
Abstract
Innate immunity, the front line of our defence against pathogens, relies, to a great extent, on the production of antimicrobial peptides (AMPs). These peptides exhibit antimicrobial activity and immunomodulatory properties. In humans, AMPs include the defensins (α- and β-families) and the cathelicidin, LL-37. Bacterial resistance against antibiotics is a growing concern, and novel antimicrobial strategies are needed urgently. Hence, the concept of strengthening immune defences against infectious microbes by inducing AMP expression may represent novel or complementary pharmaceutical interventions in the treatment or prevention of infections. We have developed and validated a robust cell-based reporter assay for LL-37 expression, which serves as a marker for a healthy epithelial barrier. This reporter assay can be a powerful tool for high-throughput screenings. We first employed our assay to screen a panel of histone deacetylase inhibitors and derivatives, and then the Prestwick Chemical Library of Food and Drug Administration-approved compounds. After hit confirmation and independent validation in the parental cell line we identified five novel inducers of LL-37. This reporter assay will help to identify novel drug candidates for the treatment and prevention of infections. Importantly, the pattern of hits obtained may suggest cellular pathways and key mediators involved in the regulation of AMP expression.
Introduction
Innate immunity constitutes the front line of our defence system against microbes. Antimicrobial peptides (AMPs) are crucial components of innate defences that are synthesised constitutively and/or induced at epithelial surfaces, where the initial contact with microbes takes place. 1 AMPs are widespread in nature, present in fungi, plants, invertebrates and vertebrates, confirming an evolutionarily-conserved system. AMPs possess broad activity against various pathogens, for example viruses, bacteria, fungi and parasites. 2 There are two major classes of AMPs in mammals, the defensins (α- and β-families) and the cathelicidins.3,4 Besides the microbicidal activity, 5 these peptides have been shown to act as chemo-attractants for cells of both the adaptive and innate immunity, and to modulate immune responses.6–8 Thus, AMPs constitute a link between the innate and adaptive immunity.
The expression of AMPs can be induced by different compounds. The short chain fatty acid butyrate (BA) was found to induce cathelicidin expression in epithelial cells. 9 Moreover, sodium butyrate counteracted pathogen down-regulation of AMP expression, resulting in pathogen elimination from epithelial surfaces in vivo in a rabbit model of shigellosis. 10 Phenylbutyrate (PBA), an analogue of butyrate, was shown to up-regulate the expression of LL-37, the sole cathelicidin in humans, in epithelial cell lines and in monocytes. 11 The active form of vitamin D3, 1,25-dihydroxyvitamin D3, was also reported to enhance the expression of LL-37 in keratinocytes, immune cells and epithelial cells.12–14 Interestingly, PBA and 1,25-dihydroxyvitamin D3 were found to up-regulate LL-37 in a synergistic manner. 11
The continual emergence of antibiotic resistance among bacterial pathogens poses a great challenge to public health. The pipeline of new antibiotics in drug development has yet to match this threat, as only a few novel agents have been developed in the last few decades.15,16 Strengthening immune defences against pathogens by boosting the expression of our own ‘natural antibiotics’ may represent novel or complementary pharmaceutical interventions in infectious diseases. Importantly, the multiplicity of AMPs with overlapping antibacterial mechanisms secures minimal risk of microbial resistance.17,18 Therefore, the goal of our research was to find novel inducers of the AMP system to be developed into anti-infective drugs.
Cell-based assays are a powerful tool in drug discovery; as an example, a luciferase reporter assay led to the finding that the amino acid
This reporter assay affords a new tool to identify novel inducers of AMPs expression and to gain insight into the mechanisms behind LL-37 induction.
Materials and methods
Vector construction
The CAMP gene, encoding LL-37, including 3027 bp upstream of translation initiation and without the endogenous stop codon in exon 4, was amplified by PCR from the BAC clone RP11-502L5 (BACPAC, Oakland, CA, USA) using the following primers: forward 5′- Schematic representation of the vector construct and confirmation of the stable transfection of HT-29 parental cells. (A) The CAMP gene, shown in blue (the CAMP gene exons are indicated in orange, E1, E2, E3 and E4), was inserted into the pGL-4.26 vector in frame with the Firefly Luciferase gene (shown in yellow). The vector also contains a hygromycin-resistance gene, shown in red, and ampicillin-resistance gene in green. The primers utilised for cloning of the CAMP gene into the pGL4-26 vector are indicated in the figure: A1 and A2 for the CAMP gene (red); A3 and A4 for the pGL4-26 plasmid (green). Primers used to assess the construct integration of the entire CAMP gene were B1 and B2 (blue). (B) The genomic insert is transcribed into CAMP–luciferase fusion mRNA that is translated into the fusion protein proLL37-Luciferase. (C) Measurement of the luciferase activity (RLU) in the clone MN8 in basal conditions and upon stimulation for 24 h with vitamin D3 (Vit D; 100 nM) and sodium phenylbutyrate (PBA; 2 mM), in combination. (D) Integration of the pGL-CampLuc vector construct into the genome of the MN8CampLuc cell line was demonstrated by PCR, with primers B1 and B2. SpeI-digested vector construct was used as positive control and DNA extracted from the HT-29 parental cell line as negative control.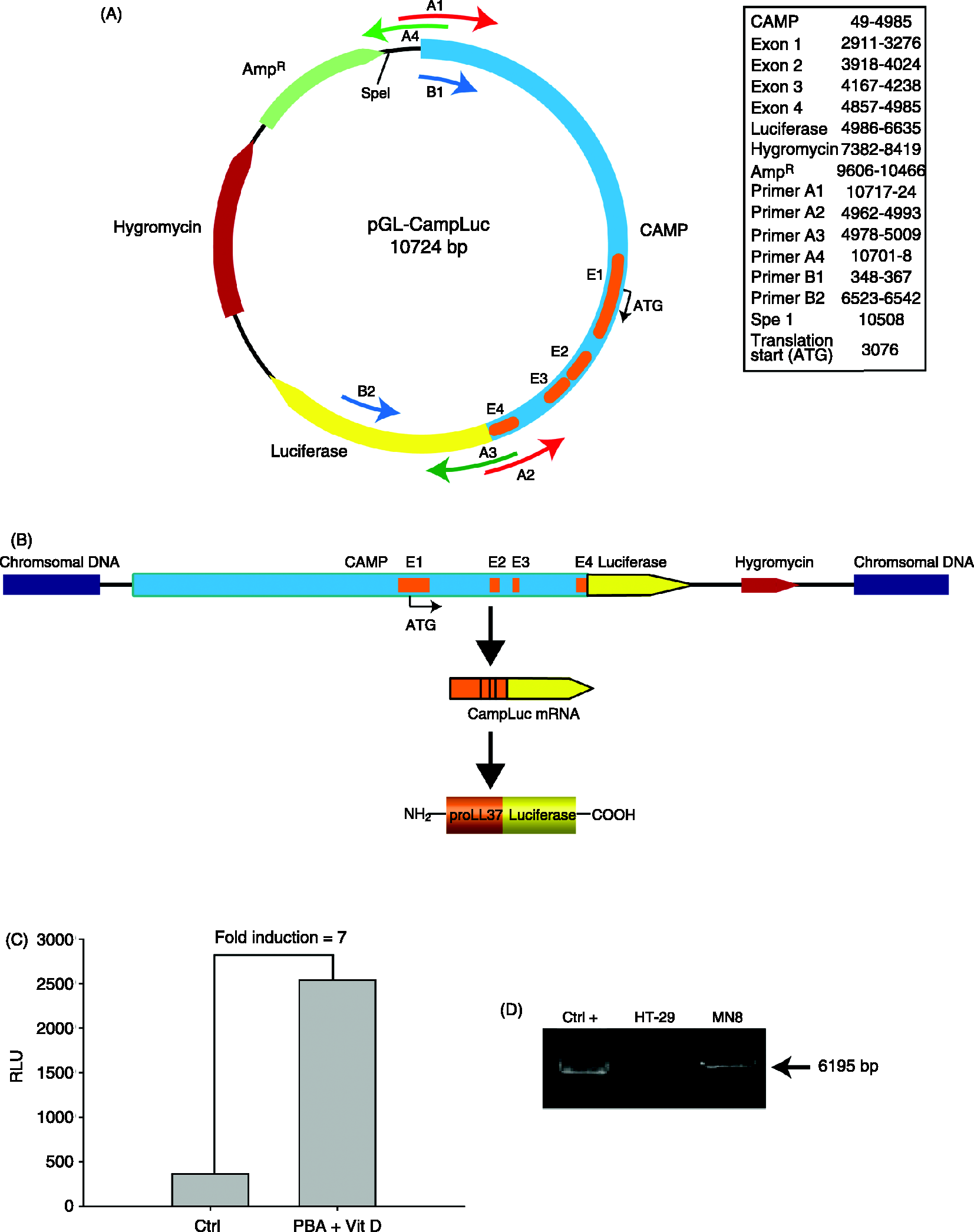
The plasmids were propagated in Escherichia coli XL-1 blue cells (Agilent, Santa Clara, CA, USA) and sequenced at Eurofins MWG Operon (Ebersberg, Germany). One plasmid construct with the correct sequence (named pGL-CampLuc; see Figure 1A) was linearised using the SpeI restriction enzyme (Fermentas, St. Leon-Rot, Germany) prior to stable transfection. This ensured the integrity of the CAMP gene and the hygromycin cassette, thus increasing the chances of a successful integration into the genome.
Cell cultures and stable transfection
The human colonic epithelial cell line HT-29 was cultured in RPMI 1640 (Gibco, Carlsbad, CA, USA), supplemented with 10% FCS (Gibco), 100 µg/ml streptomycin and 100 U/ml penicillin (Invitrogen), in a 5% carbon dioxide atmosphere at 37℃.
For stable transfection, HT-29 cells were grown to 70% confluence in a 10 cm dish and transfected with 15 µg of linearised plasmid using Turbofect (Fermentas), following the manufacturer’s instructions. After 24 h, the transfection medium was replaced with fresh medium for 3 d prior to exposure to selection medium containing 800 µg/ml hygromycin. Clones resistant to the selection medium were isolated, propagated and screened for luciferase activity. These clones were subsequently cultured in RPMI 1640 containing 400 µg/ml hygromycin for maintenance of the insert.
Assessment of the construct integration
In order to assess the integrity of the transfected construct, genomic DNA was extracted from luciferase-expressing clones and PCR was performed either with a forward primer (5′-AGGTAGTGGACACCGACCTG, named B1 in Figure 1A) located at 2728 bp upstream of the CAMP start codon ATG in Exon 1 corresponding to position + 1 (numbers refer to previous mapping of the gene), 20 or the reverse primer, which was located at 1557 bp in the luciferase gene: 5′-GTCCACGAACACAACACCAC (primer B2). The PCR products generated by these primers would be 6195 bp. PCR products were run on a 0.8% agarose gel and visualised with Gel-Red Nucleic Acid Gel Stain (Biotium, VWR International, Hayward, CA, USA).
Luciferase activity assay
To identify clones expressing a functional luciferase protein, 4 × 105 cells/well were seeded in 24-well plates in culture medium in the absence of hygromycin. After 24 h, cells were incubated with either vehicle or the previously described CAMP inducer PBA (2 mM; Santa Cruz Biotechnology, Santa Cruz, CA, USA) combined with 1,25-dihydroxyvitamin D3 (100 nM; Sigma-Aldrich, St. Louis, MO, USA). After 24 h, intracellular luminescence was assayed with the Luciferase assay kit (Promega) in accordance with manufacturer’s instructions. The fold of induction was calculated as the ratio between the signal [expressed as relative light units (RLU)] measured after stimulation with PBA and 1,25-dihydroxyvitamin D3, and the signal during basal conditions.
For the cell-based reporter assay, MN8CampLuc-transfected cells were seeded into 96-well plates at a density of 6 × 104/well and cultured for 48 h in normal growth medium. Afterwards, cells were exposed to either vehicle (control) or test compounds at different concentrations (as specified below). These compounds were either synthesised (see Supplementary Material) or purchased from commercial sources. All stimulations were performed in the absence of hygromycin in the culture medium. After stimulation for 24 h cells were harvested and lysates were assayed for luciferase activity as described above.
Z′-Factor measurement
For assay quality assessment, the Z’ factor of our cell-based assay was calculated according to the method described.
21
Briefly, transfected MN8CampLuc cells were seeded into a 96-well plate, at a density of 6 × 104 cells/well. After 48 h, cells were incubated with vehicle (negative control) or with PBA in combination with 1,25-dihydroxyvitamin D3 (positive control) for 24 h. Technical replicates of 12 wells per condition were prepared. The luciferase activity was recorded as described above in the ‘Luciferase activity assay’ section. The Z’ factor was calculated according to the following formula
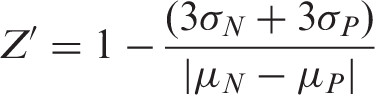
Prestwick Chemical Library screening
The Prestwick Chemical Library of 1200 FDA-approved compounds was kindly provided by Chemical Biology Consortium Sweden and is commercially available from Prestwick Chemical (Illkirch, France). The library compounds were provided in a DMSO solution and were added at a final concentration of 10 µM. The final concentration of DMSO was 0.1%. The luciferase activity was measured in the cell lysates after 24 h, as described above. Four vehicle (0.1% DMSO) and four positive (2 mM PBA and 100 nM Vitamin D3, in combination) controls were run on each plate. The fold induction was calculated as the ratio between the signal recorded in each well and the average signal obtained for the vehicle controls of the corresponding plate. The statistical robustness of the screen was assessed by calculating the Z’-factor of each plate run in the screening.
For hit confirmation, MN8CampLuc cells were treated with each of the positive hits obtained from the initial screen at 11 concentrations, ranging from 100 µM to 1.7 nM (serial dilutions of 1 to 3). The final concentration of DMSO was adjusted to 1% in all wells. The protocol was the same as for the screening.
Real-time PCR
For evaluation of induction at the mRNA level, HT-29 parental cells were seeded either in 6 - or 24-well plates (at a cell density of 1.5 × 106 cells/well or 3 × 105 cells/well respectively) and grown for 24 h before incubation with stimulants or vehicle for an additional 24 h. Sample preparation and quantitative real-time PCR (qRT-PCR) using iQ SYBR Green (Bio-Rad) were performed according to protocol in Steinmann et al. 11
Western blot analysis
Cells were harvested and lysed with RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0). Whole cell lysates were dissolved in lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) containing 50 mM dithiothreitol (Sigma-Aldrich) and incubated for 10 min at 70℃. Afterwards, samples were subjected to gel electrophoresis and Western blot analysis as described elsewhere. 22
Results
Generation and validation of the MN8CampLuc stable cell line
In order to generate a cell model to screen for CAMP gene inducers, the HT-29 human colonic epithelial cell line was transfected with a plasmid containing the CAMP gene, including the upstream promoter region of approximately 3000 bp, fused with the Firefly Luciferase gene (Figure 1A). The clones obtained constitutively express proLL37 as a fusion protein with luciferase (Figure 1B), the activity of which is easily measured in the cell lysate. Using this cell line, compounds that induce the CAMP gene expression are readily identified owing to an increase in cellular luminescence.
Forty-four clones growing in the selective medium were designated with the acronym MN followed by consecutive numbers (i.e. MN1, MN2, etc.). All these clones were tested for their ability to express the fusion protein proLL37-luciferase by measuring the intracellular luminescence (expressed as RLU) both in basal conditions and after co-stimulation with the CAMP inducers PBA (2 mM) and 1,25-dihydroxyvitamin D3 (100 nM) for 24 h. The clones selected exhibited different characteristics in terms of constitutive expression of the fusion protein and response to known inducers. From the 10 clones displaying appropriate baseline level and inducibility, genomic DNA was isolated and subjected to PCR validation. The validated clone having the whole construct integrated in the genome and that showed the highest reproducibility in different experiments with PBA and Vitamin D3 (Figure 1C, D) was chosen as a reporter cell line to develop a cell-based reporter assay: this clone, MN8, was then designated MN8CampLuc. MN8CampLuc cells contains only one copy of the transgene randomly integrated in the genome (see Supplementary Material). The transgene integration was stable even after a high number of passages, proving that the phenotype can be maintained with the appropriate antibiotic pressure (see Supplementary Material).
Development and validation of the CampLuc reporter assay
All steps of the reporter assay were optimised, i.e. number of plated cells, incubation time, cell lysis and luciferase activity measurement, and a standard protocol was set up. To decide if our cell-based assay may be a useful tool to screen for novel inducers of AMP expression we analysed whether it confirmed the results previously observed with traditional techniques. Thus, mRNA and protein levels were assessed by qPCR and Western blot in the parental HT-29 cells stimulated with a number of known inducers (Figure 2A, B). The assay was performed in parallel in MN8CampLuc cells treated under the same conditions as in the HT-29 cell line. BA and PBA have been observed to increase LL-37 expression at both the mRNA and protein level9,11 (here shown in Figure 2A, B). These compounds significantly induced luciferase expression in MN8CampLuc cells and showed synergy with vitamin D3 (Figure 2C). A recent finding from our group showed that lactose stimulated LL-37 expression in HT-29 cells and that the co-incubation with either PBA or BA enhanced this induction in a synergistic way
23
(Figure 2A, B). Accordingly, lactose also increased the fusion protein expression in the cell-based assay, in a synergistic way with PBA or BA (Figure 2C). The expression of LL-37 has also been reported to increase upon induction of the endoplasmic reticulum (ER) stress pathway.
24
We confirmed this result by employing the ER-stress inducer tunicamycin. The parental cell line (HT-29) also showed enhanced transcription of the CAMP gene when stimulated with tunicamycin (11-fold induction; Figure 2A); however, we were not able to detect an increase at the protein level in HT-29 cells (Figure 2B). Similar induction by tunicamycin was observed in MN8CampLuc cells in terms of luciferase activity (Figure 2C).
Validation of MN8CampLuc cells with known inducers of LL-37 expression. The parental HT-29 cell line and MN8CampLuc cells were stimulated for 24 h with compounds known to induce CAMP gene expression. Expression of the CAMP gene was measured (A) at the mRNA level by qRT-PCR in HT-29 and in MN8CampLuc cells, and (B) at protein level in HT-29 cells by Western blot. (C) The response to the known inducers was verified in MN8CampLuc cells incubated for 24 h under the same conditions and then assayed for luciferase activity. Graphs are representative of at least three experiments. Ctrl: control; Vit D: vitamin D3 (100 nM); PBA: sodium phenylbutyrate (2 mM); BA: sodium butyrate (2 mM); Lac: lactose (60 g/l); Tu: tunicamycin (300 nM).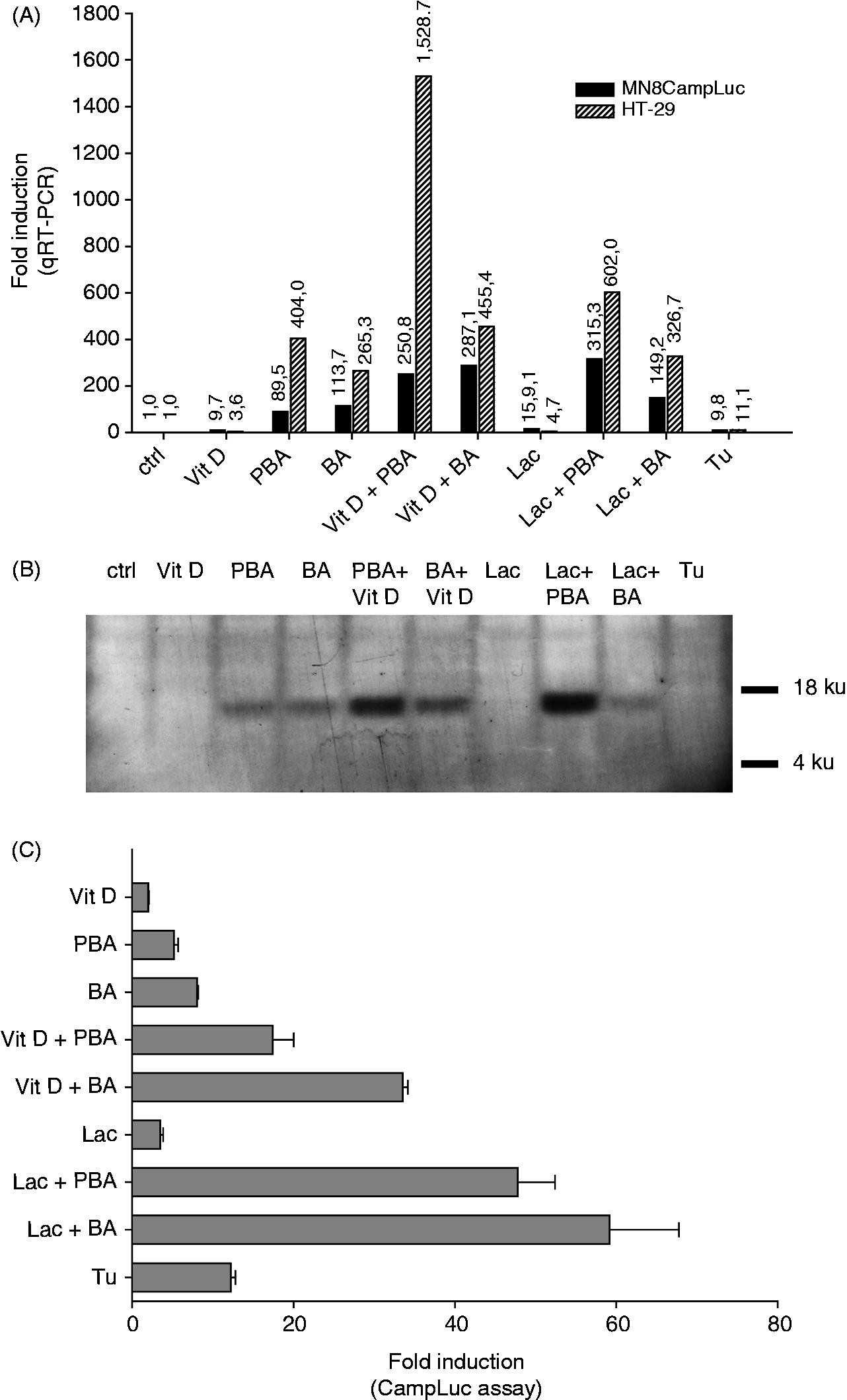
In accordance with the aim to use our cell-based reporter assay to screen for novel inducers of antimicrobial peptides, we evaluated its suitability for high-throughput screenings. This was done by calculating the Z’-factor of the assay, which is a dimensionless screening window coefficient used for assay quality assessment. 21 The Z’-factor was found to be higher than 0.7 (data not shown), where values between 0.5 and 1 predict good performance in high-throughput screenings.
Initial screen of a panel of histone deacetylase inhibitors and structurally-related compounds
We used MN8CampLuc cells to screen a panel of analogues of PBA and BA (synthesised as described in the Supplementary Material). The compounds were tested at different concentrations according to the expected potency and toxicity. All the compounds tested were able to induce proLL37-luciferase to the same level or higher than the positive control, PBA (Figure 3A). Notably, 0.5 mM isovaleric acid and 2 mM isobutyric acid enhanced the fusion protein expression more than PBA, vitamin D3 and the combination of these compounds. We confirmed by qRT-PCR the ability of isovaleric and isobutyric acids to increase the CAMP gene expression in the parental HT-29 cell line. As shown in Figure 3B, both compounds showed higher induction than PBA in the parental cells.
Initial pre-screening of a panel of HDAC inhibitors and derivatives thereof. (A) MN8CampLuc cells were stimulated for 24 h with a panel of nine HDAC inhibitors and structurally related compounds. The assay was carried out in a 96-well format, and each condition was performed in triplicate. Untreated cells were used as negative control (ctrl), while the inducers PBA (2 mM) and vitamin D3 (Vit D; 100 nM), were used as internal positive controls, both alone and in combination. Results are expressed as fold induction of each condition versus the negative control. (B) Confirmation of LL-37 induction in HT-29 parental cells by isovaleric acid (0.5 mM) and isobutyric acid (2 mM) at mRNA level, measured by qRT-PCR. Results are expressed as fold induction and are presented as mean ± SD.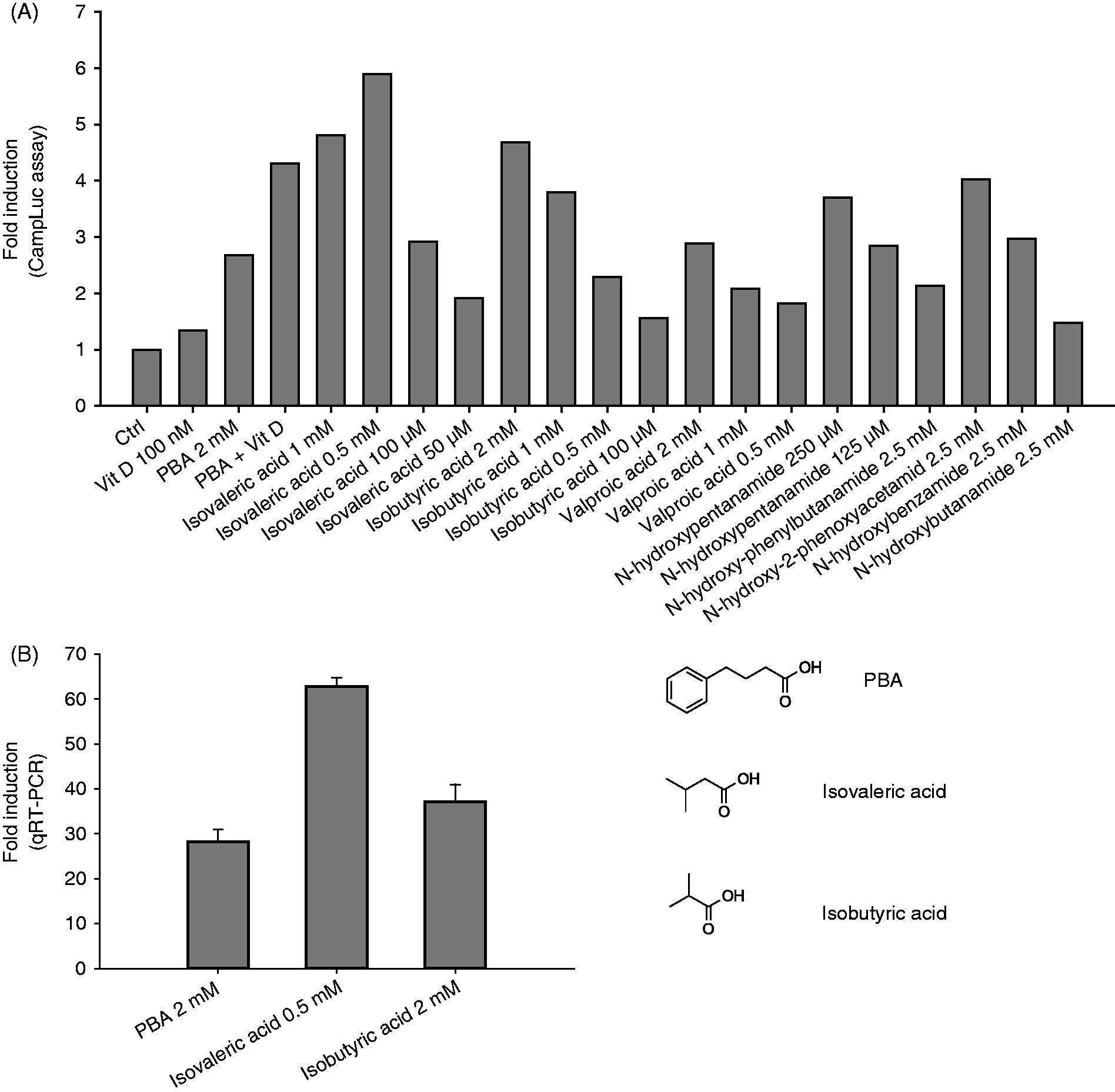
Screening of Prestwick Chemical Library
The CampLuc reporter assay was used to screen the Prestwick Chemical Library of 1200 approved drugs and natural compounds. The positive controls, PBA and vitamin D3 in combination, ranged from a six-fold to an 8.1-fold induction. The threshold for the hits was calculated as µ ± 3σ, giving a 99.7% confidence interval.
21
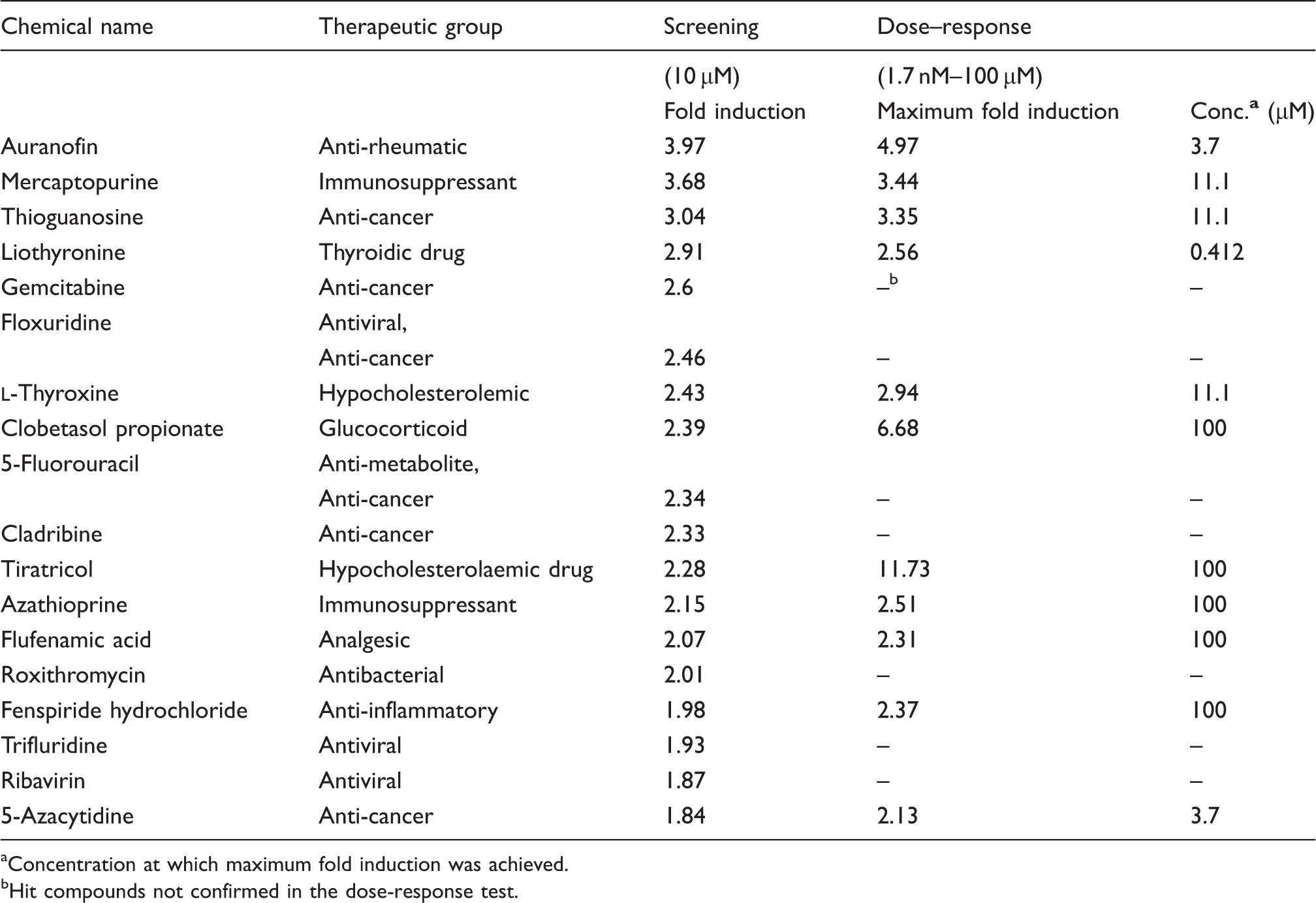
By applying this formula, the threshold for the positive hits was set at 1.83-fold induction, whereas compounds showing less than 0.17-fold induction were considered negative hits. Out of 1200 compounds, 18 positive hits were obtained (1.5% of the library) with fold induction up to almost four (Figure 4 and Table 1). We also found 14 negative hits, corresponding to 1.17% of the whole library (data not shown). The statistical robustness of the screen was assessed by measuring the Z’-factor of each plate: values ranged from 0.7 to 0.91, indicating a very good quality of the assay.
Screen of the Prestwick chemical library. The 1200 compounds of the Prestwick Chemical Library were screened as described in the ‘Material and methods’. The screen spanned 15 plates and in each plate four vehicle controls (0.1 % DMSO) and four positive controls [vitamin D3 (Vit D; 100 nM)] in combination with sodium PBA (2 mM) were included. The signal recorded in each well was normalised to the average vehicle control of the corresponding plate. Using the formula μ ± 3σ, the hit thresholds were calculated (gray lines), giving 18 positive and 14 negative hits. The Z’-factor was calculated for each plate, giving the range of 0.70–0.91. Summary of positive hits and activity. The 18 positive hits are presented with a summary of the fold induction in luciferase activity obtained in the screen and in the dose-response test, as well as concentrations giving optimal activity. aConcentration at which maximum fold induction was achieved. bHit compounds not confirmed in the dose-response test.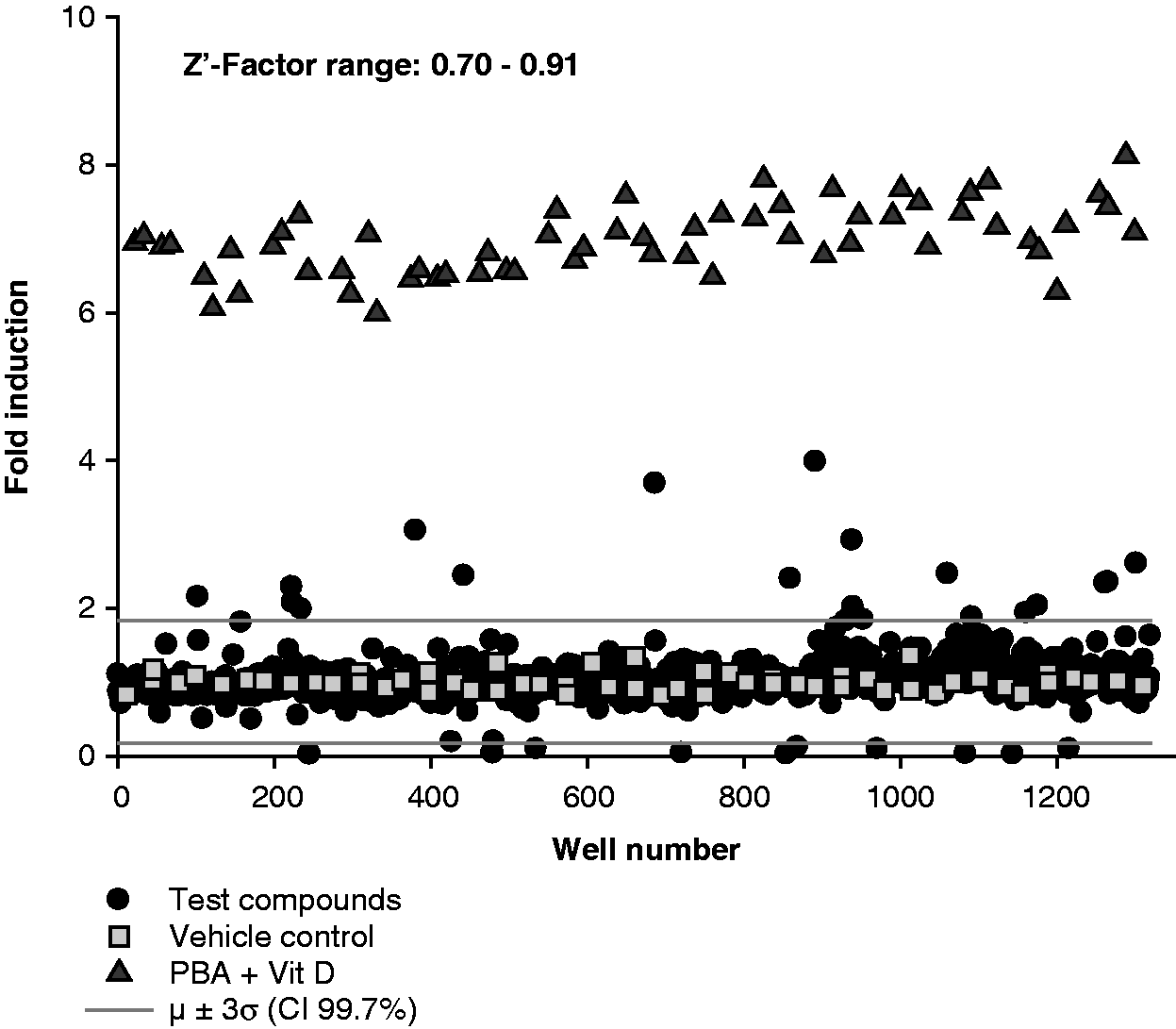
To confirm the positive hits, dose–response was analysed using 11 concentrations for each compound, from 100 µM to 1.7 nM. Out of the 18 compounds tested in this experiment, 11 induced the fusion protein proLL37-luciferase and were confirmed as hits. Ten of the compounds analysed showed dose-dependent expression, whereas liothyronine did not show dose-dependence in the range of concentrations tested (Figure 5). The confirmed hits included 10 approved drugs and one natural compound, the hormone Hit confirmation with dose–response analyses. Dose–response curves were obtained for each of the 18 hit compounds by stimulating MN8CampLuc cells for 24 h with 11 serial dilutions (from 100 µM to 1.7 nM). The assay was carried out in a 96-well format, as described in the ‘Materials and methods’. In the figure the dose–response curves of the 11 compounds that were confirmed as hits are shown. Results are expressed as fold induction versus the vehicle control (1% DMSO).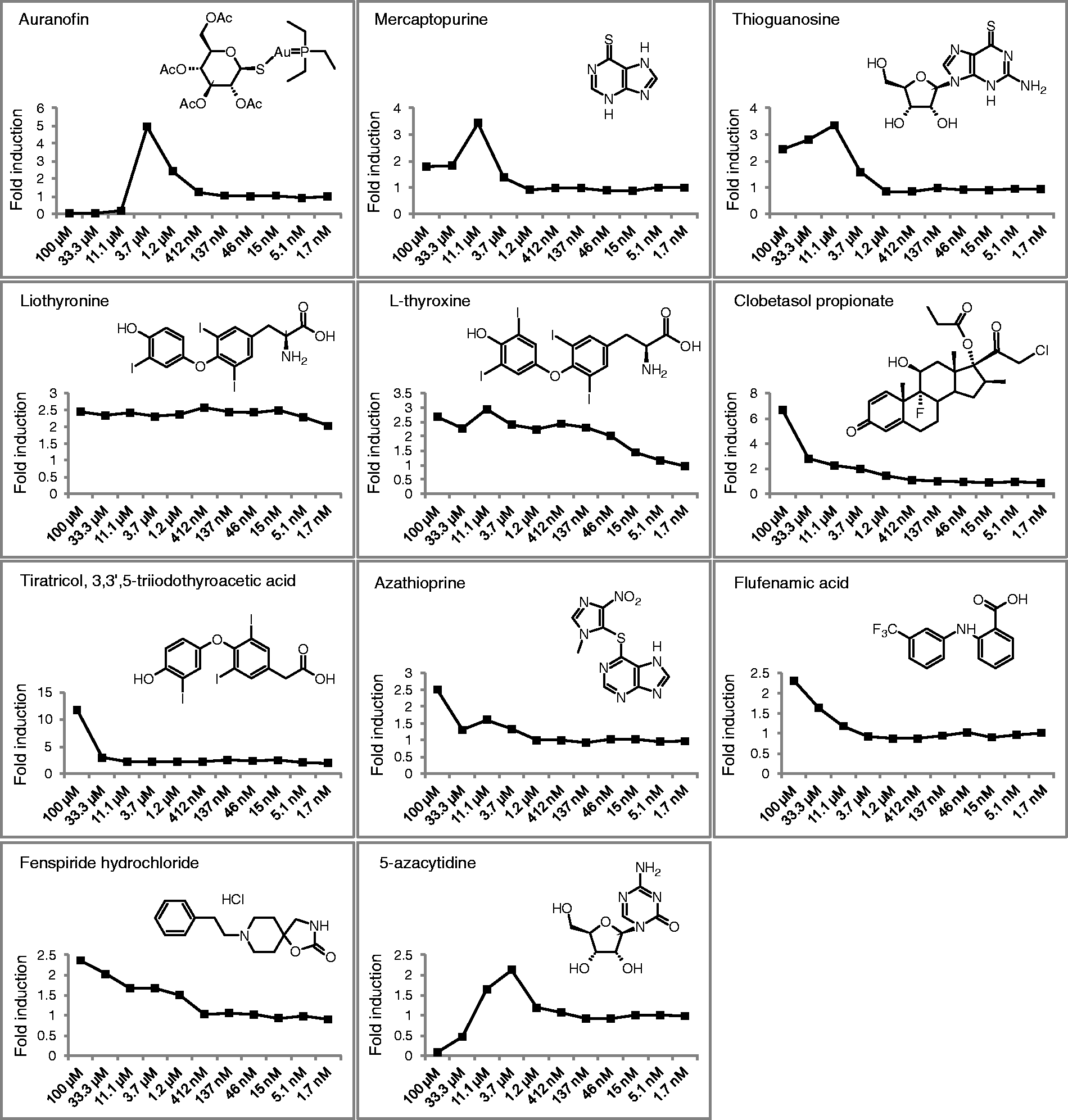
Based on their safety profile, 7 out of the 11 confirmed hits (tiratricol, liothyronine,
For the independent validation, the seven hit compounds were tested at different concentrations, depending on the maximal induction and the cellular toxicity observed in the dose–response analyses. As shown in Figure 6, the FDA-approved drugs clobetasol propionate, auranofin and tiratricol significantly increased the expression of the CAMP gene in HT-29 cells after 24 h of stimulation. The thyroid hormone analogue tiratricol was able to induce only at 100 µM, whereas a lower expression was observed at the other concentrations tested (10 nM, 100 nM and 1 µM). The same decrease of expression of the CAMP gene was measured for the thyroid hormone analogue liothyronine and for the synthetic form of thyroid hormone Independent validation of the confirmed hits. HT-29 parental cells were grown in 24-well plates and stimulated for 24 h with seven compounds of interest from the Prestwick Chemical Library, at various concentrations (determined from the dose–response experiment). The CAMP gene induction was assessed by mRNA level by qRT-PCR. Results are expressed as fold induction and are presented as mean ± SD.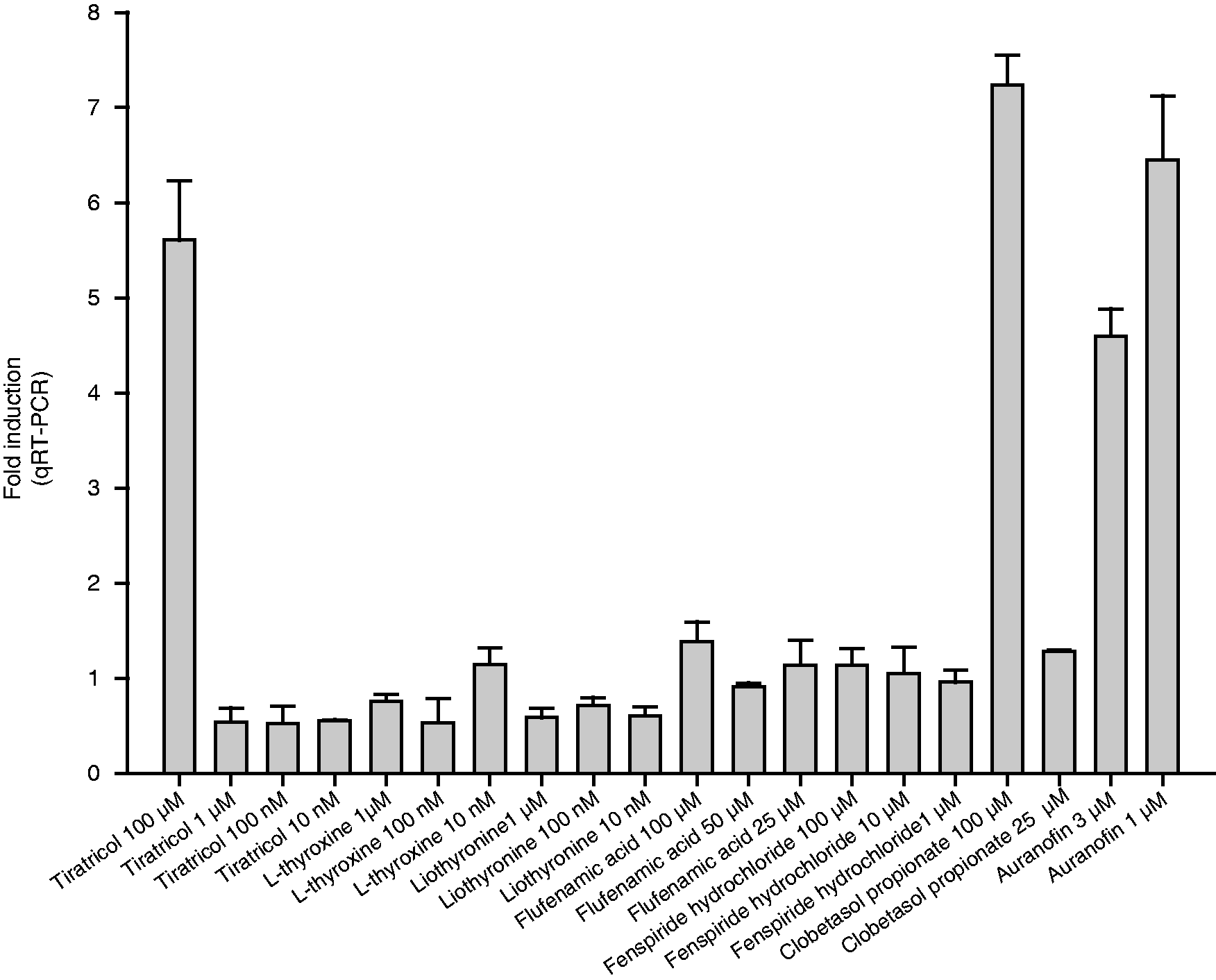
Discussion
In this study we describe the development of the CampLuc assay, a cell-based reporter assay for the expression of the antimicrobial cathelicidin LL-37. The CampLuc reporter assay was validated with known inducers of LL-37 expression, confirming its reliability. This reporter system was first utilised in a small screen of a panel of histone deacetylase (HDAC) inhibitors and derivatives thereof, and then to screen the Prestwick Chemical Library of approved drugs and natural compounds. After initial screenings, hit confirmation and independent validation we identified five hit compounds that might be of interest for the development of novel antimicrobial strategies.
Cell-based reporter assays are useful tools in drug discovery, allowing a rapid screen of large chemical libraries. Such high-throughput screenings call for testing each compound in single wells. Thus, the reproducibility of the assay response to test compounds is crucial. The CampLuc reporter assay performed well in tests of robustness, with a Z’-factor higher than 0.7, proving its suitability for high-throughput screenings. 21 It is to be noted that the pre-proLL37 contains a signal sequence targeting it for secretion. However, no luciferase activity was detected in the supernatants of MN8CampLuc cells, indicating intracellular sequestration of the fusion protein rather than secretion in the extracellular milieu. The fusion protein proLL37-luciferase is most likely retained in intracellular vesicles and cannot follow the normal exocytotic trafficking pathway.
Two of the five hit compounds, isovaleric and isobutyric acids, were identified in the screening of analogues of BA and PBA, compounds that we have identified as CAMP inducers.9,11 PBA and BA are short-chain fatty acids that inhibit class I HDAC and hence promote the maintenance of the histone acetylated state. Histone acetylation plays a critical role in gene transcriptional regulation by causing chromatin relaxation and allowing the binding of transcription factors to DNA regulatory elements. 25 Yet, the molecular mechanism for BA- and PBA-elicited induction of the CAMP gene is not clear, as it has been shown to be secondary and to depend on the de novo synthesis of unknown specific factor(s). 11 Analogues of PBA and BA were tested in the CampLuc assay, and all were able to induce the CAMP gene at least to the same level as the positive control PBA. Interestingly, we did not observe a clear correlation between HDACi activity and induction of LL-37, as suggested by the fact that PBA is 100-fold less potent compared with its hydroxamic acid N-hydroxy-4-phenylbutanamide and with valproic acid as an HDAC inhibitor, 26 and yet the increase in proLL37-luciferase was comparable for these compounds. However, isovaleric acid and isobutyric acid enhanced LL-37 expression more than the combination of PBA and vitamin D3 in the CampLuc assay, suggesting additional mechanism(s) for the induction by these compounds. Nonetheless, taking into account the similarity in structure with BA and PBA, 26 we cannot rule out the possibility that isovaleric and isobutyric acid might inhibit HDAC activity.
Compounds that stabilise luciferase and increase its intracellular levels can lead to false-positives in screening assays utilising luciferase as a reporter enzyme. 27 Therefore, it is crucial to independently confirm the effect of the hit compounds on the expression of the CAMP gene. We did this for isovaleric and isobutyric acid, assessing mRNA levels by qRT-PCR in HT-29 parental cells, and, indeed, we confirmed the induction exerted by these BA analogues. Despite their ability to induce LL-37 expression, in our opinion isovaleric and isobutyric acid are not of interest for clinical applications owing to their unpleasant odor. However, the mechanism behind the induction of the CAMP gene by isovaleric and isobutyric acids is still unknown and studies of the transcriptional regulation are ongoing. This might add relevant insight into the regulation of the CAMP gene and, perhaps, on the expression of additional antimicrobial peptides, leading to a better understanding of innate immunity.
Interestingly, isovaleric acid is found at high concentrations in the blubber of dolphins. 28 About 40% of adult bottlenose dolphins show marks from shark attacks that leave the dolphins with a great deal of tissue injury and, remarkably, with no sign of infection. The wound healing observed in the dolphins resembles tissue regeneration rather than healing, and the first part of this process covers the wound with blubber. Isovaleric acid is believed to control local microbial growth, as it exhibits antimicrobial activity per se. 28 From the results of our work we propose that isovaleric acid also acts as an activator of the AMP system and works in two ways to halt infection.
The screening of the Prestwick Chemical Library yielded 18 hit compounds. The Prestwick Chemical Library consists of 1200 small compounds with high chemical and pharmacological diversity. The number of hits as percentage of the library was 1.5%, comparable with other published screens,29–31 suggesting that the assay was specific and would yield a low number of false-positives. Nonetheless, only 11 of the initial 18 hits were confirmed in the dose–response analyses, and this was expected owing to experimental variation. However, this can also reflect the different experimental conditions in which the two experiments were run: the screening was performed with a 0.1% final concentration of DMSO, whereas the dose–response curves were conducted using 1% DMSO concentration. The CAMP gene was shown to be induced by ER stress, 24 and some of the hits found in the screening might affect the transcription of the CAMP gene via ER stress. In this regard, the concentration of DMSO in the set-up of our assay might be crucial, as DMSO is known to prevent ER stress. 32 To validate this theory we re-analysed the hit compounds under the original conditions (10 µM, 0.1% DMSO) and they all induced again (data not shown). Although giving further strength to the assay, we do not believe that using ER stress to induce AMPs is the way forward and we therefore discontinued all work with these compounds.
The active compounds of the Prestwick Library are all FDA-approved drugs and natural compounds, presenting the greatest possible degree of drug likeliness. Information about bioavailability and safety in humans are available for the Prestwick compounds. Based on their safety profile and side effects, we chose seven of the 11 confirmed hits for independent validation in HT-29 parental cells. Of these seven clobetasol propionate, auranofin and tiratricol were shown to enhance the endogenous transcription of the CAMP gene after 24 h.
The inducing effect of clobetasol propionate was seen at a concentration of 100 μM. clobetasol propionate is used topically in the treatment of skin disorders, such as psoriasis and eczema. 33 It is difficult to directly compare in vitro and in vivo doses; however, the concentration of clobetasol propionate needed to achieve an induction of the CAMP gene seems to lie far above the therapeutic dose used in humans. Nonetheless, the observation that clobetasol propionate enhances the expression of the antimicrobial peptide LL-37 adds information about the transcriptional regulation of the CAMP gene, as it is a potent corticosteroid and is known to activate the glucocorticoid receptor signalling. 34 The prolonged treatment with corticosteroids has been associated with increased risk of bacterial and fungal infections.35,36 It is worth noting that 17 compounds displaying corticosteroid activity are present in the Prestwick Chemical Library. Among them, only clobetasol was able to induce LL-37 expression in the CampLuc assay. This might indicate that the use of this drug would be preferable to other drugs with the same therapeutic indication. However, further investigations in a clinical setting are needed.
Auranofin is an organo-gold compound utilised in rheumatoid arthritis to ameliorate the symptoms, even though little is known about the mode of action. 37 Auranofin is known to regulate the transcription factors Nrf2 and NF-κB, which are responsible for the antioxidant and anti-inflammatory responses respectively.38–40 More studies are needed to address the question of which molecular mechanisms are responsible for the induction of the CAMP gene by auranofin.
Induction of the CAMP gene was also confirmed in HT-29 parental cells for the thyroid hormone analogue tiratricol, but only at the highest concentration tested (100 μM), whereas at lower concentrations (1–10 nM) the mRNA levels were lower than in the control after 24 h of incubation. The same reduction versus control was observed for the synthetic thyroid hormone
Footnotes
Conclusions and future perspectives
In this article we have described a cell-based reporter assay for inducers of LL-37 expression, which we see as a marker for the entire AMP system. This assay can be a powerful screening tool for drug discovery. By employing this cell-based system we have found several novel inducers of LL-37. The next step will be to test some of these compounds in preclinical trials for their ability to up-regulate AMP expression and prevent or treat infections in mouse and rabbit models of infectious diseases. Drugs enhancing the endogenous AMP system and thus strengthening immune defences against pathogens could be applied to several infectious diseases, i.e. tuberculosis, shigellosis, enteropathogenic E. coli and other enteric infections,41,42 and the field is open for additional models of diseases. As bacterial resistance is an increasing burden for healthcare and society, alternative therapy against infections is urgently needed and novel treatment regimens based on this concept can emerge for the treatment of infectious diseases. In this respect, our results offer great promise for the rapid development of novel effective strategies for infectious diseases.
Funding
The authors are supported by the Swedish Research Council (56X-1217-01-6 and 20854, a Linneus grant CERIC), The Swedish Strategic Foundation: RBd08-0014, Swedish Cancer Society: CAN 2011/559, Karolinska Institutet, the Icelandic Centre for Research (RANNIS) and the University of Iceland Research Fund. Gudmundur H. Gudmundsson is a visiting scientist at Karolinska Institutet supported by the Wenner-Gren foundation. The Prestwick Chemical Library and plating thereof was provided by the Chemical Biology Consortium Sweden (CBCS).
Acknowledgments
We are grateful to Hanna Axelsson, at CBCS and Karolinska Institutet, for discussions and advice regarding assay formatting and data analysis.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.