxide-treated diabetic group (STZ + CO; n = 12). To evaluate any effects of inhaled CO, the STZ + CO group was treated with 250 ppm CO daily for 3 h after STZ injection in specialized chambers. CO was mixed with compressed air using a gas mixer to deliver 250 ppm. At 30 d after the STZ injection, mice were sacrificed and their kidneys were immediately fixed in PBS containing 10% formaldehyde for histological analysis.
Histological analysis
The kidneys were removed from each mouse and fixed in PBS containing 10% formaldehyde and embedded in paraffin. Each sample was then cut into 5 -µm sections, and the sections were mounted on glass slides and depleted of paraffin with xylene. The sections were subjected to standard hematoxylin and eosin (H&E) staining. All renal H&E sections were observed with a light microscope (400× magnification).
Statistical analyses
Data were expressed as mean tioD. Statistical analysis was performed by nonparametric t-tests for paired data using GraphPad Prism (GraphPad Software, La Jolla, CA, USA). A P-value of < 0.05 was considered to represent a statistically significant change.
Results
ER stress induces IL-1β production in vitro and in vivo
To determine whether disturbance of ER homeostasis can induce the release of the pro-inflammatory cytokine IL-1β from leukocytes, the human leukemic monocyte cell line (U937) was treated with the chemical ER stress-inducing compounds TM (10 µg/ml), Tg (10 µM) or Hcys (10 mM), and the secretion of IL-1β was quantified by ELISA. Treatment with ER stress-inducing compounds time-dependently increased the levels of secreted IL-1β in U937 cells (Figure 1A; Supplementary Figure 1A, B). Treatment of mouse primary peritoneal macrophages (Figure 1B), primary BMDM (Figure 1C) and differentiated 3T3-L1 cells (Supplementary Figure 1C) with TM induced the secretion of IL-1β in a dose- (Figure 1C; Supplementary Figure 1C) or time-dependent manner (Supplementary Figure 1D and E). Treatment with TM also caused a time-dependent increase in TNF-α secretion in U937 cells and primary peritoneal macrophages (Supplementary Figure 1F, G). In contrast, mouse embryonic fibroblasts exposed to TM did not secrete IL-1β, but were able to secrete TNF-α (data not shown).
ER stress induces the production of IL-1β in vitro and in vivo. (A) Human leukemic monocyte cells (U937) were treated with the chemical ER-stress inducing compounds TM (10 µg/ml) for the indicated time intervals and the secretion of IL-1β was quantified by ELISA. Mouse primary peritoneal macrophages (B) and primary BMDM (C) were treated with TM for 18 h at the indicated concentrations and the secretion of IL-1β was quantified by ELISA. (D) C57BL/6 mice (n = 6) were injected with TM (3 mg/kg, i.p.). Mice were sacrificed, and then blood was collected. Serum IL-1β was quantified by ELISA. Liver tissue was collected for the analysis of CHOP mRNA by RT-PCR. 18s rRNA served as the standard (lower panel). U937 cells (E) or BMDM (F) were treated with palmitate for 24 h at the indicated concentrations or with 200 µM at the indicated times, and the secretion of IL-1β was quantified by ELISA. Data represent mean ± SD of three independent determinations. *P < 0.05, **P < 0.01, ***P < 0.001. Blots shown are representative of three independent experiments.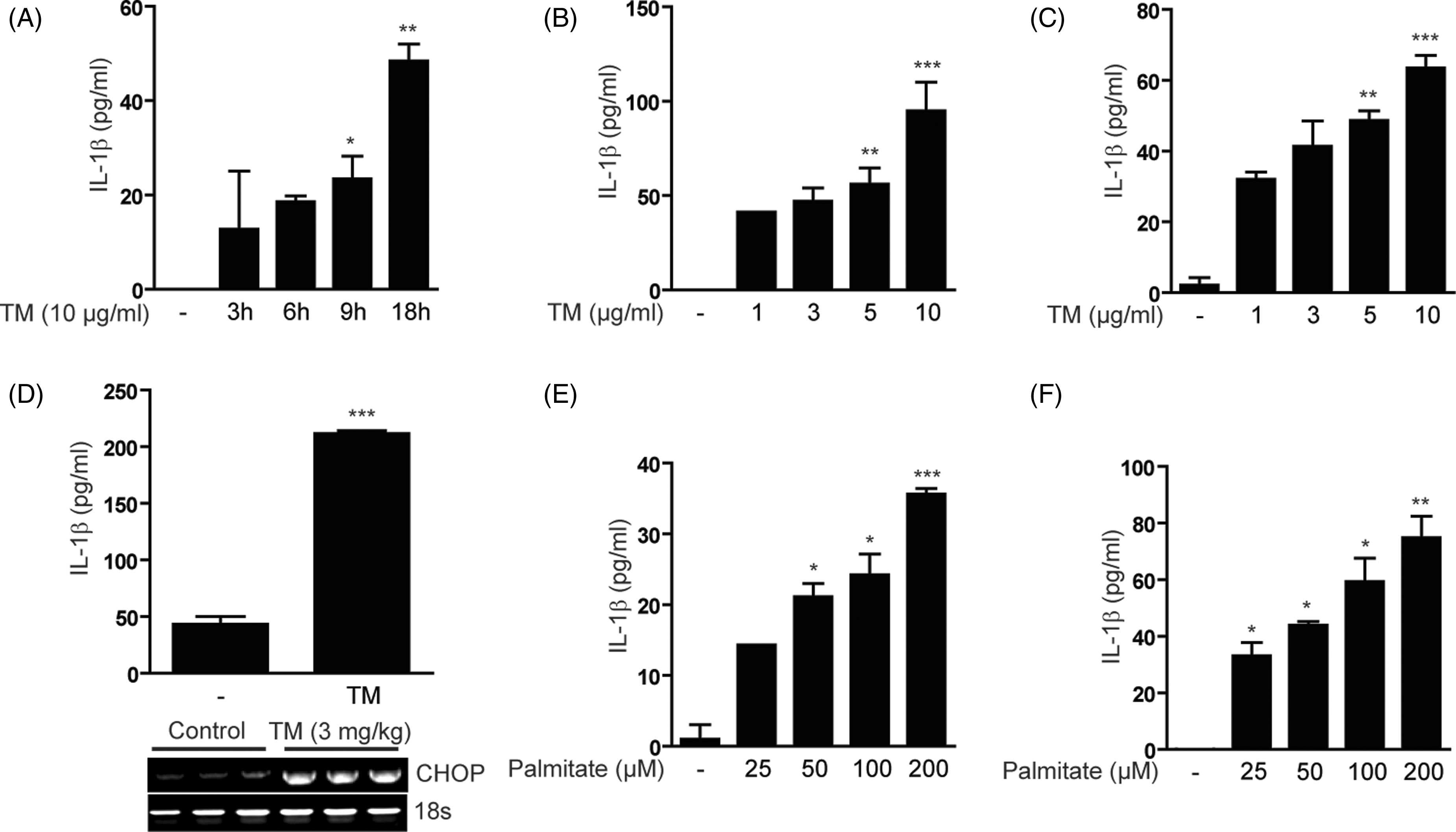
To determine the effect of ER stress on IL-1β production in an in vivo model, C57BL/6 mice were injected with TM (Figure 1D). Mice were sacrificed, and then blood was collected for the analysis of serum IL-1β, and liver tissue was collected for the analysis of ER stress markers. Significant amounts of secreted IL-1β were detected in the serum of TM-injected mice compared to control mice. Furthermore, the expression of CHOP mRNA, a marker of ER stress, was increased in the liver of mice injected with TM (Figure 1D, lower panel). Serum IL-1β was also elevated in mice injected with Hcys (Supplementary Figure 1H).
Saturated fatty acids (i.e. palmitate) have been recently shown to cause ER stress by disrupting the structure and integrity of the ER.32,33 We examined the physiological effects of ER stress induced by palmitate on IL-1β secretion in U937 and BMDM cells. Palmitate treatment dose- and time-dependently increased IL-1β secretion from U937 cells (Figure 1E; Supplementary Figure 1I). Dose-dependency of this response was also observed in BMDM (Figure 1F). Together, these data suggest that immune cells subjected to ER stress triggered by fatty acids can produce IL-1β.
ER stress induces the expression of pro-IL1β mRNA via activation of NF-κB
The production of IL-1β requires two steps of priming and activation, including the expression of pro-IL-1β and the activation of inflammasome.3,4 The first step, which induces the transcription of pro-IL-1β, can be achieved by activation of NF-κB and MAPK signaling in cells primed with various pro-inflammatory stimuli, including TNF-α. To test whether ER stress can induce pro-IL-1β expression, U937 cells, primary peritoneal macrophages, BMDM and differentiated adipocytes were treated with TM. In all cell types tested, pro-IL-1β mRNA expression was induced by TM in a dose-dependent manner (Supplementary Figure 2A, C, D, F, G). The levels of pro-IL-1β expression reached a peak at 3 h with treatment of 10 µg/ml of TM (Figure 2A; Supplementary Figure 2B, E, H, I). In addition, in various macrophages cells and adipocytes, NLRP3, CHOP and sXBP-1 mRNA expression was significantly increased by TM in a fashion similar to that of pro-IL-1β expression (Figure 2B–D; Supplementary Figure 2A-I). Similar to TM treatment, Tg and Hcys treatment induced pro-IL-1β, NLRP3 and CHOP mRNA expression (Figure 2E; Supplementary Figure 2J). In order to confirm the physiological effects of ER stress-induced priming, RAW 264.7 cells were treated with palmitate. Pro-IL-1β mRNA expression was time- and dose-dependently increased in palmitate-treated RAW 264.7 cells (Figure 2F; Supplementary Figure 2K–M). Consistent with the stimulation of ER stress, the mRNA levels of CHOP were dramatically increased by palmitate treatment in a time- and dose-dependent fashion (Supplementary Figure 2K–M). However, palmitate cannot reflect physiological concentrations of fatty acids because plasma contains a variety of long-chain fatty acids, such that about 35% are saturated and 65% are unsaturated. Thus, macrophage cells were treated with fatty acid cocktail.
31
The fatty acid cocktail increased the production of IL-1β in macrophage cell lines (Supplementary Figure 2N). Also, pro-IL-β and CHOP mRNA expression were increased by fatty acid cocktail (Supplementary Figure 2O). towing to the fact that, in many cases, oleate prevents the adverse effects of palmitate, we also investigated the effect of oleate on inflammasome activation. In contrast to the effects of palmitate on inflammsome, oleate did not induce IL-1β expression. Thus, saturated fatty acids (palmitate) but not unsaturated fatty acids (oleate) can induce IL-1β production (Figure 2F).
ER stress induces the expression of pro-IL-1β mRNA via the activation of NF-κB. RAW 264.7 cells (A–D) were treated with TM, and then mRNA expression for pro-IL-1β, NLRP3, CHOP and XBP-1(s) was determined by quantitative real-time PCR. (E) Primary BMDM were treated with TG or Hcys for 6 h at the indicated concentrations, and then mRNA expression for pro-IL-1β, NLRP3 and CHOP were determined by PCR. (F) RAW 264.7 cells were treated with the saturated fatty acids palmitate (16:0) or equivalent monounsaturated fatty acids (oleate, 18:1) for 6 h at the indicated concentration and mRNA expression for pro-IL-1β was determined by quantitative real-time PCR (right panel). (G) U937 leukemia cells were treated with TM (10 µg/ml), and then the level of RelB/p65 and p50 in cytosolic (upper panel) and nuclear (lower panel) extracts was determined by Western immunoblot analysis. β-Actin was served as the standard. Lamin A/C was served as the standard for nuclear extracts. (H) Peritoneal macrophages were treated with TM (10 µg/ml) for 6 h. The expression of RelB/p65 and p50 subunits of NF-κB was determined in cytosolic and nuclear extracts. β-Actin was served as the standard. Lamin A/C was served as the standard for nuclear extracts. (I) U937 (left panel) and THP-1 leukemia cells (right panel) were treated with TM (10 µg/ml) in the absence or presence of the NF-κB translocation inhibitor SN50 (20 µM). The expression of pro-IL-1β mRNA was determined by RT-PCR. GAPDH or 18s rRNA was served as the standard. Blots shown are representative of three independent experiments. NE: nuclear extracts; CE: cytoplasmic extracts.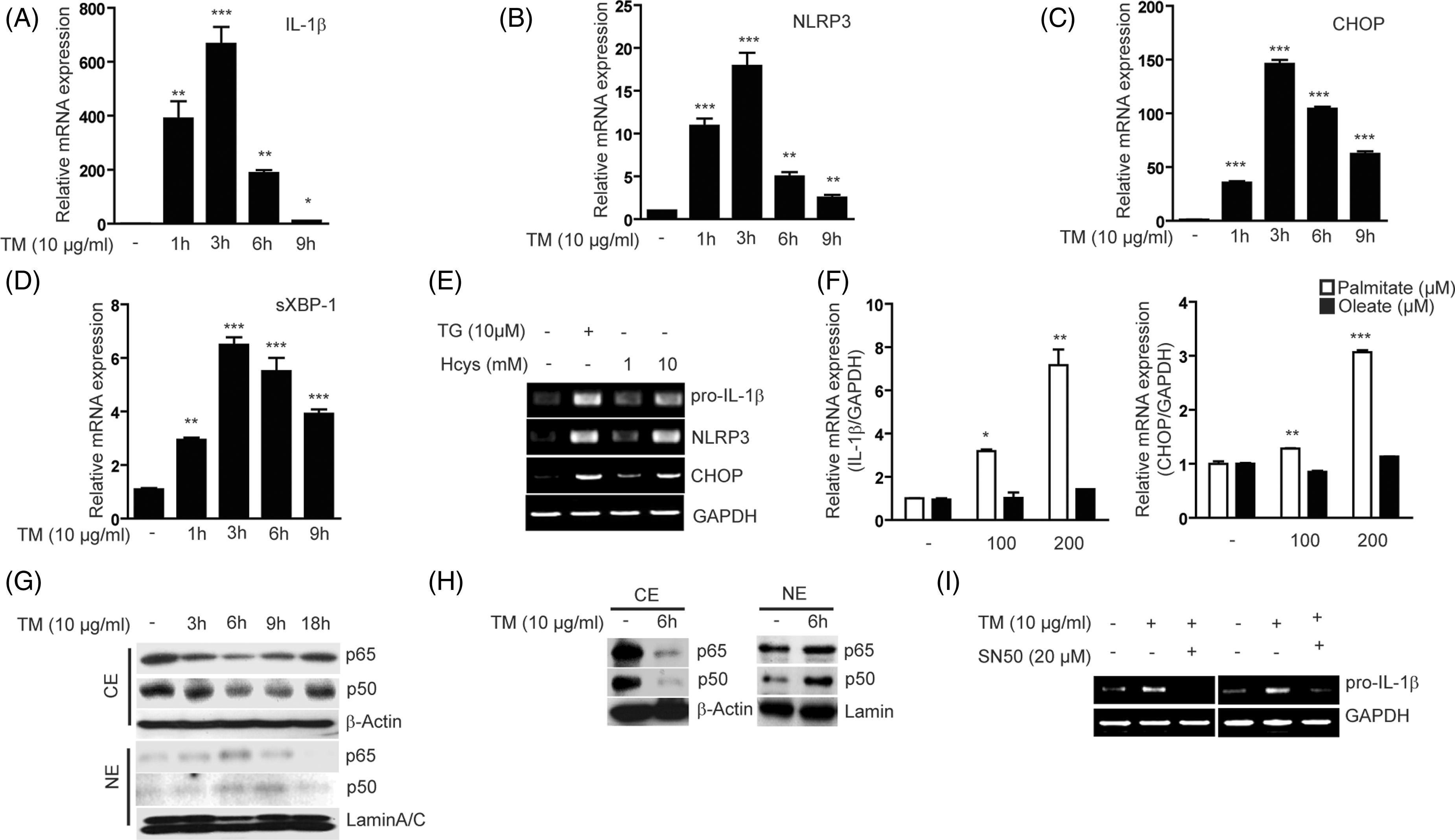
The UPR can activate NF-κB by reducing the level of IκB (inhibitor of NF-κB) during UPR activation. Therefore, we examined whether TM treatment reduces the level of IκB in leukemia cell lines and macrophages. As expected, the level of IκB in U937 cells was substantially reduced within 3 h of TM treatment (Supplementary Figure 2P). Consistent with this result, the relative nuclear translocation of RelB/p65 and p50, a subunit of NF-κB, was increased (Figure 2G, lower panel), whereas the cytoplasmic levels of RelB/p65 and p50 were decreased in TM-treated U937 cells (Figure 2G, upper panel). In addition, the nuclear translocation of the RelB/p65 and p50 subunits of NF-κB was also detected in peritoneal macrophages treated with TM for 6 h (Figure 2H). Finally, the expression of pro-IL-1β mRNA by TM treatment was reduced to the basal level by pre-incubation with SN50, a cell-permeable specific inhibitor of NF-κB translocation, in both U937 and THP-1 cells (Figure 2I). Thus, these data suggest that ER stress can induce the expression of pro-IL-1β mRNA through the activation of NF-κB.
ER stress stimulates pro-caspase-1 activation
The second step for IL-1β production requires activation of the inflammasome complex to cause the autocatalytic cleavage of pro-caspase-1 to caspase-1, which can convert inactive pro-IL-1β into its bioactive and secreted form.3,4 We next investigated whether treatment with ER stress inducers can activate the inflammasome complex which results in the autocatalytic cleavage of pro-caspase-1 to caspase-1. Treatment of U937 cells, THP-1 cells, adipocytes and primary macrophages with TM increased the amount of cleaved caspase-1 in a time- or dose-dependent manner (Figure 3A–C; Supplementary Figure 3). Together, these data suggest that the ER stress can stimulate pro-caspase-1 activation.
ER stress stimulates pro-caspase-1 activation. U937 leukemia cells (A), differentiated adipocytes (B) and primary macrophages (C) were treated with TM (10 µg/ml) for the indicated times or at the indicated doses for 18 h. The expression of pro-caspase-1 and the formation of cleaved caspase-1 were determined by Western immunoblot analysis. β-Actin (A) or α-tubulin (B, C) served as the standard. Additionally, IL-1β secretion was determined in the supernatant of U937 cells. Non-specific immunoreactivity was used as the standard for secreted protein (A). Blots shown are representative of three independent experiments. SN: supernatant.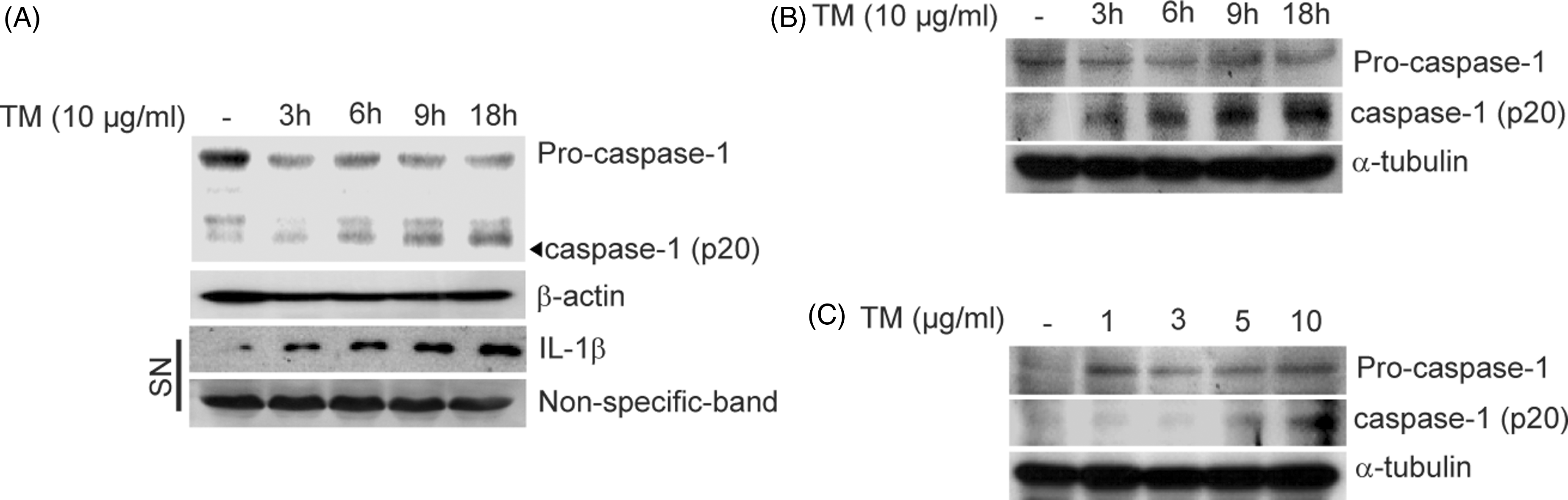
Chemical chaperone treatment attenuates ER stress-mediated IL-1β production
Chemical chaperones, such as 4-PBA and tauroursodeoxycholic acid (TUDCA) can attenuate the UPR and thereby alleviate ER stress-mediated toxicity in cultured cells and whole animals.
34
Therefore, we investigated whether chemical chaperone treatment can reduce ER stress-dependent IL-1β production. The effect of a chemical chaperone, 4-PBA, was analyzed by determining the level of pro-IL1β mRNA, the nuclear translocation of NF-κB, and the amount of secreted IL-1β in TM-treated U937 cells. Co-incubation of 4-PBA (0.5 mM) with TM overnight reduced the expression of pro-IL-1β transcripts to a barely detectable level in U937 cells in which expression of CHOP transcripts was also significantly reduced (Figure 4A). Consistent with the prevention of pro-IL-1β mRNA expression, 4-PBA treatment significantly blocked the nuclear translocation of the RelB/p65 and p50 subunit of NF-κB in a dose-dependent manner (Figure 4B). As expected, 4-PBA (0.5 or 1 mM) treatment decreased ER stress induced IL-1β secretion (Figure 4C). Moreover, treatment with another chemical chaperone, TUDCA (0.1 mM) caused a stronger inhibitory effect on IL-1β production in TM-treated U937 cells (Figure 4D). We also determined whether antioxidant treatment could affect the production of IL-1β from cells undergoing ER stress. Treatment with the general antioxidant N-acetyl- Chemical chaperone treatment attenuates ER stress-mediated IL-1β production. U937 cells (A–C) were treated with TM (10 µg/ml) for 18 h in the absence or presence of 4-PBA (0.5 mM). (A) The level of pro-IL-1β and CHOP mRNA was analyzed by PCR. GAPDH served as the standard. (B) The nuclear translocation of the RelB/p65 and p50 subunit of NF-κB was determined by Western immunoblot analysis. β-Actin served as the standard. Lamin A/C was served as the standard for nuclear extracts. (C) IL-1β secretion in U937 supernatants was determined by ELISA. (D, E) U937 cells were treated with TM (10 µg/ml) for 18 h, in the absence or presence of TUDCA (0.1 mM) or NAC (25 mM). IL-1β secretion in U937 supernatants was determined by ELISA. (F) C57BL/6 mice were pre-injected with TUDCA (n = 10, 500 mg/kg/d i.p.) or pre-treated with 4-PBA (n = 10, 1 µg/kg/d oral gavage) for 20 d. At the end of the 20 d pre-treatment, mice were injected 20 d later with TM (3 mg/kg, i.p.). Mice were sacrificed at 6 h post-injection and then blood was collected for the analysis of serum IL-1β (left and center panels). Liver tissue was collected for the analysis of pro-IL-1β, NLRP3 and CHOP mRNA expression levels in the liver by RT-PCR. 18s RNA was served as the standard (Figure 4F, right panel). U937 cells were pre-treated for 1 h with (G) 4-PBA (1 mM), (H) TUDCA or for 5 min with NAC (I) at the indicated concentrations, and then treated with palmitate (200 µM) for 18 h. IL-1β secretion in U937 supernatants was determined by ELISA. Data represent mean ± SD of three independent determinations. *P < 0.05, **P < 0.01. Blots shown are representative of three independent experiments. NE: nuclear extracts; CE: cytoplasmic extracts.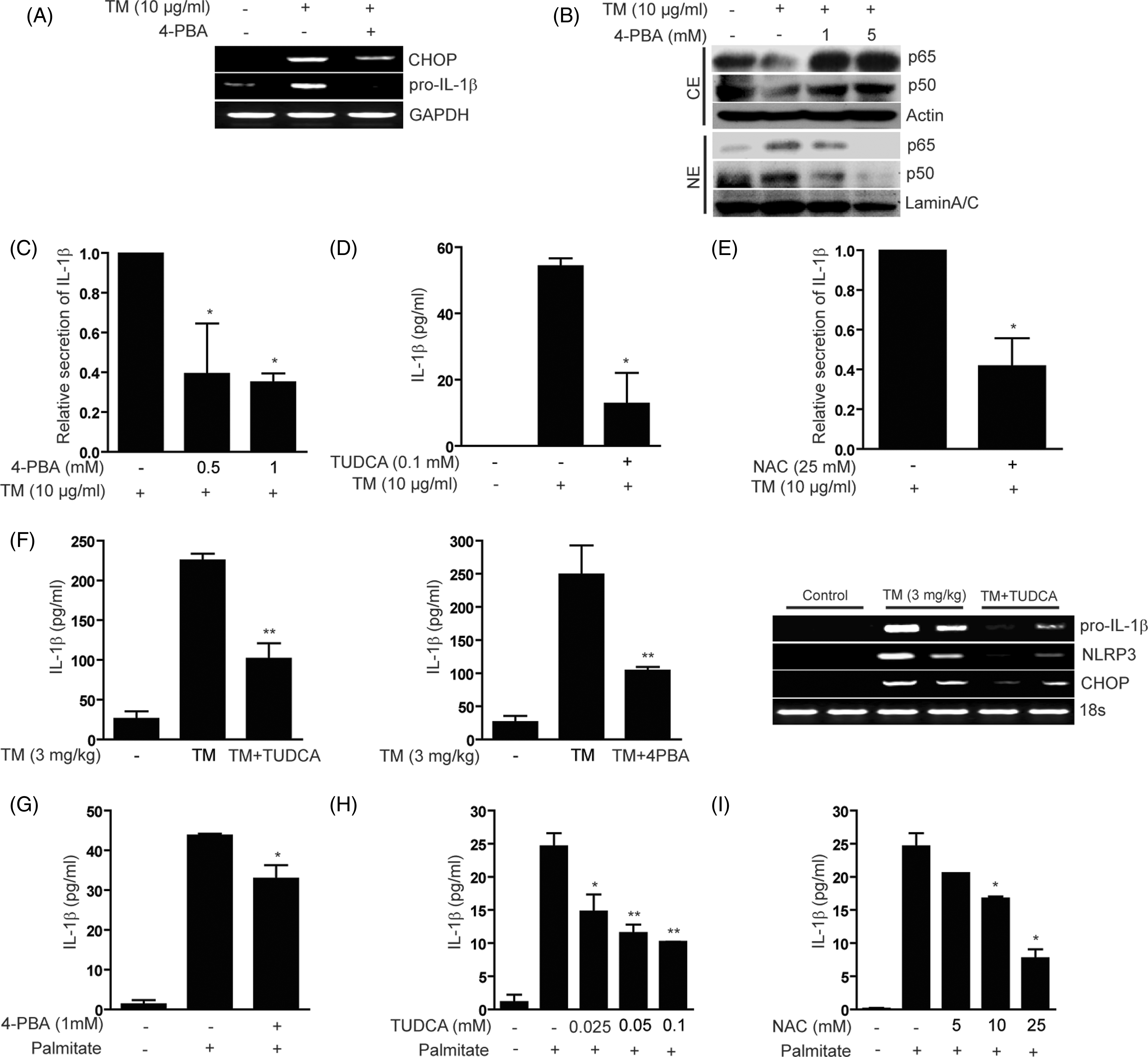
To determine the inhibitory effect of chemical chaperones on IL-1β production in an in vivo model, C57BL/6 mice were pre-injected with 4-PBA or TUDCA, and then injected 20 d later with TM. Mice were sacrificed at 6 h, and then blood was collected for the analysis of serum IL-1β, and liver tissue was collected for the analysis of pro-IL-1β, NLRP3 and ER stress markers. Mice injected with TM displayed increased secretion of lL-1β. Pre-injection with chemical chaperones inhibited IL-1β production in the serum induced by TM (Figure 4F, left and center panels). Furthermore, pro-IL1β, NLRP3 and CHOP mRNA expression levels in the liver were inhibited by pre-injection of chemical chaperones (Figure 4F, right panel). Moreover, pre-treatment with chemical chaperones (Figure 4G, H) or the antioxidant NAC (Figure 4I) significantly inhibited palmitate-induced secretion of IL-1β from U937 cells in vitro. These results collectively affirmed that ER stress can act as a primer for IL-1β activation in both in vitro and in vivo systems.
ER stress-mediated ROS accumulation leads to activation of NLRP3 inflammasome through binding of TXNIP
ER stress can produce ROS as byproducts of the oxidative protein folding pathway during disulfide bond formation within the ER,35,36 and through the stimulation of mitochondrial respiration by increased mitochondrial loading of calcium released from the ER.37,38 In addition, activation of the NLRP3 inflammasome is dependent on the generation of ROS.9–14 As the consequence of ROS accumulation, TXNIP is released from oxidized TRX and, in turn, binds NLRP3 to stimulate activation of pro-caspase-1.
14
As described above, treatment with the antioxidant NAC suppressed IL-1β production in monocytes after treatment with TM or palmitate (Figure 4E, I). To further explore the mechanisms by which ER stress induces IL-1β production, we investigated whether ER stress-mediated ROS production can induce TXNIP binding to NLRP3 in order to induce maturation and secretion of IL-1β. To confirm the production of ROS by ER stress inducers, TM-treated U937 cells were evaluated by confocal microscopy (Figure 5A), microplate assays (Supplementary Figure 4A) and flow cytometry (Figure 5C). ROS production in U937 cells was time-dependently increased by TM (Figure 5A, B; Supplementary Figure 4A). Next, we examined whether CO treatment could modify ROS production induced by TM. Pre-treatment with CORM-2 inhibited ER stress-induced ROS production (Figure 5B, right panel).
ER stress-mediated ROS accumulation leads to the activation of NLRP3 inflammasome through TXNIP. (A, B) U937 cells were treated with TM (10 µg/ml) for the indicated times and then stained with DCF-DA. ROS production was evaluated using DCF-DA fluorescence measured by confocal microscopy (A) or flow cytometric assays (B). U937 cells were pre-treated with CORM-2 (40 µM) or NAC (5 mM), and then treated with TM (10 µg/ml) for the indicated times (B, left panel) or for 9 h (B, right panel). (C, D) U937 cells were treated with TM for the indicated times (C), or for 9 h in the absence (C) or presence (D) of NAC (5 mM). Lysates were immunoprecipitated with anti-TXNIP and then immunoblotted with anti-NLRP3. IgG was served as the immunoprecipitation control. (E) U937 cells were transfected with specific siRNA against TXNIP or control siRNA (scramble), and the level of endogenous TXNIP was measured by Western blot analysis (upper panel). Under these conditions, the amount of secreted IL-1β was measured from cultured media of U937 cells in the absence or presence of TM treatment (10 µg/ml) (lower panel). (F) Peritoneal macrophages isolated from TXNIP knockout mice (Txnip-/-) were subjected to TM treatment (10 µg/ml), and the production of IL-1β was measured by ELISA. (G) NLRP3-/- mice (n = 6) were exposed to TM (3 mg/kg i.p.). Serum was collected 6 h post-injection and evaluated for the secretion of IL-1β by ELISA. Data represent mean ± S.D of three independent determinations. *P < 0.05, **P < 0.01. Blots shown are representative of three independent experiments.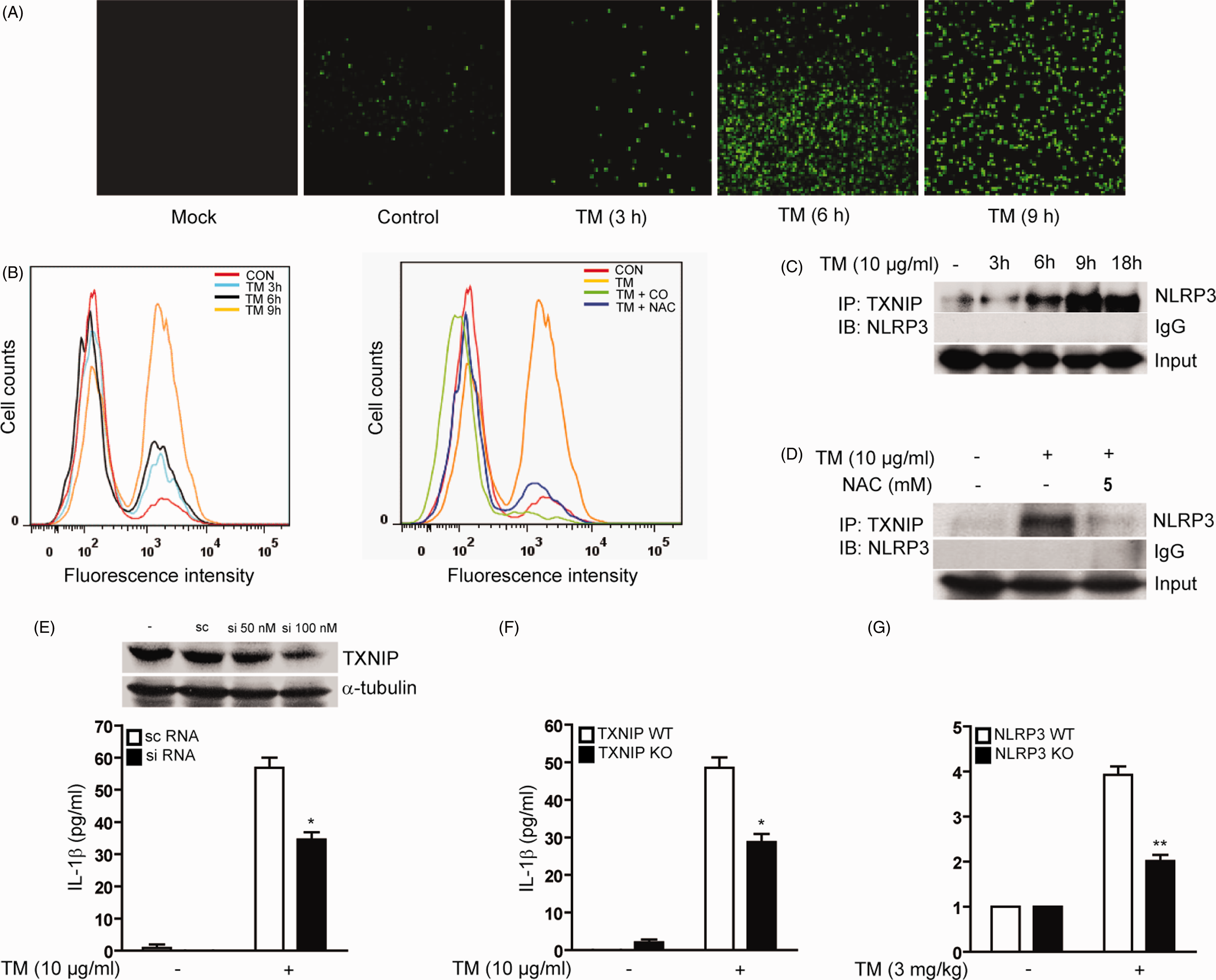
To determine whether the binding of TXNIP to NLRP3 occurs during ER stress conditions, a co-immunoprecipitation assay was performed using a specific Ab against TXNIP to immunoprecipitate lysates from U937 cells treated with TM for the indicated time points (Figure 5C). The immunoblot analysis with NLRP3 Ab revealed that TM treatment induced the binding of TXNIP to NLRP3, which gradually increased to reach saturation at 9 h following ER stress activation (Figure 5C). Next, to address the involvement of ER stress-mediated ROS generation in the interaction between TXNIP and NLRP3, we tested the effect of the antioxidant NAC on the binding of TXNIP to NLRP3 in TM-treated cells. Pretreatment with NAC (5 mM) substantially reduced the binding of TXNIP to NLRP3 induced by prolonged treatment of TM (Figure 5D). In agreement with this result, activation of pro-caspase-1 to cleaved caspase-1 by TM treatment was completely inhibited by incubation with NAC (Supplementary Figure 4B). Taken together, these results suggest that ROS accumulation in ER stressed cells has an essential role in the assembly and activation of the NLRP3 inflammasome that regulates IL-1β secretion.
TXNIP is an important factor for activation of the NLRP3 inflammasome during ER stress
To further verify the requirement of TXNIP in the activation of NLRP3 inflammasome by ER stress, we investigated the effect of the siRNA-dependent knockdown of endogenous TXNIP on IL-1β production in TM-treated cells. By transfection of specific siRNA against TXNIP, the levels of endogenous TXNIP were reduced by ∼ 50% compared with scramble siRNA-transfected U937 cells (Figure 5E, upper panel). Under these conditions, the amount of secreted IL-1β was measured from cultured media of U937 cells in the absence or presence of TM treatment. Knockdown of endogenous TXNIP by siRNA transfection significantly reduced the level of secreted IL-1β by ∼ 67% compared to scramble siRNA transfected cells (Figure 5E, lower panel). Finally, we confirmed the requirement for TXNIP in the activation of ER stress-mediated NLRP3 inflammasome activation using peritoneal macrophages isolated from TXNIP knockout mice (Txnip-/-). During ER stress, TXNIP deficiency in macrophages was associated with the reduced production of IL-1β by ∼ 60% compared to that observed in wild type macrophages (Figure 5F). Thus, these data demonstrate that during ER stress, TXNIP works as an important factor in activating the NLRP3 inflammasome to produce the pro-inflammatory cytokine IL-1β. To validate the specific role of NLRP3 in the responses, we exposed NLRP3-/- mice to TM (3 mg/kg, i.p.) and then isolated the liver or serum after 6 h. Hepatocytes from TM-treated NLRP3-/- mice displayed equivalent expression of pro-IL-1β and CHOP mRNA in the liver under basal conditions and after treatment with TM (Supplementary Figure 4C). In contrast, the activation of caspase-1 following TM stimulation was dramatically reduced in NLRP3-/- hepatocytes relative to wild type hepatocytes (Supplementary Figure 4D). Furthermore, the serum level of IL-1β in response to TM was also reduced in NLRP3-/- mice relative to wild type mice (Figure 5G).
The heme oxygenase-1/CO system inhibits both pro-IL-1β induction and triggering of inflammasome signaling for mature IL-1β excretion
CO arises endogenously as the by-product of the cytoprotective heme oxygenase (HO; E.C. 1:14:99:3) enzyme system. 39 When applied at low concentration, CO can exert anti-inflammatory, anti-proliferative and anti-apoptotic effects in a variety of models of cellular injury. 40 To investigate whether the CO/HO-1 system could suppress IL-1β production in response to ER stress inducers, U937 cells were treated with ER stress in the presence of the HO-1 inducer cobalt-protoporphyrin-IX (CoPP).
CoPP treatment markedly inhibited the secretion of IL-1β in U937 cells (Figure 6A), THP-1 cells (Supplementary Figure 5A) and peritoneal macrophages (Supplementary Figure 5B) in response to TM, as determined by ELISA. Consistent with the prevention of IL-1β secretion, CoPP treatment significantly inhibited pro-IL-1β (Figure 6B), NLRP3 (Figure 6C) and CHOP (Figure 6D) mRNA expression in a dose-dependent manner in U937 cells, and pro-IL-1 and NLRP3 mRNA expression in peritoneal macrophages (Figure 6E). CoPP treatment inhibited the activation (cleavage) of caspase-1 (Figure 6F) and dose-dependently inhibited the production of IL-1β (Supplementary Figure 5C) in response to TM treatment. CoPP treatment also dose-dependently inhibited TNF-α production in U937 and THP-1 cells (Supplementary Figure 5D, E).
The HO-1/CO system inhibits pro-IL-1β synthesis and IL-1β maturation. U937 cells (A, F), Raw264.7 cells (B–D), or peritoneal macrophages (E) were treated with TM (10 µg/ml for 18 h) in the absence or presence of CoPP (1–10 µM, as indicated). (A) The secretion of IL-1β was determined by ELISA; (B) pro-IL-1β, (C) NLRP3 and (D) CHOP mRNA expression was determined by quantitative real-time PCR, with GAPDH as the standard; (E) pro-IL-1β and NLRP3 mRNA expression was determined by RT-PCR, with 18s rRNA as the standard; (F) the activation (cleavage) of caspase-1, with β-actin as the standard, were determined by Western analysis. U937 cells (G, L, M), Raw264.7 cells (H–J), or peritoneal macrophages (K) were treated with TM (10 µg/ml for 18 h) in the absence or presence of CORM-2 (0, 20, 40, 80 µM, as indicated). (G, M) The secretion of IL-1β was determined by ELISA; (H) pro-IL-1β, (I) NLRP3 and CHOP (J) mRNA expression was determined by quantitative real-time PCR, with GAPDH as the standard; (K) pro-IL-1β and NLRP3 mRNA expression was determined by RT-PCR, with 18s rRNA as the standard; (L) The activation (cleavage) of caspase-1, with β-actin as the standard, was determined by Western analysis.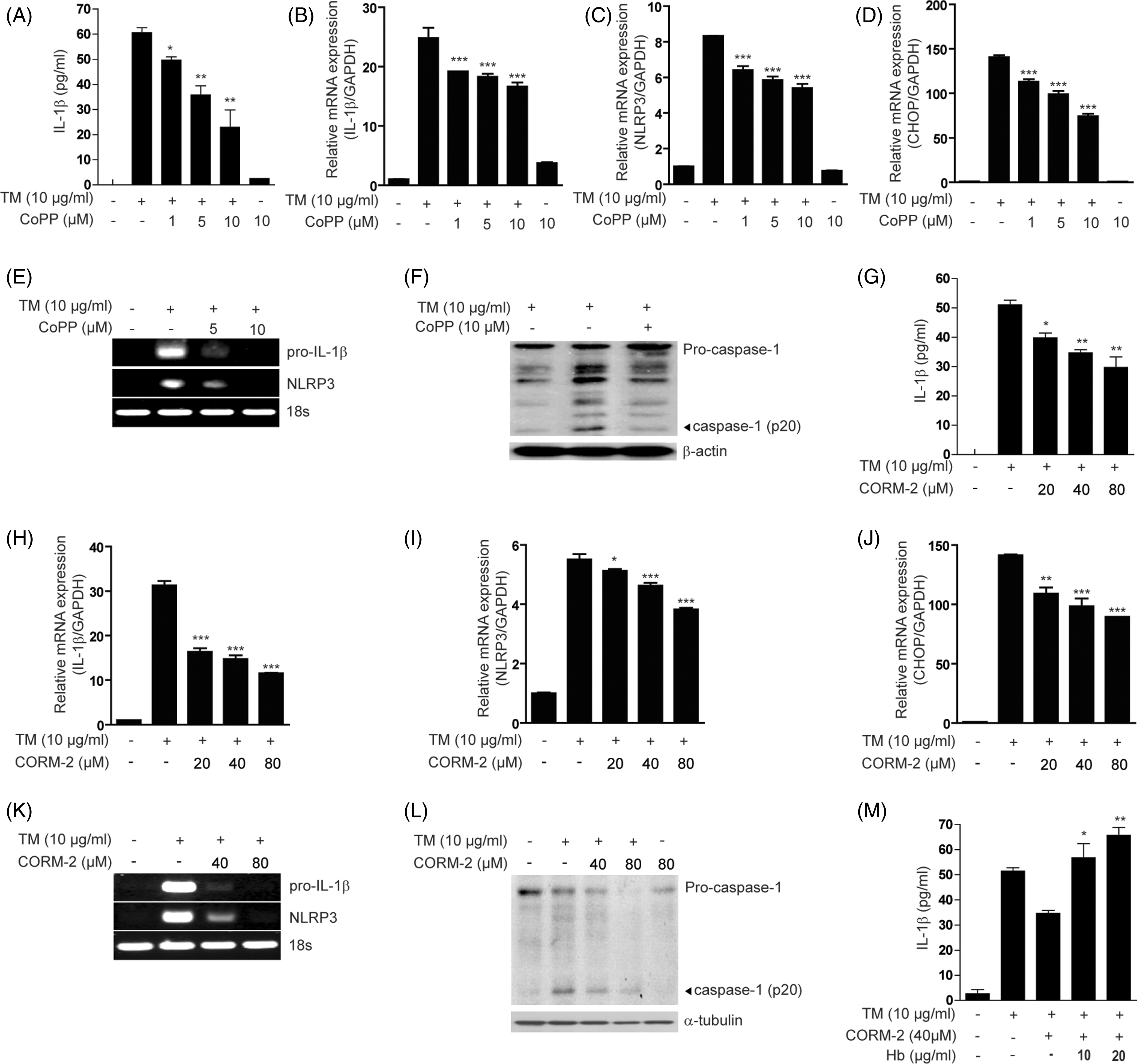
Next, we investigated the effect of pharmacological application of CO using the CO-releasing molecule (CORM-2) on the priming and triggering steps of IL-1β secretion.
CORM-2 treatment markedly inhibited the secretion of IL-1β and TNF-α following TM treatment in U937 cells (Figure 6G; Supplementary Figure 5F) and peritoneal macrophages (Supplementary Figure 5G). CORM-2 treatment significantly inhibited pro-IL-1β (Figure 6H), NLRP3 (Figure 6I) and CHOP (Figure 6J) mRNA expression in a dose-dependent manner in U937, and pro-IL-1 and NLRP3 mRNA expression in peritoneal macrophages following TM treatment (Figure 6K). Furthermore, CORM-2 treatment inhibited the activation of caspase-1 (Figure 6L) induced by ER stress conditions.
Finally, the inhibitory effect of CORM-2 on the IL-1β secretion induced by TM was reversed by pre-incubation with hemoglobin, a CO scavenger (Figure 6M). Taken together, these data suggest that the HO-1/CO system inhibits IL-1β production by ER stress.
CO inhalation inhibits inflammasome-associated cytokine production and reduces inflammation in HFD mice and STZ-induced diabetic model
We next sought to determine whether CO could modulate inflammasome cytokine production in HFD mice and in an in vivo model of STZ-induced diabetes.33,41–43
First, we determined whether CO could reverse HFD-induced ER stress and associated production of IL-1β. CHOP expression, a marker of ER stress, was increased in the liver tissue of C57BL/6 mice on HFD compared with mice on a normal diet. In contrast, CO-treatment reduced CHOP expression in liver tissue of HFD mice. Furthermore HFD caused a decrease in the expression of HO-1 in liver tissue, which was significantly increased in mice subjected to CO inhalation (Figure 7A). Significant amounts of pro-IL-1β and TNF-α were detected in the liver tissue of HFD mice compared with mice on a normal diet. In contrast, CO-treatment inhibited IL-1β and TNF-α production in liver tissue of HFD-induced mice (Figure 7B, C). Furthermore, the activation of caspase-1 in liver tissue of HFD-induced mice was dramatically reduced in the liver of mice that inhaled CO (Figure 7D). Next, to determine the effect of CO inhalation in the STZ-induced diabetic model, mice were treated with CO after STZ injection. Mice were sacrificed, and then blood was collected for the analysis of serum IL-1β and kidney tissue was collected for the analysis of IL-1β mRNA levels, as well as histological evaluation. The expression of CHOP mRNA, a marker of ER stress, was increased in the kidneys of mice injected with STZ (Figure 7E). CO is an effector molecule capable of inducing HO-1 expression. We examined whether CO inhalation can increase the expression of HO-1. As expected, the mRNA expression of HO-1 was increased in the in mice subjected to CO inhalation (Figure 7E).
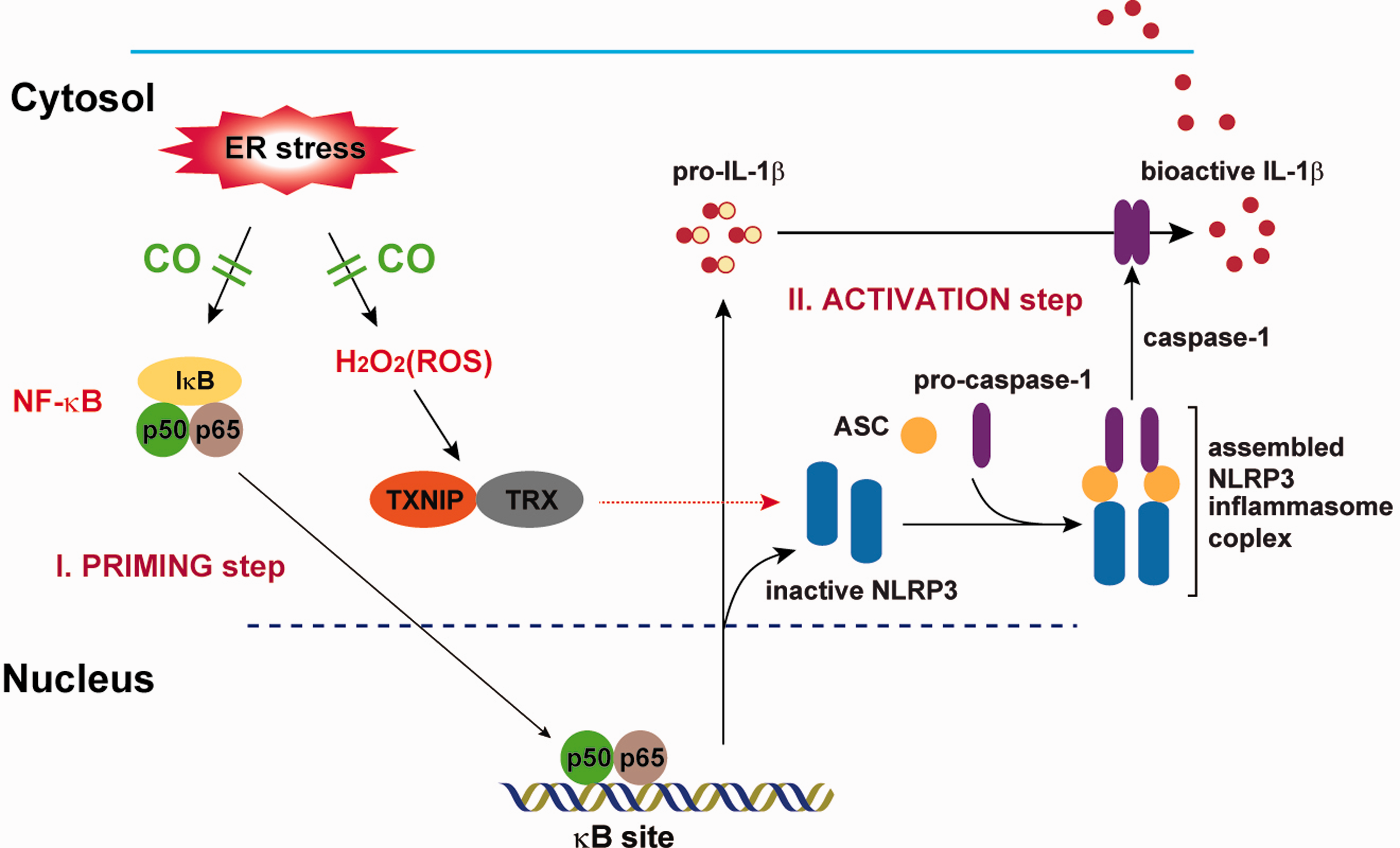
CO inhalation inhibits inflammasome formation in HFD mice and a STZ-induced diabetic model. C57BL/6 mice were fed a HFD or normal diet for 16 wks. Thirty mice were divided into four groups: ND group (n = 10); HFD group (n = 10); HFD + CO or H + CO (n = 10); and N + CO. Animals in the HFD + CO and N + CO groups were treated with CO (250 ppm) daily for 2 h for 10 wk in specialized chambers. Liver tissue was collected for the analysis of pro-IL-1β, TNF-α, CHOP, and HO-1 mRNA expression and caspase-1 activation. (A) CHOP and HO-1 mRNA expression was determined by RT-PCR, with 18 S rRNA as the standard. (B, C) pro-IL-1β and TNF-α mRNA expression was determined by qRT-PCR. (D) The activation (cleavage) of caspase-1, with β-tubulin as the standard, was determined by Western blot analysis. C57BL/6 mice were given a daily i.p. injection with STZ (40 µg/g body mass in 0.1 M citrate buffer) for 5 d to induce diabetes.40–42 Control mice were injected with buffer alone. Thirty mice were divided into three groups: CON (n = 6); STZ (n = 12); and STZ + CO (n = 12). Animals in the STZ + CO group were treated with CO (250 ppm) daily for 3 h after STZ injection in specialized chambers. CO was mixed with compressed air using a gas mixer to deliver 250 ppm. At 30 d after the STZ injection, mice were sacrificed, and their blood was collected for the analysis of serum IL-1β. Kidney tissue was collected for the analysis of pro-IL-1β, TNF-α, CHOP and HO-1 mRNA expression, and for histological analysis. (E) CHOP mRNA expression was determined by RT-PCR, with 18S rRNA as the standard. (F) The secretion of IL-1β was determined by ELISA; (G, H) pro-IL-1β and TNF-α mRNA expression was determined by quantitative real-time PCR. (I) Blood Glc levels were examined 3 d after the final STZ injection by obtaining blood from the lateral saphenous vein. (J) Representative photomicrographs (magnification 400×) of hematoxylin and eosin staining in the kidney sections from mice treated with control, STZ or CO + STZ. The kidney sections from STZ-treated diabetic mice showed marked degenerated glomeruli infiltrated by inflammatory cells and thickening of the basement membrane (arrow), and edematous changes with tubular simplification of the proximal convoluted tubules (arrowheads). CO treatment reduced morphological changes evident in the STZ-treated diabetic mice. Data represent mean ± SD of three independent determinations. ***P < 0.001. Blots shown are representative of three independent experiments. Schema representing ER stress-induced priming for the production of pro-IL-1β via NF-κB activation and activation of the NLRP3 inflammasome for the cleavage of pro-IL-1β via cleaved caspase-1. ER stress conditions result in pro-IL-1β synthesis through the activation of the NF-κB signaling pathway (priming) and the generation of ROS through the oxidative protein folding pathway during disulfide bond formation within the ER. During oxidative stress, TXNIP dissociates from TRX. The unbound TXNIP interacts with the leucine-rich region of NLRP3 (activation). The interaction between NLRP3 and TXNIP facilitates inflammasome formation and caspase-1 activation, which leads to IL-1β maturation. ER stress-induced inflammasome activation can be blocked by the CO/HO-1 system via inhibition of NF-κB activation and cellular ROS production.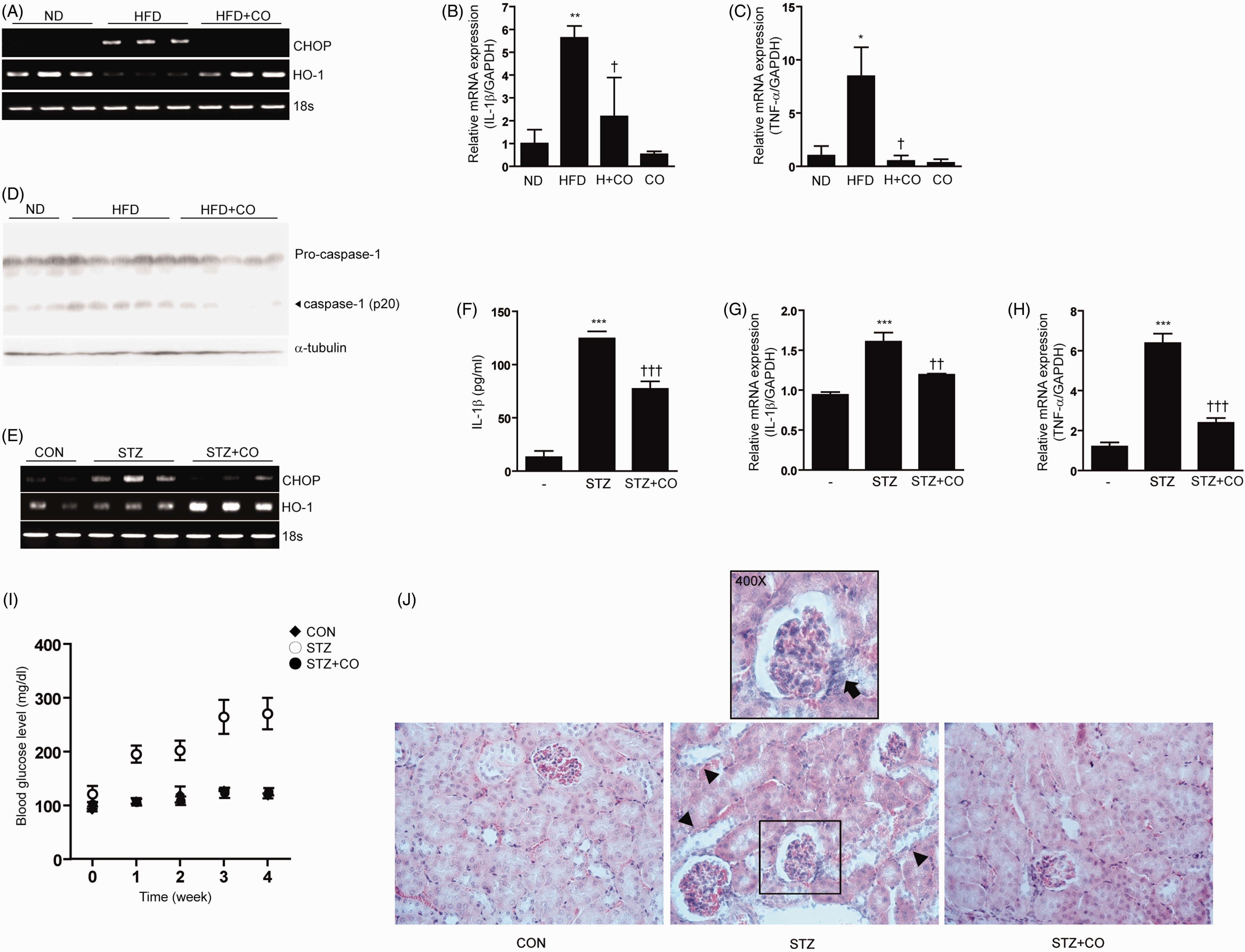
Furthermore, significant amounts of secreted IL-1β were detected in the serum of STZ-injected mice compared with control mice (Figure 7F). In contrast, CO-treatment inhibited IL-1β production in the serum and kidney tissue of STZ-induced mice. Furthermore, CHOP and TNF-α mRNA expression levels in the kidney were inhibited by CO treatment (Figure 7E, H).
STZ-induced diabetic mice were shown to have significant increases in blood Glc levels in comparison to control mice. In contrast, CO-treated STZ mice did not show significant changes in blood Glc levels relative to control mice (Figure 7I).
Histological studies of the normal kidney tissue of non-diabetic mice revealed normal glomeruli surrounded by the Bowman’s capsule, proximal and distal convoluted tubules without evidence of inflammatory changes. The kidneys of STZ-treated diabetic mice showed degenerated glomeruli infiltrated by inflammatory cells and thickening of the basement membrane. The proximal convoluted tubule exhibited edematous changes with tubular simplification. All these morphological changes were found to be absent in the diabetic mice treated with CO (Figure 7J). Taken together, these data suggest that CO inhibits IL-1β production in the STZ-induced diabetic model and reduces inflammatory tissue damage.
Discussion
ER stress may represent an underlying pathogenic component of many diseases that also involve inflammation. For example, ER stress has been implicated in metabolic syndrome and obesity; diabetes mellitus; neurodegenerative diseases; ischemia/reperfusion injury; cardiovascular diseases, such as atherosclerosis; and hepatic disorders, such as chronic alcohol exposure and hepatitis infections.44–48 In the ob/ob mouse model of leptin resistance and obesity, treatment with chemical chaperones that preserve protein folding was shown to ameliorate hepatic ER stress and reduce fatty deposits, normalize blood Glc and restore insulin sensitivity. 49 Thus, a link between ER stress and metabolic disorders is well established, though the specific impact of ER stress on inflammation is less well defined. 47
In the current study, we investigated the relationship between ER stress and the regulation of inflammatory processes. We demonstrate that ER stress can induce both the priming and activation steps of IL-1β production. We show that ER stress stimulates the production of immature pro-IL-1β through the activation of the NF-κB signaling pathway. Furthermore, we show that ER stress activates the NLRP3 inflammasome, which directs the caspase-1-dependent cleavage of pro-IL-1β production to generate its mature secreted form. Thus, our findings provide evidence for previously uncharacterized functions for ER stress in the regulation of the immune response.
In addition to chemical inducers of ER stress, such as TM, we also found that palmitate, one of the most abundant saturated fatty acids in plasma, can also trigger IL-1β production in the context of ER stress. Consistent with prior observations, 49 4-PBA, a chemical chaperone that is known to ameliorate ER stress, also attenuated secretion of IL-1β induced by palmitate or TM treatment. All three UPR pathways induced by the ER sensors IRE1α and PERK, and ATF6 can potentially contribute to activate NF-κB during ER stress. 26 During UPR activation the ER-resident IRE1α activates NF-κB through the IκB kinase (IKK) complex recruited by the formation of a complex between IRE1α and TRAF2.27,28 The kinase activity of IRE1 α phosphorylates IKK, leading to degradation of I-κB and subsequent activation of NF-κB. 27 PERK-induced phosphorylation of a subunit of the eukaryotic initiation factor 2-alpha (eIF2α) decreases the level of IκB protein by repression of I-κB translation, facilitating the nuclear translocation of NF-κB to regulate the transcription of target genes. 29 In addition to these mechanisms, NF-κB may be activated by Ca2+-mediated ROS production during ER stress.50,51 Here we have shown that NF-κB activation associated with ER stress participates in the priming step of IL-1β production. Further studies will be required to determine the precise mechanisms by which UPR pathways interact to induce immature pro-IL-1β production through the NF-κB signaling pathway. We have also shown that ER stress activates the NLRP3 inflammasome by inducing the binding of TXNIP, a ROS-sensitive NLRP3 ligand, in several types of immune cells, as well as in adipocytes. We demonstrated that ROS reduction by treatment with the antioxidant NAC or depletion of TXNIP prevented the assembly and activation of the NLRP3 inflammasome for IL-1β secretion during ER stress. The sources of ROS for NLRP3 activation by pro-inflammatory stimuli remain incompletely understood, though recent reports implicate mitochondrial dysfunction as the major contributor to this process.12,13 Also, the role of autophagy in regulating the triggering step of inflammasome activation by various stimulators has been investigated.13,52 Although ER stress induces autophagy proteins, the mechanisms by which autophagy regulates inflammasome activation remain unclear. At present, further studies are needed to investigate the mechanisms by which autophagy can regulate ER stress-induced inflammasome activation. Menu et al. 30 have recently reported that in macrophages primed with LPS or PMA, ER stress activates the NLRP3 inflammasome through a mechanism dependent on potassium efflux and ROS. In contrast, our results provide a direct link between ER stress and IL-1β production in the absence of other priming stimuli. Furthermore, our results suggest that ER stress-mediated ROS production is an important factor for IL-1β secretion. Although we did not determine the precise mechanism for ROS production by ER stress in the current study, several lines of evidence indicate that both the activation of the oxidative protein folding pathway in the ER and mitochondrial deregulation by the release of Ca2+ from the ER are responsible for ROS production during ER stress.35–38 ER Ca2+ is released to the cytosol during ER stress through activity modulation of the inositol 1,4,5-triphosphate receptor or the sarco(endo)plasmic reticulum Ca2+-ATPase1 (SERCA1) pump, and the induced expression of the truncated isoform (ST1) of SERCA1 pump.53–56 The released cytosolic Ca2+ is mainly taken up by mitochondria. Subsequently, the Ca2+-overloaded mitochondria produce ROS by hyperactivation of the citric acid cycle and oxidative phosphorylation.35,38 In addition, ER stress can produce ROS through the oxidative protein folding pathway during disulfide bond formation within the ER.35–37 Thus, ER stress can cause both NF-κB activation and ROS generation, which are required for production of the pro-inflammatory cytokine IL-1β.
CO arises as a reaction product of HO enzymatic activity. 39 HO-1, the inducible form of the enzyme, catalyzes the degradation of heme, generating CO, biliverdin-IX and ferrous iron. 39 HO-1 is considered as a general cellular defense mechanism against oxidative stress, though less is known about the function of this protein in ER stress. Although CO is considered a toxic waste product of heme catabolism, recent studies suggest that this gas may act as a potential physiological and cellular signaling molecule. Furthermore, CO, when applied as a pharmacotherapeutic agent, can modulate the pathways that regulate inflammation, apoptosis and cellular proliferation. 40 Recent studies have shown that ER stress-induced endothelial cell apoptosis can be blocked by CO via the inhibition of CHOP expression. 53 In the current study, we examined the hypothesis that CO may also inhibit ER stress-induced inflammasome activation. CO inhibited cellular ROS production in response to ER stress treatments, consistent with down-regulation of inflammasome activation. We find that the HO-1-inducing compound CoPP or CO delivered by the pharmacological CO donor compound CORM-2 could inhibit ER stress-dependent activation of caspase-1. CO treatments also inhibited mature IL-1β production and TNF-α secretion in response to ER stress. These results implicate a role for HO-1 and/or CO in the modulation of both NF-κB and inflammasome-dependent inflammatory responses. Furthermore, these results suggest a novel therapeutic target of the HO-1/CO system. We examined these relationships in an in vivo model of STZ-induced type 1 diabetes, which is associated with oxidative stress, inflammation and ER stress.33,41–43 STZ treatment is known to cause the development of diabetic kidney injury with hyperuricemia, hyperlipidemia and inflammation. 57 Furthermore, the activation of NLRP3 inflammasome increases renal inflammation with up-regulation of IL-1β and IL-8, which leads to chronic kidney disease. 58 Thus, the effects of CO on NLRP3 inflammasome activation were investigated in STZ-treated mice. In this model, we found that inhaled CO reduced indicators of ER stress and inhibited IL-1β production. These effects were associated with the amelioration of inflammation-associated kidney tissue injury.
In conclusion, we show that ER stress alone is a sufficient signal for IL-1β maturation. ER stress promotes IL-1β synthesis through stimulation of the NF-κB pathway, and also stimulates inflammasome-dependent responses leading to caspase-1 dependent IL-1β maturation. We have shown that these responses can be modulated by chemical chaperones and the candidate anti-inflammatory therapeutic CO. We demonstrate the effectiveness of CO at ameliorating inflammasome-associated cytokine production in a rodent model of diabetes. Our findings suggest potential therapeutic opportunities for inhibiting inflammation through targeting the ER stress pathway in diseases such as metabolic syndrome and type 1 diabetes.
Footnotes
Funding
This study was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MEST) (2012M3A9C3048687).
Conflict of interest
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.