
Abstract
A host type I IFN response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP (c-di-GMP) by STING (stimulator of IFN genes). Because the STING, an adaptor protein, links the cytosolic detection of DNA by the cytosolic DNA sensors such as the IFN-inducible human IFI16 and murine p202 proteins to the TBK1/IRF3 axis, we investigated whether c-di-GMP-induced signaling could regulate expression of IFI16 and p202 proteins. Here, we report that activation of c-di-GMP-induced signaling in human and murine cells increased steady-state levels of IFI16 and p202 proteins. The increase was c-di-GMP concentration- and time-dependent. Unexpectedly, treatment of cells with type I IFN decreased levels of the adaptor protein STING. Therefore, we investigated whether the IFI16 or p202 protein could regulate the expression of STING and activation of the TBK1/IRF3 axis. We found that constitutive knockdown of IFI16 or p202 expression in cells increased steady-state levels of STING. Additionally, the knockdown of IFI16 resulted in activation of the TBK1/IRF3 axis. Accordingly, increased levels of the IFI16 or p202 protein in cells decreased STING levels. Together, our observations identify a novel negative feedback loop between c-di-GMP-induced levels of IFI16 and p202 cytosolic DNA sensors and the adaptor protein STING.
Introduction
Infection of mammalian cells with certain bacterial pathogens induces type I IFN expression via a cytosolic signaling pathway that is independent of TLR.1–3 This response involves production of cyclic-di-GMP (c-di-GMP) by the pathogens.2–4 Studies have suggested that the production of cytosolic c-di-GMP can stimulate a variety of signaling pathways in cells that result in transcriptional activation of type I IFN genes (IFN-α/β; a type I IFN response). 2–4 Further, activation of a type I IFN response by the cytosolic c-di-GMP is similar to the response that is triggered by cytosolic DNA in mammalian cells.1,3–5 This response depends on activation of TANK-binding kinase 1 (TBK1) and IFN regulatory factor 3 (IRF3) transcription factor.3,4 In addition to TBK1 and IRF3, the response also depends upon an adaptor protein STING (stimulator of IFN genes; also called MITA or TMEM173).6–8 STING interacts with both TBK1 and IRF3 to promote activating phosphorylation of IRF3 by TBK1. 9 STING is essential for type I IFN (IFN-β) induction by cytosolic DNA and poly(dA:dT).2,8 The bulk of the STING protein is detected in the endoplasmic reticulum. 7 However, in the presence of cytosolic DNA, STING re-localizes to cytoplasmic foci that also contain TBK1.7,10
STING has been identified as an innate immune sensor for cyclic dinucleotides in cytosol. 6 The C-terminal domain of STING binds directly to c-di-GMP and one molecule of c-di-GMP binds to two molecules of STING protein. Furthermore, certain mutations in the STING selectively abrogate the response to cyclic dinucleotides without affecting the response to cytosolic double stranded DNA (dsDNA). 6 These observations support a role for STING as a direct sensor of cytosolic cyclic dinucleotides, as well as an adaptor for cytosolic DNA-induced signaling that results in induction of type I IFN.2,3,5
IFN-inducible p200 family proteins include the murine p202 and human IFI16 proteins.11–13 These proteins contain at least one HIN domain that allows binding to ds DNA non-specifically. The IFI16 protein also contains a pyrin domain (PYD) in the N-terminus.11,13 The PYD associates with the adapter protein ASC, which recruits caspase-1 to form an inflammasome. The p202 protein contains two HIN domains (HINa and HINb).12,13 Upon sensing the cytosolic DNA, both p202 and IFI16 proteins can stimulate expression of IFN-β that depends on the adaptor protein STING and activation of the TBK1/IRF3 axis.10,14,15
Promoter polymorphism-dependent increased constitutive and IFN-induced levels of p202 protein in certain strains of mice, such as NZB and (NZB/W)F1, compared with C57BL/6 mice are associated with the development of systemic lupus erythematosus (SLE).16,17 Further, age- and gender-dependent increased expression of the p202 protein in B6.Nba2 congenic strain of mice is associated with increased levels of IFN-α/β.16,17 Increased expression of the p202 protein in murine macrophage cell lines induces expression of IFN-β and activates a type I IFN response.15,17 Accordingly, p202 protein expression can activate the STING-dependent type I IFN response in transfected cells. 14 Given that increased serum levels of type I IFNs are associated with the severity of the disease in SLE patients and certain mouse models,17,18 the above observations suggest a role for the p202 protein in the regulation of the type I IFN response.
Increased levels of IFI16 mRNA have been reported in leukocytes isolated from SLE patients compared with healthy individuals. 19 Further, the IFI16 protein can activate human monocyte-derived dendritic cells (DCs), as well as primary DCs, upon sensing cytosolic DNA. 20 Increased levels of IFI16 protein in a variety of cells are associated with senescence-associated permanent cell growth arrest and activation of an inflammatory response.11,21 In phorbol 12-myristate 13-acetate (PMA)-treated human monocytic THP-1 cells, increased levels of IFI16 protein mediate the anti-inflammatory actions of the type I IFN. 22 Further, expression of IFI16 protein is up-regulated by IFN-β in a variety of cells (including THP-1 cells),11,22,23 and reduced levels of IFI16 protein in senescent (versus young or old) human WI-38 fibroblasts are associated with increased levels of STING. 24 Given that both p202 and IFI16 proteins can sense the cytosolic DNA and can activate a type I IFN response in immune cells,10,14,15 we investigated the potential role of the IFI16 and p202 proteins in c-di-GMP-induced signaling. Here we report that c-di-GMP treatment of human and murine cells increased steady-state levels of IFI16 and p202 proteins. Further, knockdown of IFI16 or p202 protein expression in cells increased levels of the adaptor protein STING and activated the TBK1/IRF3 axis. To our knowledge, our observations demonstrate, for the first time, that the c-di-GMP-induced levels of IFI16 and p202 cytosolic DNA sensors suppress the expression of human and murine STING.
Materials and methods
Mice and isolation of immune cells
Age-matched C57BL/6J and B6.Nba2 congenic mice 16 were purchased from Jackson Laboratory (Bar Harbor, ME, USA). These mice were housed in specific pathogen-free animal facilities at the University of Cincinnati (Cincinnati, OH, USA). The institutional animal care and use committees approved the protocol to use mice in this study. Total splenocytes were prepared from mice as described. 25 From total splenocytes, CD19+ cells were purified using magnetic beads (purification kit purchased from Miltenyi Biotech, Auburn, CA, USA) allowing the positive selection of CD19+cells. Similarly, from bone marrow cells isolated from mice, CD11b+ cells were purified using magnetic beads using a kit from Miltenyi Biotech. The purified (> 90% pure) cells were cultured in RPMI 1640 supplemented with 10% FBS.
Cells and treatments
Human monocytic THP-1 cells [purchased from the American Type Culture Collection (ATCC), Manassas, VA, USA] were maintained (CO2, 5.5%) in RPMI-1640 culture medium (cat # 30-2001; ATCC) with high Glc. 22 The culture medium was supplemented with 10% FBS and antibiotics (Invitrogen, Carlsbad, CA, USA). Murine macrophage cell lines (RAW264.7 and J774.A1) were purchased from the ATCC and maintained in culture, as suggested by the supplier. 15 When indicated, THP-1 cells were treated with 100 nM PMA for 18–24 h (to differentiate cells). 22 When indicated, sub-confluent cultures of cells were incubated with the desired concentrations of c-di-GMP (KeraFAST, Winston-Salem, NC, USA) for the indicated time.
Nucleofections
Actively proliferating THP-1 cells were nucleofected as described. 22 In brief, ∼ 2 × 106 cells were nucleofected with 2 µg of empty vector pCMV or pCMV–IFI16B plasmid using the Nucleofector-II device (Amaxa Biosystems, Natthermannalle, Germany), nucleofection kit-V and program U-001. This nucleofection program resulted in ∼ 50% cell survival after 24 h of nucleofections. Twenty-four hours after nucleofections, cells were lysed and cell lysates containing equal amounts of proteins were used for immunoblotting.
Knockdown of IFI16 or Ifi202 expression
We have described the generation of THP-1 cell line allowing a stable constitutive knockdown of the IFI16 gene and a control THP-1 cell line. 22 In brief, THP-1 cells were either infected with control lentiviral particles (sc-108080; Santa Cruz Biotech, Santa Cruz,, CA, USA) or the lentiviral particles expressing shIFI16 RNA (sc-35633-V; Santa Cruz Biotech). Infected cells were selected with puromycin (0.5–1 µg/ml) and cells exhibiting the resistance to puromycin were pooled. Further, we have also described knockdown of IFI16 expression in human WI-38 lung fibroblasts by infecting cells with either letiviral particles expressing the shIFI16 RNA or control a control RNA. 24 Similarly, we have described the generation of J774.A1 cell line allowing a stable constitutive knockdown of the Ifi202 gene and a control J774.A1 cell line. 25 For experiments, cells were cultured without puromycin in the culture medium.
Immunoblotting
Total cell lysates prepared from cells were analyzed by immunoblotting, as described previously. 22 Abs were IFI16 (sc-8023) and ASC (sc-22514) from Santa Cruz Biotech; p202 (IMG-6670A) from Imgenex (San Diego, CA, USA); STAT1 (cat# 9172), PY-STAT1 (# 9167), TBK1 (# 3504), p-TBK1 (Ser-172; # 5483) and β-actin (cat # 4967) from Cell Signaling Technology (Danvers, MA, USA); and human STING (ab82960) and mouse STING (ab92605) from Abcam (Cambridge, MA, USA). HRP-conjugated secondary anti-mouse (NXA-931) and anti-rabbit (NA-934) Abs were from Amersham Biosciences (Piscataway, NJ).
Reverse transcriptase reaction, real-time PCR
Total RNA isolated from THP-1 cells (using Trizol reagent; Invitrogen) was subjected to cDNA synthesis. cDNA was used to perform RT-PCR and quantitative real-time TaqMan PCR as described.22,25 The following primers were used for RT-PCR: IFI16 (forward: 5′-CCAAGACTGAAGACTG AA-3′; backward: 5′-ATCGTCAATGACATCCAG-3′); human STING (forward: 5′-CTTCAGTGGCTGAATGTCCA-3; backward: 5′-GCGAAGGAGCCT GTTGATAC-3′); human IFN-β (forward: 5′- GAATGGGAGGCTTGAATACTGCCT-3′; backward: 5′- TAGCAAAGATGTTCTGGAGCATCTC-3′), and actin (forward: 5′-GCTCGTCGTCGACAACGGCTC-3′; backward: 5′-CATGATCTGGGTCATCTTCTC-3′). The following TaqMan gene-specific assays were purchased from the Applied Biosystems (Foster City, CA, USA) and used as suggested by the supplier: IFI16 (assay Id# Hs 00194261_m1), mouse STING (assay ID# Mm01158114), murine Ifi202 (assay Mm0304 8198_m1; the assay allows for the detection of both the Ifi202a and Ifi202b mRNA levels), murine actin (assay Id# Mm4352933) and the endogenous control human β-actin (assay Id# Hs99999903_ml).
Statistical analyses
GraphPad Prism software 5 (GraphPad Software, La Jolla, CA, USA) was used for statistical analyses. All values are expressed as mean ± SD. P < 0.05 was considered statistically significant.
Results
Activation of cyclic-di-GMP-induced signaling increases levels of IFI16 and p202 proteins
To explore whether c-di-GMP-induced signaling could regulate expression of IFI16 protein, we incubated PMA-treated human monocytic THP-1 cells with increasing concentrations of c-di-GMP. As shown in Figure 1, treatment of cells with 5 μM concentration of c-di-GMP for 3 h did not result in appreciable increase in STING adaptor protein levels. However, treatment of cells with 10 μM c-di-GMP moderately (∼ 1.5-fold) increased levels of STING protein. As expected,
4
the treatment increased levels of p-TBK1, p-STAT1 and STAT1 (compare lane 3 with 1) thus indicating activation of the IFN-induced signaling in cells by c-di-GMP treatment. Further, the activation of c-di-GMP-induced signaling in cells was accompanied by appreciable (2–3-fold) increases in levels of IFN-inducible IFI16 protein (compare lane 2 or 3 with lane 1). Accordingly, treatment of cells with c-di-GMP also increased steady-state levels of IFI16 mRNA ∼ twofold (Figure 1B). Similarly, treatment of murine RAW264.7 macrophage cell line with increasing concentrations of c-di-GMP for 3 h increased levels of p-STAT1 and STAT1 (compare lane 3 or 4 with lane 1). Further, the activation of STAT1 by c-di-GMP-induced signaling in cells was associated with increases in p202 protein levels. Together, these observations revealed that c-di-GMP-induced signaling in human THP-1 cells and murine RAW264.7 cells induced the expression of IFN-inducible IFI16 and p202 proteins through activation of the JAK/STAT pathway.
Activation of cyclic-di-GMP-induced signaling increases levels of IFI16 and p202 proteins. (A) PMA-treated THP-1 cells were incubated with the indicated concentrations of c-di-GMP for 3 h. At the end of the incubations, cells were harvested and cell lysates containing equal amounts of proteins were subjected to immunoblotting using Abs specific to the indicated proteins. FC indicates fold change (increases) in the levels of STING and IFI16 proteins. (B) PMA-treated THP-1 cells were either left untreated or treated with 5 μM c-di-GMP for 3 h. At the end of the treatment, cells were harvested and total RNA was prepared. The RNA was analyzed by quantitative real-time PCR using the TaqMan assay specific to the IFI16 gene. The ratio of the test gene to actin mRNA was calculated in units (one unit being the ratio of the test gene to actin mRNA). Levels of IFI16 mRNA in control cells are indicated as 1.0. The error bars represent standard deviation (**P < 0 .001). (C) Sub-confluent cultures of murine RAW264.7 macrophage cell line were either left untreated or treated with the indicated concentrations of c-di-GMP for 3 h. At the end of the treatments, cells were harvested and total cell lysates were analyzed by immunoblotting using Abs specific to the indicated proteins. FC indicates fold change in the levels of p202 protein.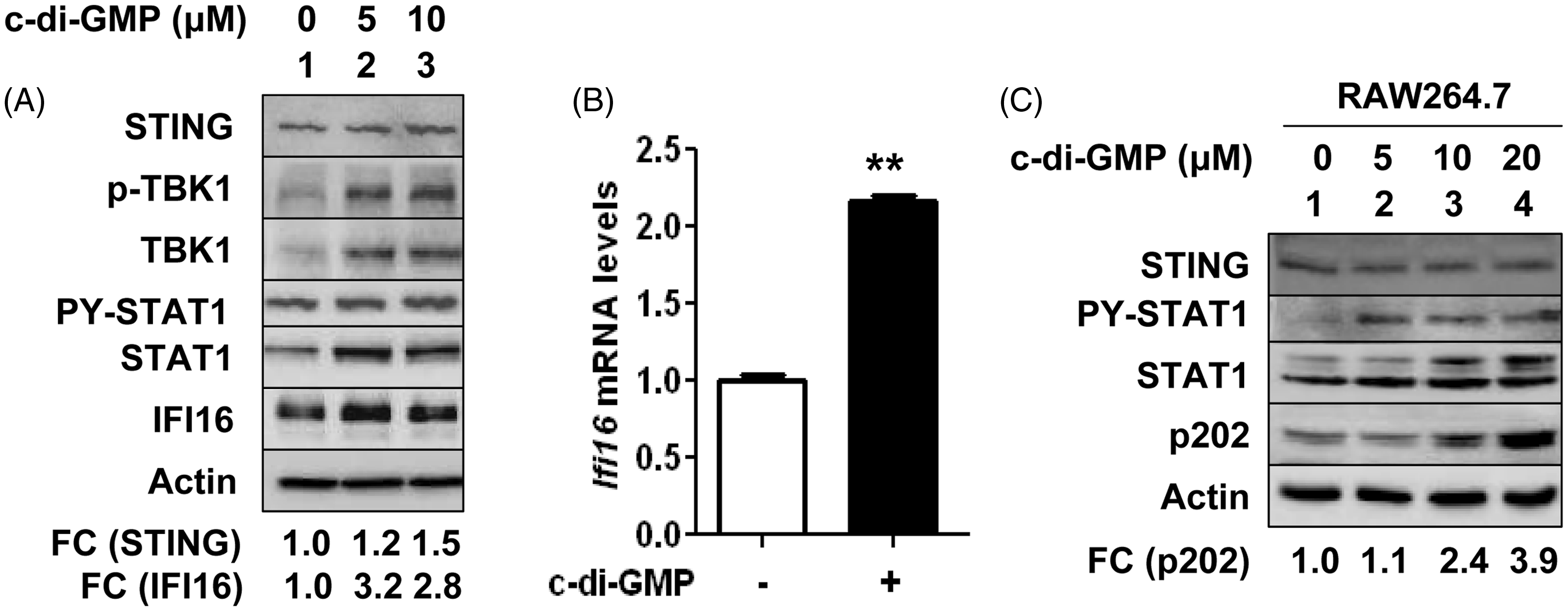
Activation of IFN signaling in cells decreases STING levels
During our experiments described above (Figure 1A), we noted that longer (>5 h) treatment of THP-1 cells with c-di-GMP, which activated TBK1 and STAT1 and increased levels of IFI16 protein (compare lanes 3 and 4 with lane 1), measurably (∼ 40–50%) decreased steady-state levels of STING adaptor protein (Figure 2A). Therefore, we explored whether activation of an IFN signaling in cells could down-regulate levels of the STING. As shown in Figure 2B, activation of IFN signaling by IFN-α, β or γ in THP-1 cells reduced steady-state levels of STING protein by ∼ 50–70%. Similarly, treatment of murine CD11b+ cells from C57BL/6 mice with IFN-β (1000 u/ml), which induced p202 protein levels (Figure 2C), decreased levels of STING ∼ 80%. Further, treatment of THP-1 cells with increasing concentrations of Trichostatin A (TSA), an inhibitor of histone deacetylases (HDAC),
26
which activated STAT1 transcription factor and increased levels of the IFI16 protein (Figure 2D), also decreased levels of the STING protein by ∼ 60– 70% (compare lane 3 with lane 1). These observations prompted us to investigate whether knockdown of IFI16 expression in cells could up-regulate levels of STING protein. For this purpose, we used WI-38 human diploid fibroblasts (HDFs; because basal levels of IFI16 protein are detectable
24
). As shown in Figure 2E, knockdown of IFI16 expression in young WI-38 HDFs, which decreased basal levels of STAT1, increased the basal levels of STING protein. These observations indicated that activation of type I IFN signaling negatively regulated the STING protein levels in cells that were tested.
Activation of IFN signaling in cells decreases levels of STING. (A) THP-1 cells in culture were either left untreated (lane 1) or treated with c-di-GMP (5 μM) for the indicated time. Total cell extracts were prepared and cell extracts containing equal amounts of proteins were analyzed by immunoblotting for the indicated proteins. (B) THP-1 cells in culture were either left untreated (lane 1), treated with universal recombinant IFN-α (1000 u/ml), human IFN-β (1000 u/ml), or human IFN-γ (10 ng/ml) for 14 h. Total cell extracts were prepared and cell extracts containing equal amounts of proteins were analyzed by immunoblotting for the indicated proteins. (C) Bone marrow-derived CD11b+ cells were either left untreated (lane 1) or treated with murine IFN-β (1000 u/ml) for 16 h. After the treatment, total cell extracts were prepared and cell extracts containing equal amounts of proteins were analyzed by immunoblotting for the indicated proteins. (D) THP-1 cells in culture were either left untreated (lane 1) or treated with HDAC inhibitor TSA (in ethanol) at the indicated concentrations for 14 h. Total cell lysates were prepared and the cell lysates containing equal amounts of proteins were analyzed by immunoblotting for the indicated proteins. (E) Total cell lysates were prepared from sub-confluent cultures of human WI-38 HDFs (at passage 16) that were infected with either control lentivirus (lane 1) or the lentivirus expressing the short hairpin (sh) RNA to IFI16 mRNA (lane 2). Cell lysates were analyzed by immunoblotting using Abs specific to the indicated proteins. FC indicates the fold change (increases) in the STING protein levels.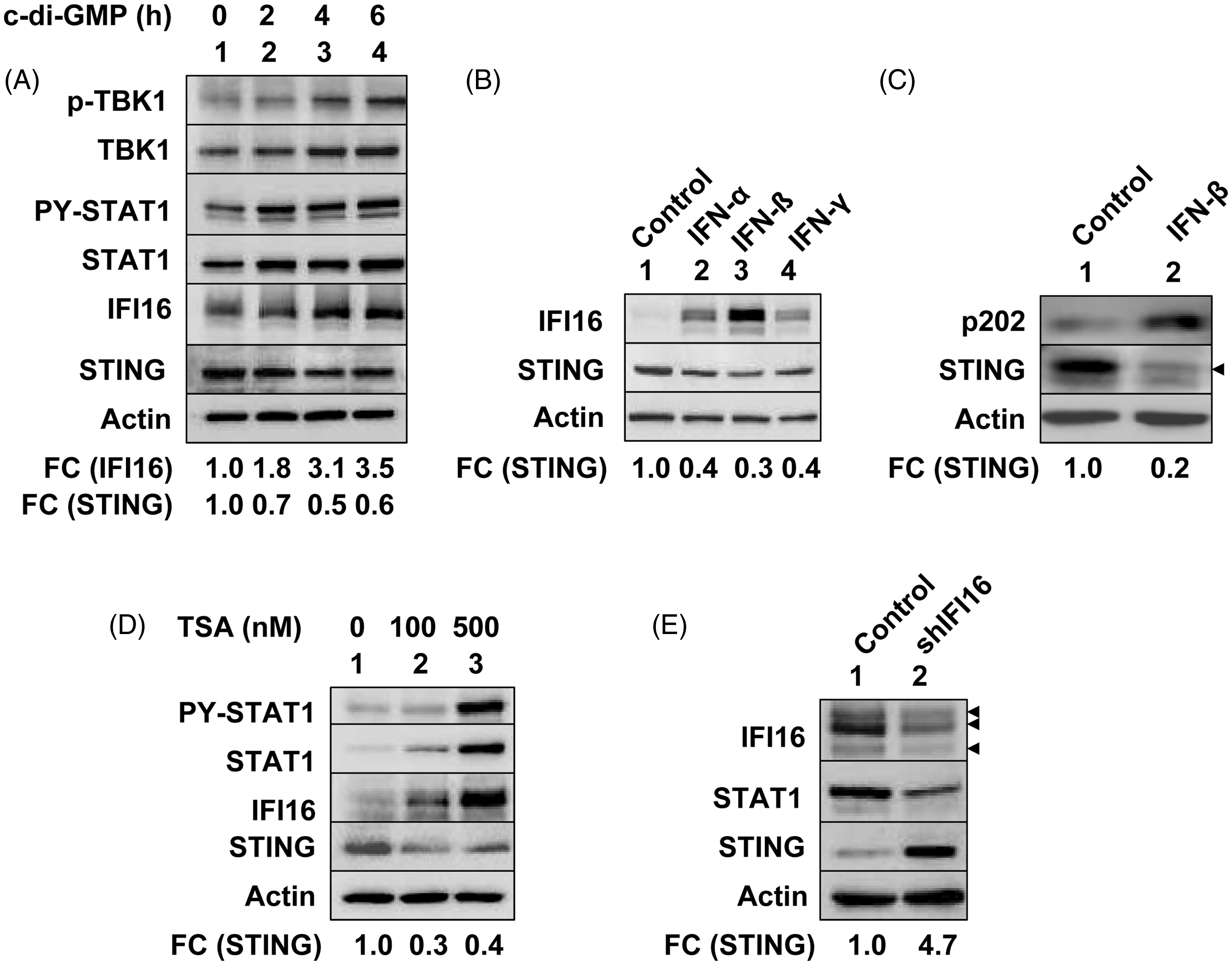
Knockdown of IFI16 expression increases levels of STING and potentiates an activation of c-di-GMP-induced signaling
To further investigate whether IFI16 protein could regulate expression of STING and STING-mediated signaling, we chose to constitutively knockdown expression of IFI16 protein in THP-1 cells. As shown in Figure 3A, the knockdown of IFI16 expression in cells increased basal steady-state levels of STING mRNA (compare lane 3 with 1). Moreover, steady-state levels of IFN-β mRNA were also higher in cells after knockdown of IFI16 expression (compare lane 3 with 1). Further, treatment of cells with c-di-GMP that expressed reduced levels of IFI16 mRNA did not result in further measurable increases in the steady-state levels of IFN-β and STING mRNAs (compare lane 4 with 3). Accordingly, we observed that the knockdown of IFI16 expression in THP-1 cells increased the basal levels of STING, p-TBK1, TBK1, p-IRF3, IRF3, p-STAT1 and STAT1 protein levels (Figure 3B; compare lane 3 with 1). Again, treatment of cells that expressed reduced levels of IFI16 with c-di-GMP did not result in further measurable increases in the levels of the above proteins (compare lane 4 with 3).
Constitutive knockdown of IFI16 expression in cells increases expression of STING and activates the TBK1/IRF3 axis. (A) PMA-treated control THP-1 cells (infected with control lentivirus; lanes 1 and 2) or cell expressing the shIFI16 RNA (lanes 3 and 4) were either left untreated (lanes 1 and 3) or treated with c-di-GMP (5 μM) for 3 h (lanes 2 and 4). At the end of the treatments, cells were harvested and total RNA was isolated. RNA was analyzed by semi-quantitative PCR using a set of primers that were specific for the indicated genes. Fold change (FC) in STING mRNA levels is indicated. (B) PMA-treated THP-1 cells infected with either a control lentivirus (lanes 1 and 2) or the virus expressing the shIFI16 RNA (lanes 3 and 4) were either left untreated (lanes 1 and 3) or treated with c-di-GMP for 3 h (lanes 2 and 4). After treatment, total cell extracts containing equal amounts of proteins (∼ 50 µg/lane) were analyzed by immunoblotting for the indicated proteins. Fold change in levels of STING protein is indicated. (C) PMA-treated THP-1 cells in culture were either nucleofected with an empty vector (pCMV; lane 1) or a plasmid encoding for IFI16B (pCMV–IFI16B; lane 2). After 24 h of nucleofections, cells were harvested and cell lysates containing equal amounts of proteins were analyzed by immunoblotting for the indicated proteins.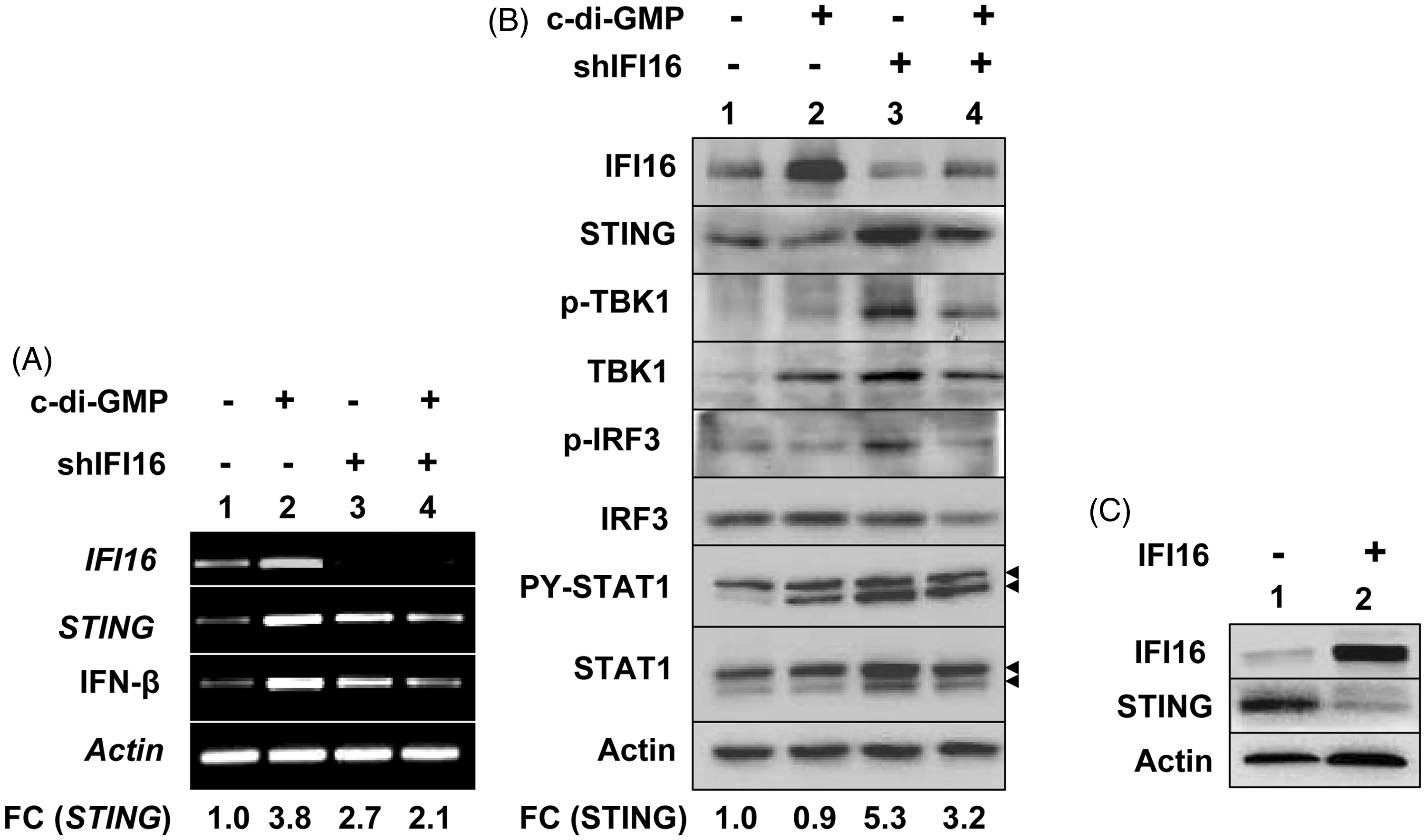
Our observations that knockdown of IFI16 expression in THP-1 cells increased the basal levels of STING protein prompted us to test whether overexpression of the IFI16 protein in cells could down-regulate the expression of STING. As shown in Figure 3C, overexpression of IFI16 protein in nucleofected THP-1 cells significantly reduced levels of STING protein compared with cells that were nucleofected with control vector (compare lane 2 with 1). Together, these observations indicated that expression of IFI16 protein in cells suppresses expression of STING and the STING/TBK1/IRF3 axis.
Knockdown of p202 expression increases levels of murine STING
We also investigated whether murine p202 protein could regulate levels of STING. For this purpose, we compared steady-state levels of STING mRNA and protein in J774.A1 murine macrophage cell line in which we constitutively knocked down the expression of p202 protein. As shown in Figure 4A, the knockdown of p202 expression in cells increased levels of STING mRNA ∼ 1.7-fold. Accordingly, levels of the STING protein were ∼ 1.8-fold higher in cells that expressed reduced levels of p202 (Figure 4B; compare lane 2 with 1). Further, murine splenic B cells (CD19+) from lupus-prone B6.Nba2 mice that express increased levels of p202 compared with non-lupus-prone C57BL/6 (B6) female mice expressed reduced levels of STING (Figure 4C; compare lane 2 with 1). Accordingly, we noted reduced levels of STING mRNA in the B6.Nba2 splenic B cells than the age and gender-matched B6 mice (Figure 4D). These observations indicate that expression of p202 protein in immune cells negatively regulates the expression of STING.
Knockdown of p202 expression increases levels of murine STING. (A) Total RNA isolated from murine macrophage J774.A1 cells either stably infected with a control lentivirus (Vect) or the virus expressing the shIfi202 RNA (sh202) was analyzed by quantitative real-time PCR using assays specific to the murine STING gene. The ratio of the test gene to actin mRNA was calculated in units (one unit being the ratio of the test gene to actin mRNA). Levels of STING mRNA in control cells are indicated as 1.0. The error bars represent SD (*P < 0.01). (B) Total cell extracts were prepared from murine macrophage J774.A1 cells that were either infected with a control lentivirus (Cont) or the virus expressing the shIfi202 RNA (sh202). The extracts containing equal amounts of proteins were analyzed by immunoblotting using Abs specific to the indicated proteins. (C) Total cell extracts were prepared from purified splenic B cells (CD19+) that were isolated from age and gender-matched C57BL/6 (B6) or B6.Nba2 female mice. The extracts containing equal amounts of proteins were analyzed by immunoblotting using Abs specific to the indicated proteins. FC indicates fold change (decreases) in STING protein levels. (D) Total RNA was prepared from purified splenic B cells (CD19+) that were isolated from age- and gender-matched C57BL/6 (B6) or B6.Nba2 female mice. The RNA was analyzed by quantitative real-time PCR using the assay specific to the STING gene. The ratio of the test gene to actin was calculated in units (one unit being the ratio of the test gene to actin mRNA). Levels of STING mRNA in B6 cells are indicated as 1.0. The error bars represent SD (*P < 0.05).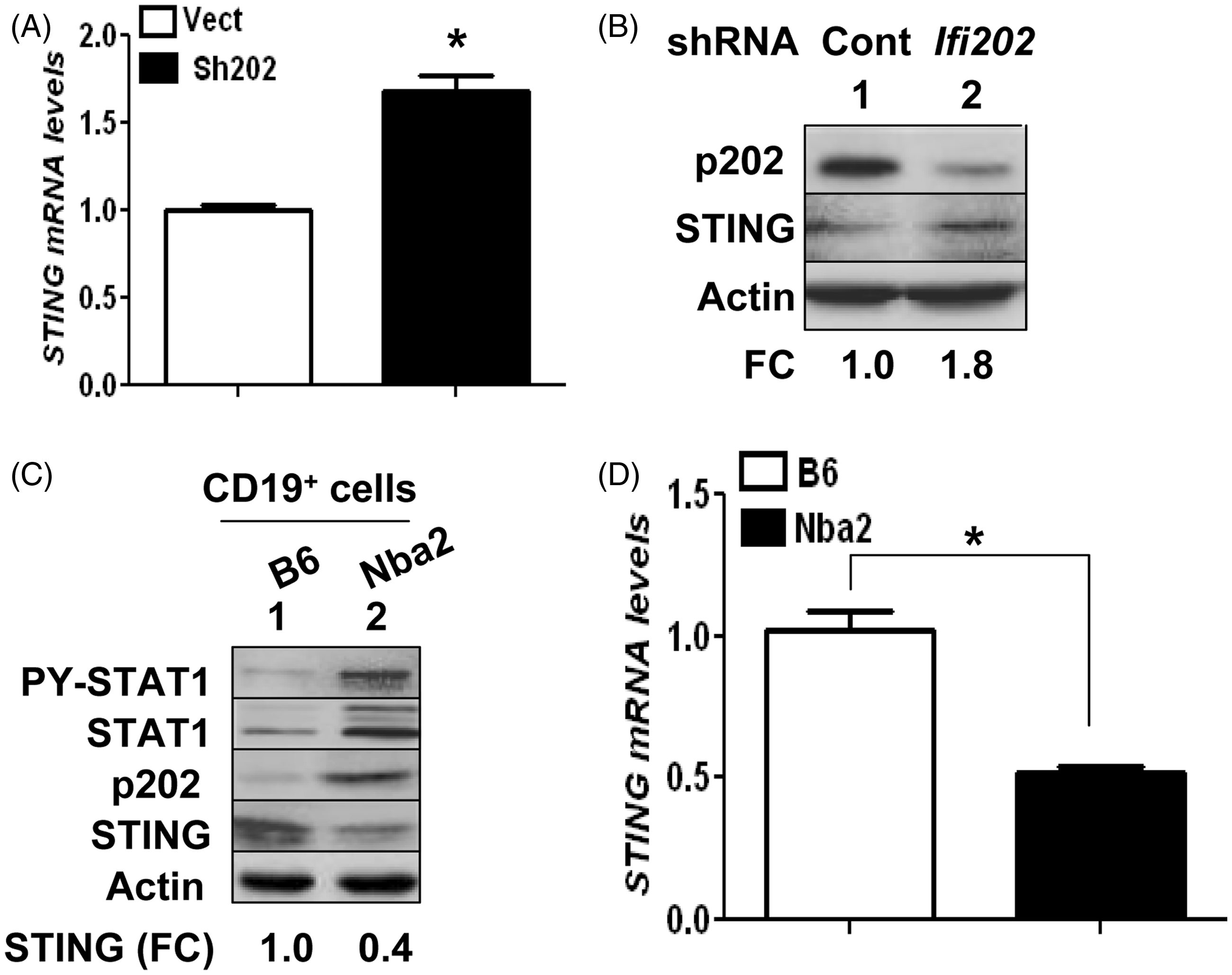
Discussion
Production of c-di-GMP, a second messenger in mammalian cells, by pathogenic bacteria influences several processes, including bacteria–host interactions.2,4 Sensing of c-di-GMP by STING in bone marrow-derived macrophages induces expression of IFN-α/β and transcriptional activation of the IFN-inducible genes.4,6,27 Moreover, human immature DC, when cultured in the presence of c-di-GMP, exhibit increased expression of co-stimulatory molecules (CD80/CD86) and cytokines and chemokines (IL-12, IFN-γ, IL-8, MCP-1 and RANTES). 28 These studies demonstrate that c-di-GMP-induced signaling enhances innate immune responses and, thus, influences bacteria–host interactions. Therefore, our observations that c-di-GMP-induced signaling increases levels of the IFI16 and p202 cytosolic DNA sensors in innate immune cells suggest a role for these proteins in the regulation of bacteria–host interactions.
STING is essential for the production of type I IFNs (IFN-α and IFN-β) by cytosolic DNA in a variety of cell types, including mouse embryonic fibroblasts, macrophages and DCs.2–5 Moreover, STING-deficient mice are sensitive to lethal infection with HSV-1.
8
Upon sensing the cytosolic DNA, both IFI1610 and p20214 proteins can activate a type I IFN response that depends on the adaptor protein STING. However, it remains unclear which domain(s) in the IFI16 and p202 proteins interact with the STING adaptor protein. The ability of one of the HIN200 domain (the HIN200A) in the IFI16 protein to bind p53 in vitro
29
raises the possibility that one of the HIN200 domain could bind to STING. Given that c-di-GMP-induced signaling increased levels of IFI16 and p202 proteins (Figure 1), an activation of the IFN signaling in human and murine cells measurably reduced basal levels of the STING protein (Figure 2), and constitutive knockdown of IFI16 (Figure 3) or p202 (Figure 4) protein in cells increased basal levels of STING protein and also activated the STING/TBK1/IRF3 axis (Figure 3), our observations identify a novel negative regulatory feedback loop between the p200 family proteins (e.g. IFI16 and p202) and STING that regulates the production of type I IFNs (Figure 5).
Proposed regulatory negative feedback loop between c-di-GMP-induced IFI16 and p202 proteins and the adaptor protein STING.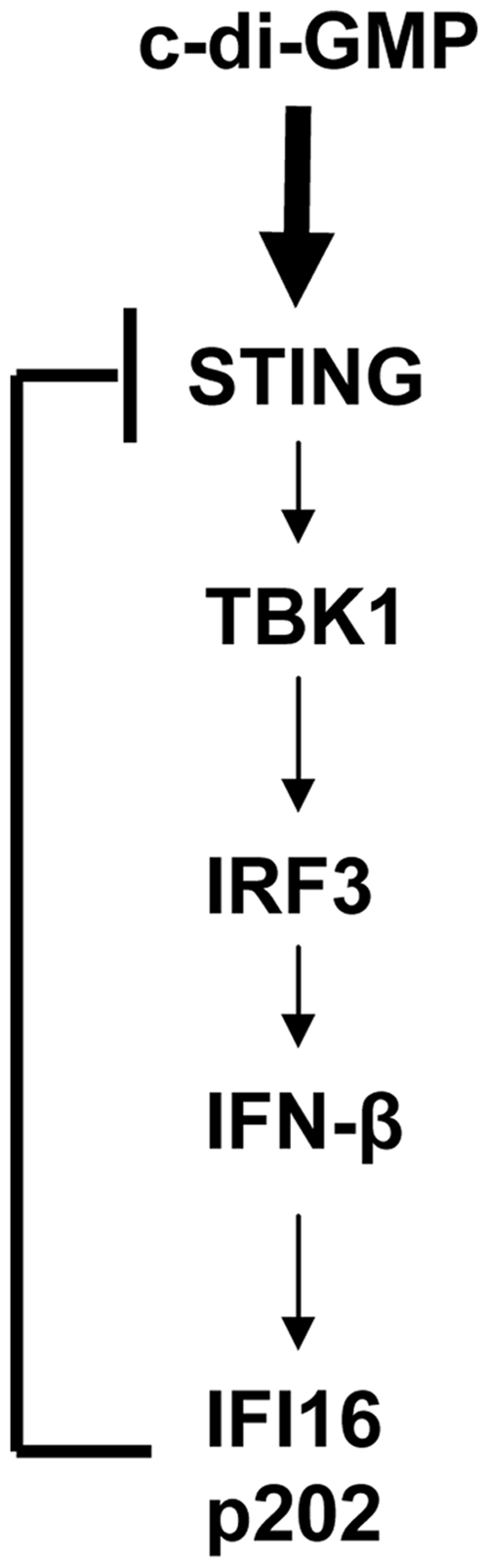
STING is a sensor of reactive oxygen species (ROS). 30 Additionally, ROS induces intermolecular disulfide bonds formation in STING homodimer; thus, ROS inhibits the STING-mediated activation of IRF3 by TBK1 and the production of IFN-β. Accordingly, we found that the accumulation of the senescent (versus old) human WI-38 fibroblasts in culture was associated with decreased levels of IFI16 protein and increased levels of IFN-β. 24 Given that increased levels of ROS activate p53, 31 a positive regulator of IFI16 protein, 32 increased levels of IFI16 protein modulate the transcriptional activity of p53, 31 and the 5’-regulatory region of the human STING gene contains a potential DNA-binding site for p53 (unpublished data), our observations that increased levels of IFI16 protein suppress the expression of STING warrant further investigation.
Our previous study revealed that increased levels of STING and p-IRF3 in old (versus young) human WI-38 fibroblast were associated with IFN-β production and increased expression of IFI16. 24 Additionally, we noted that transfection of plasmid DNA into young WI-38 cells, which activated TBK1 and IRF3, also increased the levels of IFI16 protein. Therefore, our observation that c-di-GMP-induced signaling up-regulated the expression levels of IFI16 protein in THP-1 cells (Figure 1) are consistent with our above observations. Together, these observations demonstrate that activation of the STING-TBK1-IRF3 axis by cytosolic DNA or c-di-GMP in cells up-regulates the expression of IFI16 protein.
Low levels of type I IFNs (IFN-α and IFN-β) are constitutively produced by innate immune cells.33,34 These low levels of type I IFNs play an important role through modulation of signaling intermediates that are required for responses to other cytokines.33–35 Additionally, constitutive levels of type I IFNs maintain the steady-state levels of the IFN-inducible proteins. 33 Thus, low levels of type I IFNs ‘prime’ cells for the regulation of various physiological processes, including cell cycle regulation, cell survival, cell differentiation and immunomodulation by other cytokines.34,35 However, the production of type I IFNs is induced in response to various danger stimuli, including sensing of cytosolic c-di-GMP or DNA.2–5 Sustained increased production of type I IFNs is associated with inflammatory diseases, such as SLE. 18 Given that the adaptor protein STING plays an essential role in the production of type I IFN in response to cytosolic c-di-GMP and DNA,3–5 and signaling pathways that regulate the constitutive and induced levels of STING remain unknown, our observations that knockdown of IFI16 or p202 protein in cells increases the expression of STING identify a novel negative regulatory feedback loop between the IFI16 and p202 proteins and STING. An improved understanding of the regulation of STING levels by the cytosolic DNA sensors is likely to facilitate the identification of the molecular mechanism that contribute to the development of human inflammatory diseases.
Footnotes
Funding
This work was supported by grants from the National Institutes of Health (AI066261 and AI089775) and a VA Merit Award to D.C.
Conflict of interest
The authors do not have any potential conflicts of interest to declare
Acknowledgements
We thank Dr Katherine Fitzgerald for providing thoughtful suggestions.