
Abstract
This study examines the interaction of polymyxin B and colistin with the surface and outer membrane components of a susceptible and resistant strain of Klebsiella pneumoniae. The interaction between polymyxins and bacterial membrane and isolated LPS from paired wild type and polymyxin-resistant strains of K. pneumoniae were examined with N-phenyl-1-naphthylamine (NPN) uptake, fluorometric binding and thermal shift assays, lysozyme and deoxycholate sensitivity assays, and by 1H NMR. LPS from the polymyxin-resistant strain displayed a reduced binding affinity for polymyxins B and colistin in comparison with the wild type LPS. The outer membrane NPN permeability of the resistant strain was greater compared with the susceptible strain. Polymyxin exposure enhanced the permeability of the outer membrane of the wild type strain to lysozyme and deoxycholate, whereas polymyxin concentrations up to 32 mg/ml failed to permeabilize the outer membrane of the resistant strain. Zeta potential measurements revealed that mid-logarithmic phase wild type cells exhibited a greater negative charge than the mid-logarithmic phase-resistant cells. Taken together, our findings suggest that the resistant derivative of K. pneumoniae can block the electrostatically driven first stage of polymyxin action, which thereby renders the hydrophobically driven second tier of polymyxin action on the outer membrane inconsequential.
Introduction
Klebsiella pneumoniae is an opportunistic pathogen, which has been implicated in serious nosocomial infections 1 and associated with high rates of mortality, 2 particularly in immunocompromised patients. Concerns have thus been generated in response to the escalation in the incidence of multidrug-resistant (MDR) K. pneumoniae infections,3–5 with the realization that effective therapeutic options are becoming increasingly limited. In view of the waning antibacterial development pipeline, interest in colistin (syn. polymyxin E) and polymyxin B has been renewed, and both are increasingly used as a last-line therapy. 6 Indeed, considerable in vitro activity against K. pneumoniae strains has been demonstrated; 7 98.2% of general clinical isolates of K. pneumoniae are susceptible to polymyxin.7,8 Extremely drug-resistant strains, which are also resistant to polymyxins, have emerged.9,10 These findings demand a greater appreciation of the mechanism(s) of polymyxin resistance employed by K. pneumoniae to assist targeted drug discovery strategies against these MDR Gram-negative pathogens.
The Gram-negative outer membrane constitutes a permeability barrier to various noxious substances, including numerous antimicrobials.11,12 This complex asymmetrical structure comprises an inner phospholipid leaflet, as well as an outer leaflet that contains, predominantly, LPS, proteins and phospholipids (Figure 1A). Structurally, LPS is composed of three domains: (i) the variable O-antigen chain (encompassing repeated saccharide units); (ii) a core-oligosaccharide region and (iii) the conserved lipid A moiety (Figure 1B). Lipid A is embedded within the outer membrane leaflet, functioning as a hydrophobic anchor.
13
The consensus structure of lipid A is represented by a β-(1→6)-linked (A) A schematic that depicts the gross structural organization of the Gram-negative outer membrane. (B) Silver-stained SDS-PAGE analysis of LPS from the K. pneumoniae ATCC 13883 strains examined in this study. Increasing sample amounts (0.5, 1.0, 1.5 µg) were loaded per lane to allow for visualization on minor components and owing to the differential staining of each sub-structure. The position of each LPS sub-structural component is indicated on the right ordinate. Molecular mass standards are shown on the left ordinate, in lane 1. The LPS of E. coli serotype 0111:B4 (Sigma-Aldrich) is shown as a reference in lane 2. The dashed line indicates the mass shift of the lipid A inner core between the paired strains. (Left panel) A schematic that depicts the reported sub-structures for K. pneumoniae serotype O2 LPS and K. pneumoniae lipid A (variable fatty acyl positions are indicated in bold and dashed line).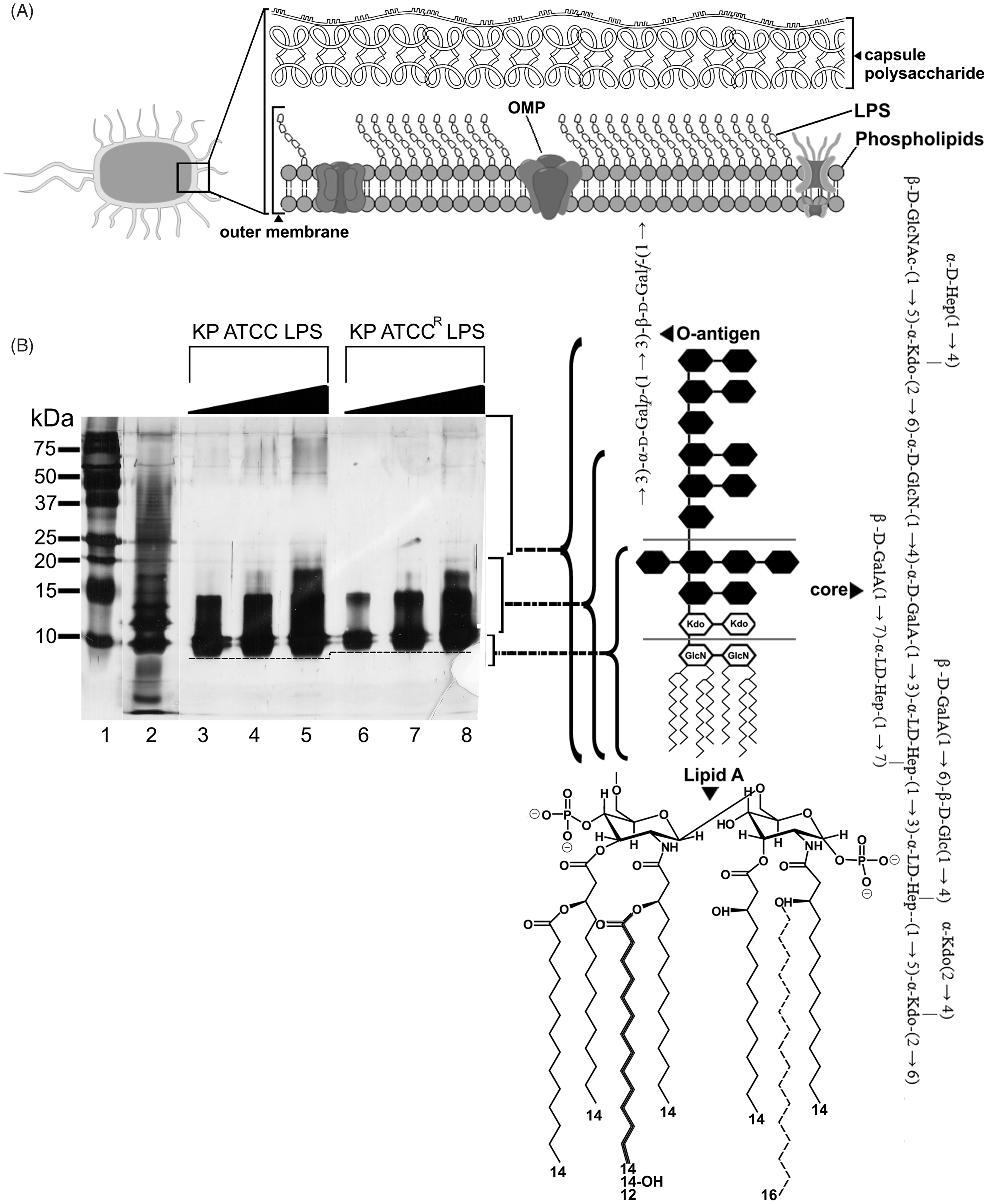
The precise mechanism of action of polymyxins remains contentious and, based on biophysical studies, a number of models have been put forward.
23
The consensus view is that polymyxins are membrane surface-active antibiotics and that lipid A is an important polymyxin binding target in the outer membrane of Gram-negative species.
23
A well-accepted model, termed the ‘self-promoted uptake’ pathway, purports that the amphipathic nature of polymyxins is crucial to enable uptake of the polymyxin molecule across the outer membrane barrier.24–27 In this model, protonation of free γ-amines present on diaminobutyric acid residues of polymyxins at physiological pH provides a means of electrostatic attraction to anionic lipid A phosphates. The resultant displacement of divalent cations that stabilize the LPS leaflet allows the hydrophobic N-terminal fatty-acyl tail, and
Polymyxin resistance in K. pneumoniae involves the multi-tier up-regulation of capsular polysaccharide expression, and the systems required for the modification of lipid A with 4-amino-4-deoxy-
In this study we employed a series of biophysical methods to probe the correlation between polymyxin resistance, and the ability of polymyxin B and colistin to interact with the bacterial surface and LPS isolated from paired wild type and polymyxin-resistant strains of K. pneumoniae American Type Culture Collection (ATCC) 13883. The obtained data shed new light on the link between bacterial killing activity and the interaction of polymyxins with the biophysical barriers and components of the K. pneumoniae surface that confer resistance.
Materials and methods
Materials
Polymyxin B (sulfate), colistin (sulfate), N-phenyl-1-naphthylamine (NPN), lysozyme and deoxycholate were obtained from Sigma-Aldrich (Sydney, NSW, Australia). All other reagents were of the highest purity commercially available. Stock solutions of polymyxin B and colistin (10 mg/l) were prepared in Milli-Q water (Millipore, North Ryde, NSW, Australia) and filtered through 0.22-µm syringe filters (Sartorius, Melbourne, VIC, Australia). Solutions were stored at 4℃ for up to 1 month. 40
Bacterial strains and growth conditions
K. pneumoniae ATCC 13883 was obtained from the ATCC (Manassas, VA, USA) and used as a reference strain [colistin minimum inhibitory concentration (MIC) = 0.5 µg/ml]. A paired colistin-resistant strain (colistin MIC > 128 µg/ml) was selected from this reference strain in the presence of 10 µg/ml colistin. All bacteria were stored at −80℃ in tryptone soya broth (Oxoid, West Heidelberg, VIC, Australia). Prior to experiments, parent strains were subcultured onto nutrient agar plates (Medium Preparation Unit, University of Melbourne, VIC, Australia). Overnight (24 h) broth cultures were subsequently grown in 5 ml of cation-adjusted Mueller-Hinton broth (CaMHB; Oxoid), from which a 1 in 100 dilution was performed in fresh broth to prepare mid-logarithmic cultures according to the OD at 500 nm (OD500nm = 0.4–0.6). All broth cultures were incubated at 37℃ in a shaking water bath (180 rpm).
Determination of MIC
MICs for each strain were determined by the broth microdilution method. 41 Experiments were performed with CaMHB in 96-well polypropylene microtiter plates. Wells were inoculated with 100 µl of bacterial suspension prepared in CaMHB [containing 106 colony forming units (cfu) per ml] and 100 µl of CaMHB containing increasing concentrations of polymyxins (0–128 µg/ml). The MICs were defined as the lowest concentration at which visible growth was inhibited following 18 h incubation at 37℃. Cell viability was determined by sampling wells at polymyxin concentrations greater than the MIC. These samples were diluted in normal saline and spread plated onto nutrient agar. After incubation at 37℃ for 20 h, viable colonies were counted on these plates. The limit of detection was 10 cfu/ml.
LPS isolation
LPS was extracted by a modified hot phenol/water procedure.
42
Overnight culture was transferred into 500 ml (1:100 dilution) of CaMHB and grown to mid-logarithmic phase. Cells were then pelleted by centrifugation at 10,000 g for 10 min at 4℃. Pelleted cells were washed in PBS (pH 7.2) and re-suspended in 40 ml PBS. Cells were lysed by ultrasonication on ice (5 × 30 s). The unbroken cells and debris were removed by centrifugation at 5400 g for 45 min at 4℃. The supernatant was removed and centrifuged for 1 h at 16,000 g. Pellets were suspended in 500 µl of 20 mM Tris-HCl buffer (pH 7.0). To ensure efficient nucleic acid digestion, DNase and RNase (Sigma-Aldrich) were added to final concentrations of 80 U/ml and 50 µg/ml respectively. The solution was incubated for 2 h at 37℃ before the addition of 50 µg/ml Proteinase K (Promega, Sydney, NSW, Australia) followed by a further incubation at 56℃ for 2 h. Samples were heated to 65℃ and mixed (1:1, v/v) with pre-heated (65℃) phenol. Samples were vortexed and incubated at 65℃ for 15 min, then cooled to room temperature (20℃) and centrifuged for 45 min at 5400 g. The aqueous phase was collected and the phenol phase was re-extracted twice. All aqueous samples were combined, dialyzed with a Slide-A-lyzer dialysis kit (Pierce, Rockford, IL, USA) and freeze-dried. The purified LPS samples were stored at −20℃. The lyophilized product was then reconstituted in Tris-HCl buffer (20 mM pH 7.0). The purity of isolated LPS samples was ascertained by SDS-PAGE. LPS samples were re-suspended in SDS-PAGE sample buffer (2.3% SDS, 0.8% Tris, 10% glycerol, 5% dithiothreitol) and boiled for 10 min. SDS-PAGE was conducted using the Laemmli buffer system
43
in 15% polyacrylamide gels, and LPS was visualized by silver staining (Figure 1B).
44
The molar concentration of 3-deoxy-
Fluorometric measurement of [Dansyl-Lys]1-polymyxin B3 binding to whole cells and isolated LPS
The fluorometric measurements were performed as previously described using the fully synthetic polymyxin probe [Dansyl-Lys] 1 PmB3. 31
Deoxycholate sensitivity induced by polymyxins
The protocol for this assay was adapted from Vaara et al. 46 with minor modifications. Mid-logarithmic phase cells (OD500nm = 0.4–0.6) were pelleted and re-suspended in 5 ml of 5-mM HEPES buffer (pH 7.2) in the presence of increasing concentrations of colistin, then incubated at 37℃ for 10 min. After the OD500nm of the cell suspensions were measured, cells were again pelleted and re-suspended in 5 ml of the same buffer containing 0.25% (w/v) deoxycholate. These suspensions were incubated at 37℃ for 10 min and the decrease in OD500nm was subsequently measured. Results were expressed as the percentage of OD of each sample in the absence of deoxycholate. All experiments were performed in triplicate using samples from independently grown cultures for reproducibility.
Fluorometric thermal shift assay
The phase transition of the fatty acyl chains of the LPS aggregates was examined in the absence and presence of polymyxin B and colistin via a fluorometric thermal shift assay. Fluorescence measurements were performed on a Cary Eclipse (Varian, Mulgrave, Victoria, Australia) spectrophotofluorometer with NPN as the probe. LPS (50 µM) dissolved in 20-mM HEPES buffer (pH 7.4) together with 5 µM NPN was heated from a starting temperature of 5℃ to a target temperature of 95℃ at 1℃ increments. Data-fitting operations were performed as previously described to obtain information on the phase transition range and its mid-point (Tm). 47 The temperature mid-point represents the gel-to-liquid crystalline transition temperature point, which occurs when 50% of the fatty acyl chains melt. 48
Permeability to lysozyme-induced by polymyxins
This method was performed as previously described. 46 Lysozyme (5 µg/ml) and colistin (9 µg/ml) were added to a mid-logarithmic phase cell suspension (OD500nm = 0.4–0.6), and the decrease in OD500nm was measured. Results were expressed as the percentage of the OD of control reactions in the absence of permeabilizing agents.
NPN uptake assay
NPN uptake was measured as previously described.
49
Briefly, mid-logarithmic phase cells were pelleted and washed twice in 5-mM HEPES buffer (pH 7.2) containing 1 mM sodium azide, then re-suspended in the same buffer (OD500nm = 0.5). The cell suspension was allowed to sit at room temperature for 30 min before analysis. NPN fluorescence was measured with the excitation and emission wavelengths set at 350 and 420 nm, respectively, with slit widths of 5 nm. The NPN uptake assay was also employed to measure the outer membrane permeabilizing activity of polymyxins. NPN was dissolved in acetone at a concentration of 500 µM and added to 1 ml of cell suspension to a final concentration of 10 µM. The initial baseline was recorded within the first 30 s of NPN addition. Upon the addition of polymyxins, the increase in NPN fluorescence intensity was monitored over 360 s. The average of the baseline was taken over 20–30 s and subtracted from the maximum response at each polymyxin concentration. Response–concentration curves were plotted from baseline corrected values. EC50 values were determined from the curves by non-linear regression fitting of the data to equation (1)
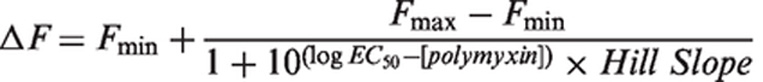
Zeta potential measurements
Zeta potential measurements were performed as previously described with minor modifications to the protocol. 50 Briefly, the bacterial surface was cleansed by washing twice with Milli-Q water, and finally resuspended in Milli-Q water at 108 cfu/ml and used to fill clear disposable folded capillary zeta cells (ATA Scientific, Taren Point, NSW, Australia). The electrophoretic mobility (EPM) of bacterial cells was measured at 25℃ with a zeta potential analyzer at 150 V (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK) before being converted to zeta potentials using the Helmholtz–Smoluchowski theory. EPM measurements were performed in triplicate on three separately prepared samples. Statistical analyses were conducted using the Student’s t-test and ANOVA using SPSS Statistics 19.0 (IBM Software, Armonk, NY, USA).
Cytoplasmic membrane depolarization assay
Cytoplasmic membrane depolarization was determined by using the membrane potential-sensitive cyanine dye diSC3-5. 51 The assay was performed as previously described 29 with slight modification. One milliliter of mid-log phase cells was harvested by centrifugation at 4000 g for 10 min at room temperature and re-suspended in 1 ml of assay buffer (5 mM HEPES, 20 mM glucose, pH 7.4). The bacterial suspension was then diluted 100-fold using assay buffer to reach an OD600 of 0.005. The cells were first treated with 0.2 mM EDTA (pH 8.0) in order to permeabilize the outer membrane to allow dye uptake. Then, 0.4 µM diSC3-5 was added into the suspension and incubated for 1 h at room temperature. The uptake of diSC3-5 dye into the bacteria was monitored using a Polarstar-Omega (BMG, Melbourne, VIC, Australia) spectrophotometer. Then, 0.1 M of KCl was added to equilibrate the cytoplasmic and external K+ concentrations. Ninety microliters of the cell suspension was placed in each well of a 96-well opti-plate (Grenier Bio-one, Wemmel, Belgium). After the fluorescence level became stable, 10 µl of a concentration series of compounds was added to each well and the fluorescent intensity (excitation/emission 620/670) was measured for 40 min. Ten microliters of vehicle solution (H2O) was used as a negative control in the assay.
1H NMR measurements
Colistin was dissolved in 0.5 ml of D2O (pH 4.0) to a final concentration of 0.1 mM. In order to examine the binding of colistin to LPS, line-broadening experiments were performed by successively adding small aliquots of a solution of LPS to colistin. One-dimensional proton NMR (1H NMR) spectra were acquired on a Varian INOVA 600 MHz spectrometer (Varian, Mulgrave, VIC, Australia) operating at 20℃. The chemical shifts (ppm) were referenced to an internal 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt. 1H NMR spectra were recorded with a total of 16.4 k data points, 64 transients and a recycle delay of 4 s. Data were processed and analyzed with MestRC software (Mestrelab Research, Santiago de Compostela, Spain).
Results
Susceptibility testing and visualization of LPS by SDS-PAGE
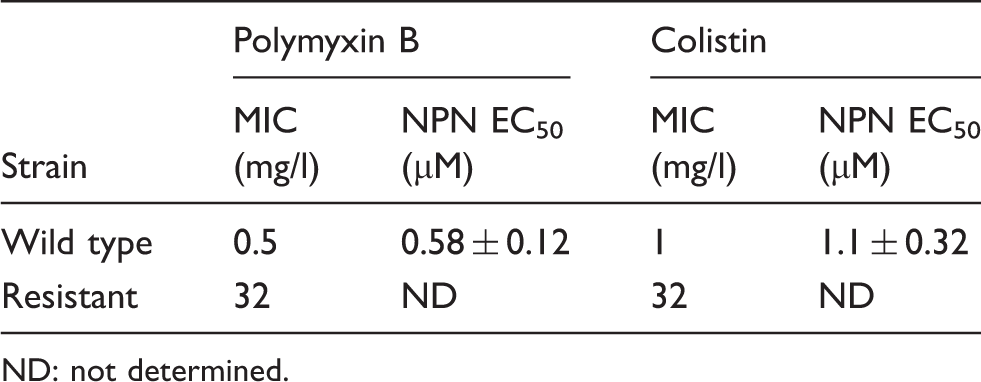
K. pneumonia ATCC 13883 susceptibility to polymyxin B and colistin and half maximal effective concentration (EC50) of polymyxins required to produce permeabilization of the outer membrane to NPN.
ND: not determined.
Fluorometric assessment of [Dansyl-Lys] 1 PmB3 binding to isolated LPS
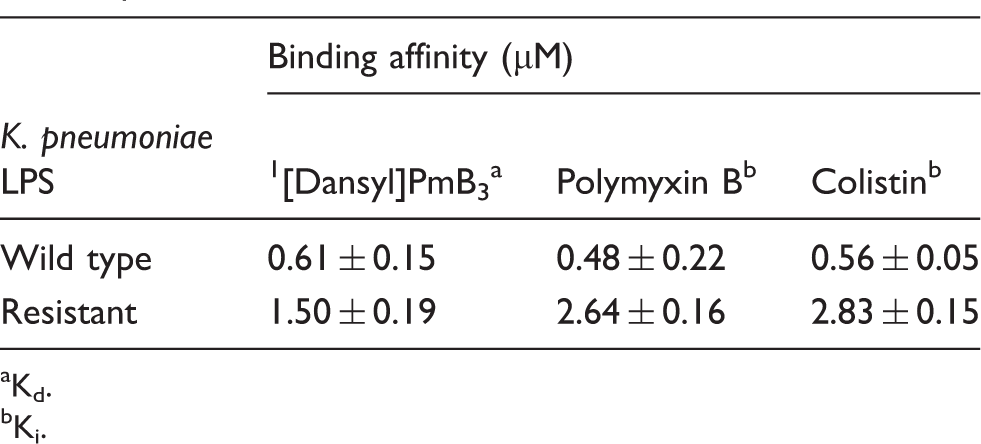
Binding affinity of polymyxins for purified LPS from each K. pneumoniae strain.
Kd.
Ki.
1H NMR analysis of the binding of colistin with LPS aggregates
Titration of LPS aggregates into a solution of colistin produces a line broadening of the colistin resonances with binding to LPS; for simplicity, only the amide region is shown in Figure 2. Line broadening was observed with an increasing colistin:LPS aggregates mass ratio with the wild type LPS aggregates (Figure 2A). Interestingly, line broadening was not observed for LPS from the resistant strain, even at the highest mass ratio (Figure 2B).
600 MHz 1H NMR spectrum of the amide region of colistin in 10% D2O pH 4.0 titrated with LPS aggregates. The ratio of LPS to colistin is indicated on the right ordinate.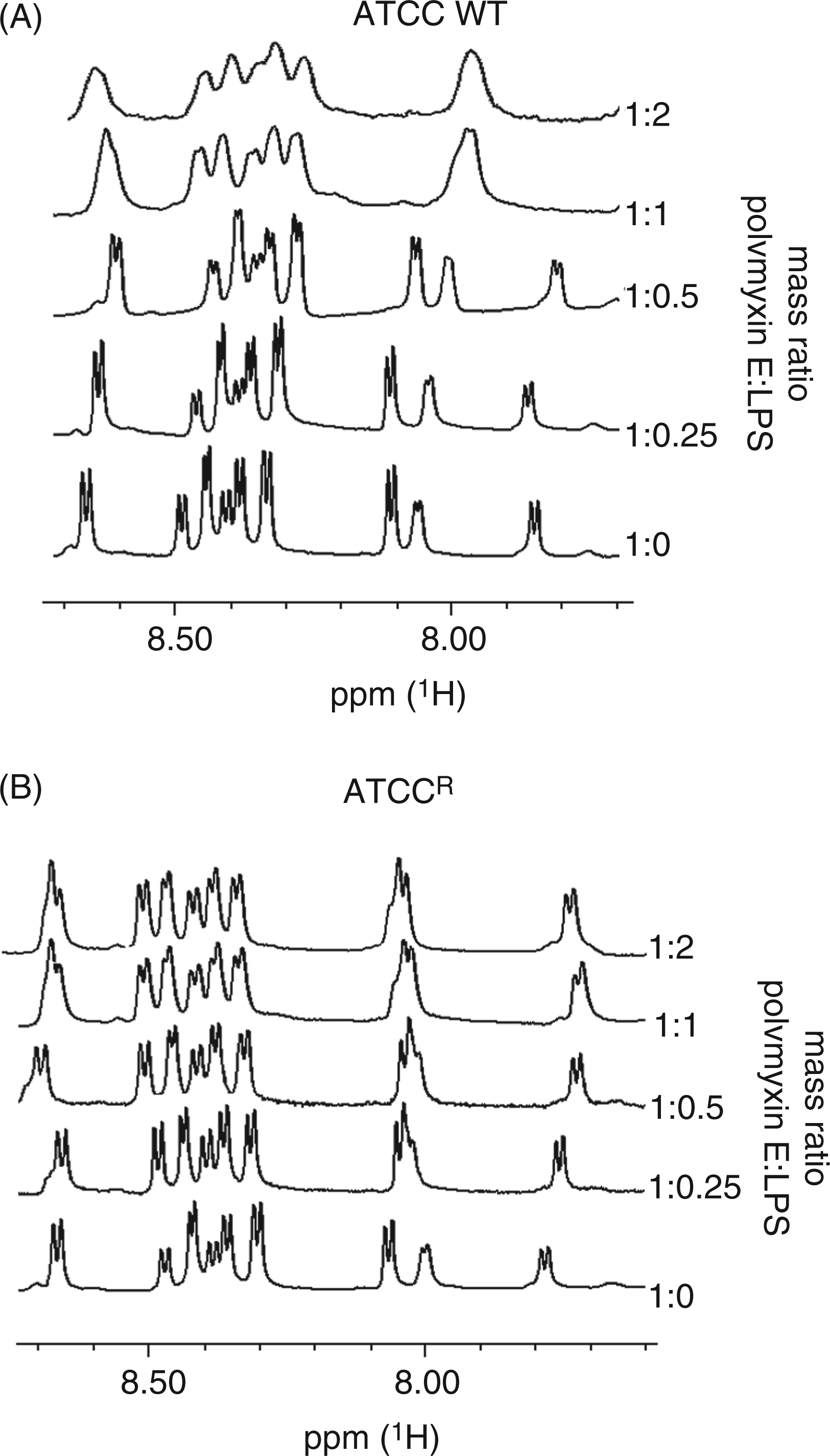
NPN uptake by K. pneumoniae cells
NPN uptake was monitored to assess the permeabilizing activity of polymyxins against the outer membrane of each K. pneumoniae strain. Figure 3A illustrates the increase in fluorescence observed upon the addition of fixed doses of NPN to a solution of K. pneumoniae cells in the absence of polymyxins. The basal levels of NPN uptake for the resistant strain were relatively higher than the wild type strain, indicative of a more permeable outer membrane structure. To test the hypothesis that the higher NPN uptake may be a result of the reduced LPS-to-LPS interactions through weaker binding of divalent cations, the effect of a chelating agent, EDTA, on NPN uptake was assessed. If this is the case then sequestration of divalent cations by EDTA should have little effect on NPN uptake. The addition of EDTA appeared to increase the kinetics of NPN entry and the maximum level of uptake for both the strains (Figure 3A). Notably, for the resistant strain the effect of EDTA on NPN permeability was far less pronounced, which is consistent with a lower dependency on divalent cation stabilization for their outer membrane integrity.
(A) NPN uptake kinetics of a suspension of mid-logarithmic phase K. pneumoniae cells. (B) Effects of increasing concentrations polymyxin B and colistin on the uptake of NPN into the outer membrane of K. pneumoniae ATCC 13883 and (C) polymyxin-resistant K. pneumoniae ATCCR. (D) The normalized peak NPN fluorescence is plotted as a function of the polymyxin B and colistin concentration. K. pneumoniae ATCC 13883, polymyxin B (blue, ▴); K. pneumoniae ATCC 13883, colistin (red, ◊).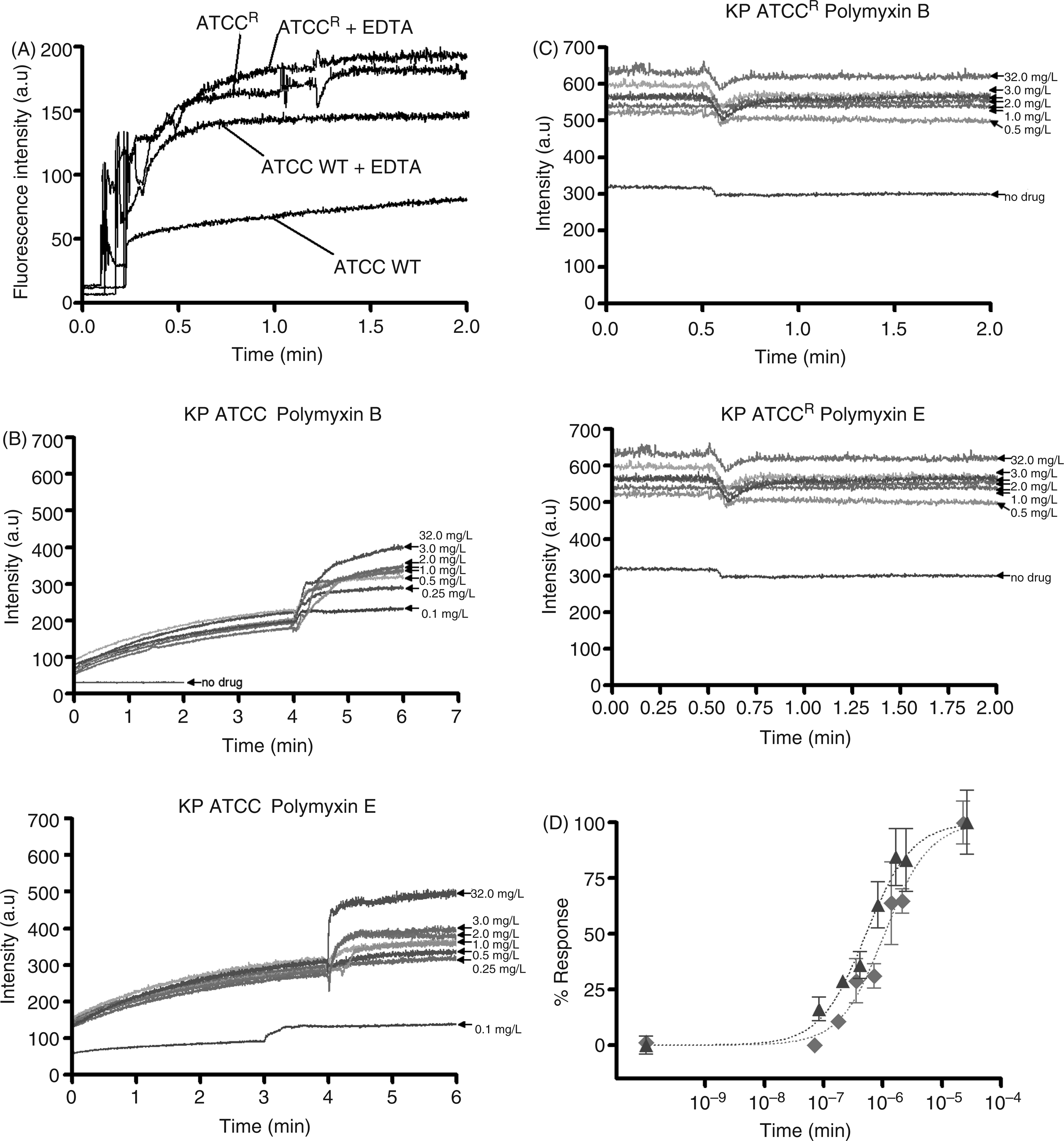
Figure 3 (B, C) shows the raw kinetics data for the change in fluorescence observed upon the addition of fixed doses of polymyxin B or colistin to a solution of K. pneumoniae cells in the presence of NPN. Polymyxin-induced NPN uptake in the wild type strain was rapid and reached a plateau level (Figure 3B). In comparison, neither polymyxin B nor colistin produced any change in the basal level of NPN uptake for the resistant strain, even at the highest polymyxin concentration that is equal to the MIC (32 µg/ml) (Figure 3C). EC50 values in Table 1 were obtained from plots of the increase in NPN fluorescence as a function of the polymyxin concentration (Figure 3D). EC50 values could not be calculated for the resistant strain, which showed a poor NPN response following polymyxin exposure.
Lysozyme and deoxycholate permeability assay
Incubation of whole-cell suspensions with deoxycholate, lysozyme or polymyxins alone had a negligible effect on the OD of the bacterial suspensions (data not shown). However, the addition of colistin to cell suspensions in the presence of lysozyme (Figure 4A) or deoxycholate (Figure 4B) produced a decrease in OD, which may be interpreted as an increase in deoxycholate or lysozyme-mediated cell lysis. For the wild type strain, the lysozyme time-course reactions indicated a rapid reduction in OD following colistin exposure, which reflects an almost instantaneous disruption of the outer membrane barrier (Figure 4A). The resistant strain was impervious to lysozyme or deoxycholate lysis at the same colistin concentration that lysed the wild type, and only showed lysis at polymyxin concentrations ≥MIC (Figure 4A, B).
(A) Colistin-induced lyozyme sensitivity. The OD500 of mid-logarithmic phase K. pneumoniae cell suspensions in the presence of lysozyme following exposure to colistin. (B) Deoxycholate induced cell lysis following exposure to increasing concentrations of colistin. ATCC 13883 (▴) and ATCCR (▪). Data points are the mean ± SD of three independent measurements.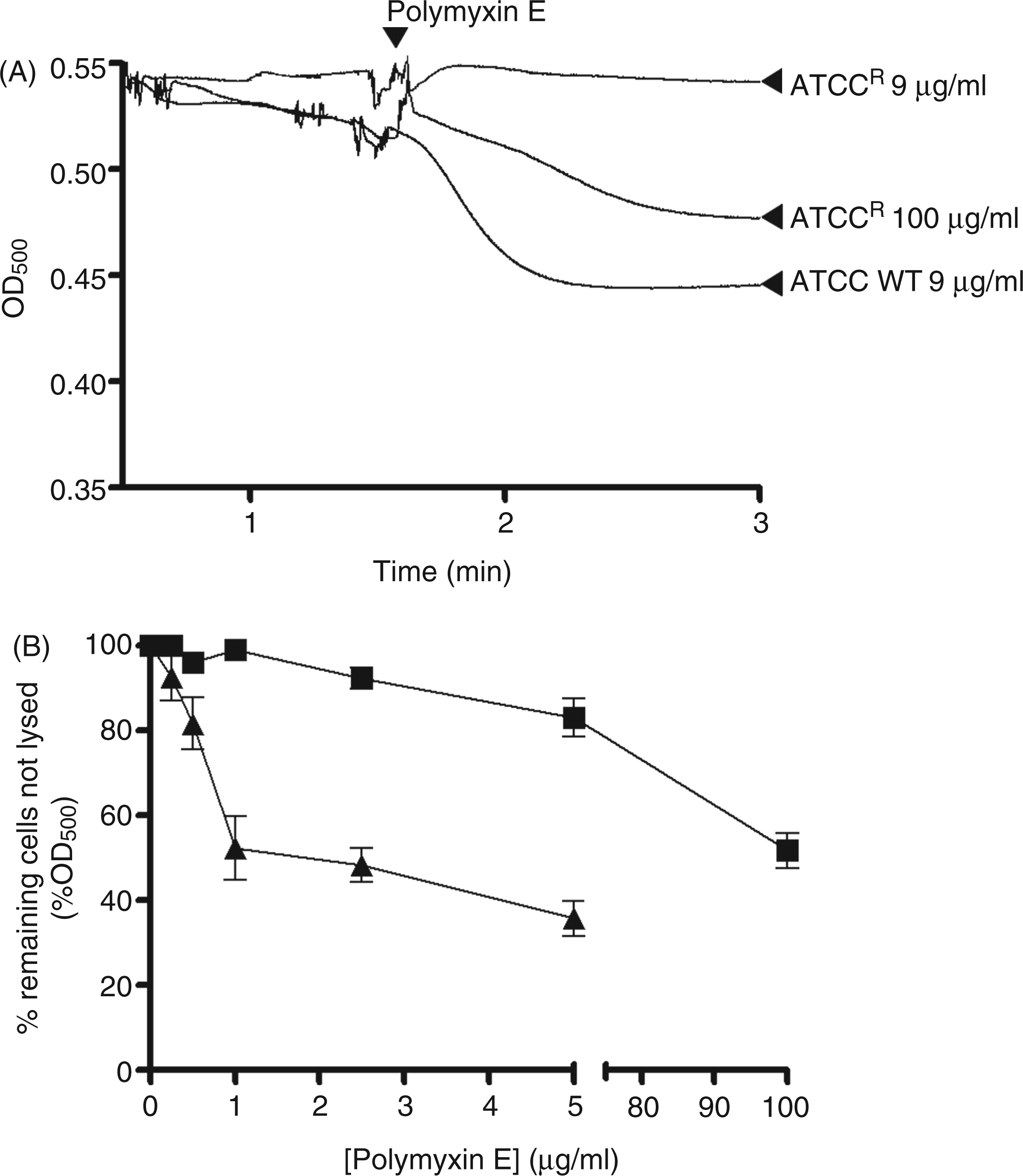
Thermal transition temperature of K. pneumoniae LPS aggregates
Figure 5 shows the phase behavior of LPS aggregates from each strain, the phase-transition temperature and its mid-point (Tm). For the wild type LPS aggregates, the gel↔liquid crystalline phase transition of the fatty acyl chains took place between 18 and 38℃, with a Tm of 26℃ (Figure 5A). In the case of the LPS aggregates from the resistant strain, the gel↔liquid crystalline phase-transition occurred in the temperature range of 16–36℃, with a Tm of 23℃ (Figure 5B). In the presence of a sub-stoichiometric colistin concentration, the phase transition temperature and Tm of all of the LPS samples were slightly shifted to higher values (wild type LPS Tm = 28℃; resistant LPS Tm = 23.5℃). In contrast, in the presence of stoichiometric levels of colistin, no phase transition was observed, indicating complete fluidization of the LPS acyl chains (Figure 5).
Fluorometric thermal shift measurements of K. pneumoniae LPS (50 µM) melting profiles in the presence of colistin. The NPN-fluorescence intensity is plotted as function of temperature. (A) ATCC 13883; (B) ATCCR.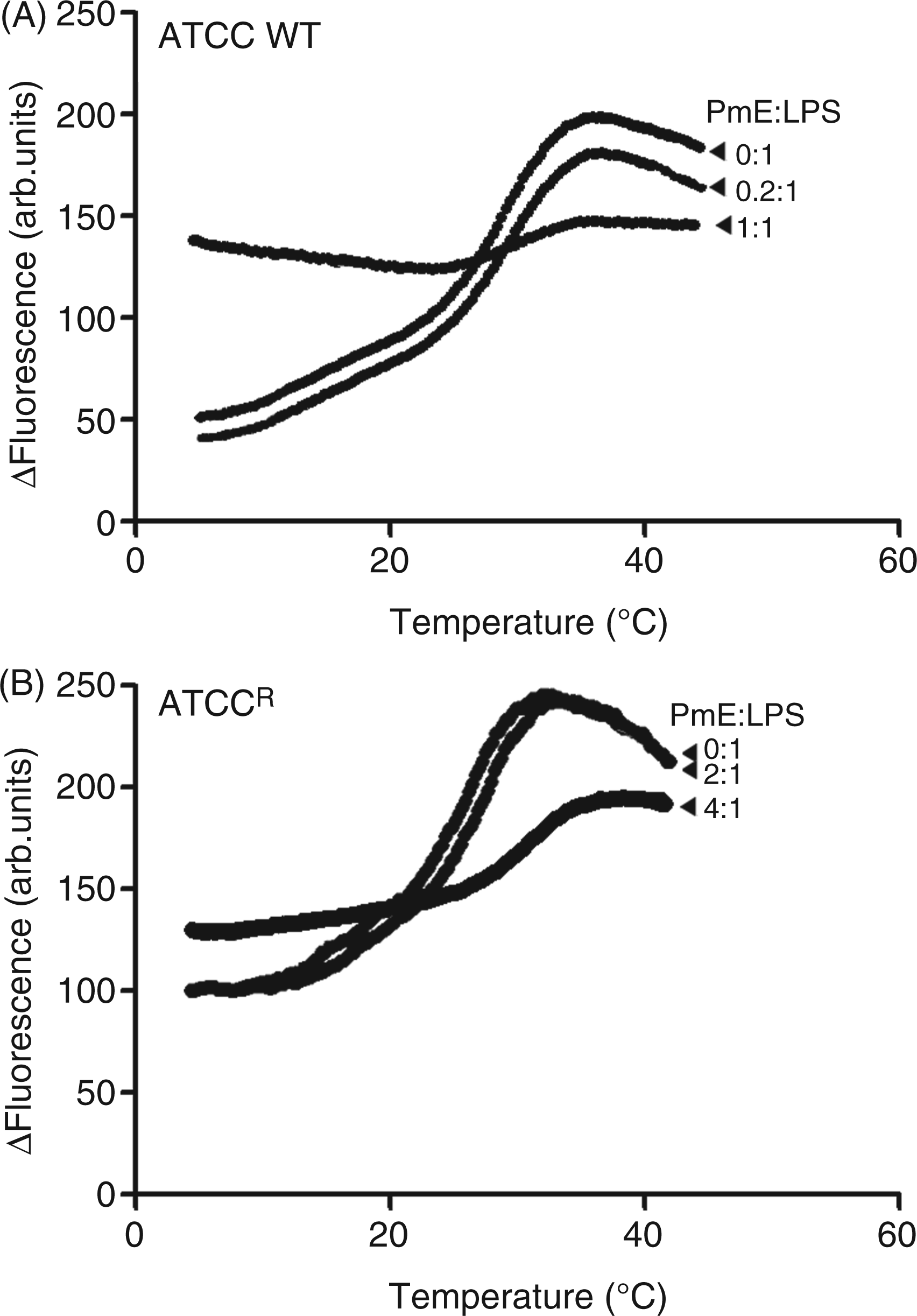
Zeta potential measurements
The zeta potential of mid-log and stationary phase cells of each strain was assessed. There was no statistically significant difference in the zeta potential between the stationary phase cells of each strain (wild type: −65.7 ± 9.8; resistant: −66.1 ± 6.6). In comparison, the mid-logarithmic phase wild type cells exhibited a greater negative charge (wild type: −62.8 ± 4.1) than the mid-logarithmic phase resistant cells (resistant: −54.4 ± 4.7).
Cytoplasmic membrane depolarization of K. pneumoniae
Figure 6 (A, B) shows the kinetics data for the change in diSC3-5 dye fluorescence upon exposure of the K. pneumoniae cells to increasing concentrations of polymyxins. Both polymyxin B and colistin induce cytoplasmic membrane depolarization in both the sensitive and resistant strain, even though a slightly decreased release of diSC3-5 dye was observed in the resistant strain. However, the depolarization was not significant at MIC concentrations in susceptible strains, which suggests that permeabilization of the cytoplasmic membrane by polymyxins does not contribute significantly to bacterial killing. Fluorescence intensity reached a plateau level 10 min after polymyxin exposure in both strains. An endpoint analysis is showed in Figure 6 (C, D. Dose-dependent depolarization effects of cytoplasmic membrane were observed in both of the strains. Overall, the data indicate that the cytoplasmic membrane of the resistant strain displays similar characteristics to that of the sensitive strain.
(A, B) Cytoplasmic membrane depolarization of K. pneumoniae cells by polymyxin B and colistin assessed by the diSC3-5 assay. (C) Effects of increasing concentrations polymyxin B and colistin on K. pneumoniae cells at a 16 min end-point. (D) Data points of 30 µg/ml of polymyxin B and colistin.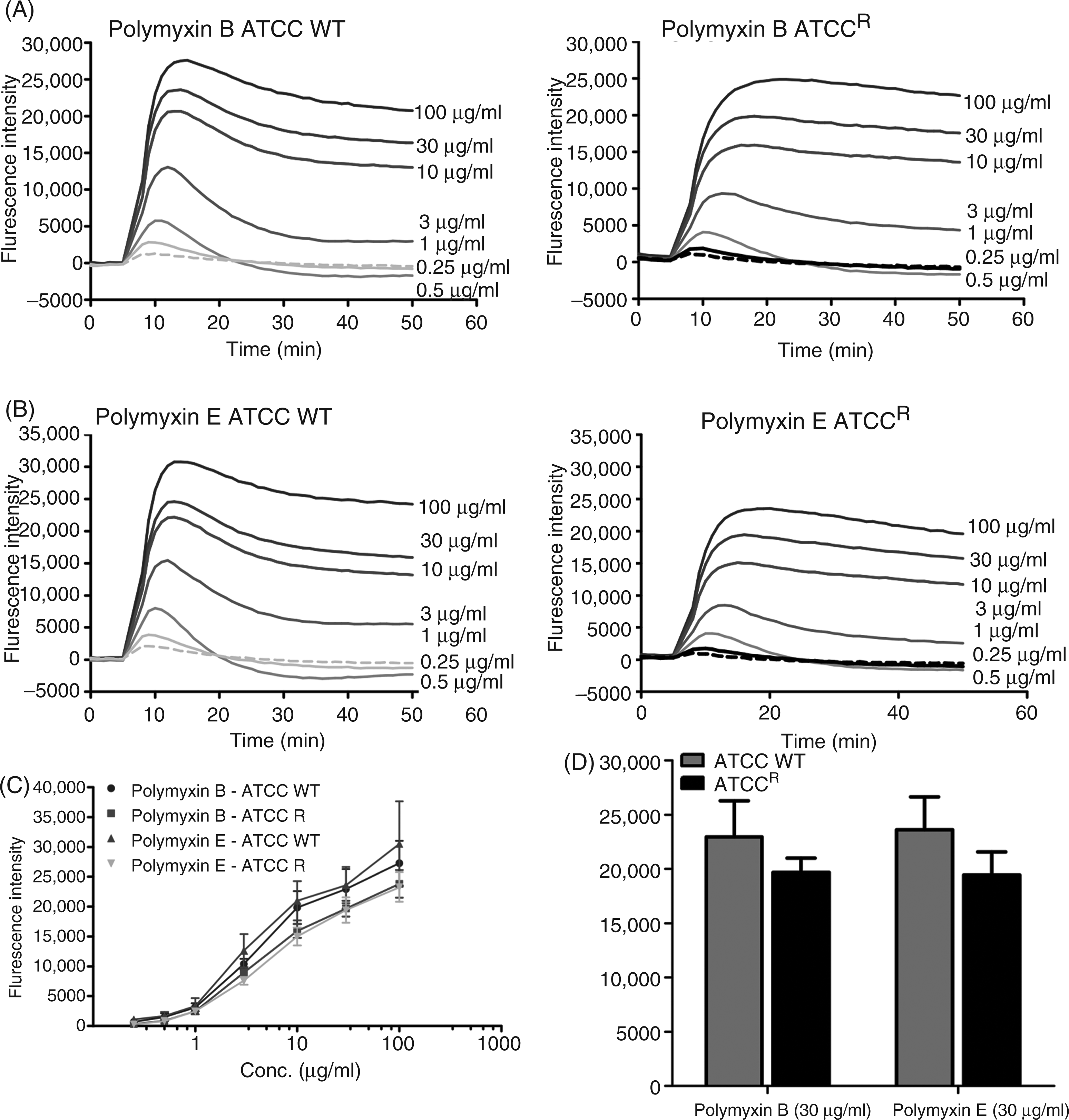
Discussion
Increasing reports of resistance to the last-line therapy polymyxins in K. pneumoniae has provided the impetus to elucidate the surface changes associated with the resistance mechanism(s) employed by this problematic pathogen. Although Campos et al. 21 suggested that resistance to cationic antimicrobial peptides in K. pneumoniae was dictated by the thickness of capsular polysaccharide material coating the surface, recent studies have revealed that polymyxin susceptibility was not altered in non-encapsulated K. pneumoniae mutants.17,22 This would suggest that, besides the capsule, additional components that constitute the outer membrane serve an important barrier function against cationic antibiotics. The present study employed a series of biophysical techniques to examine the interaction between polymyxins and the surface and LPS from paired polymyxin-susceptible and resistant strains of K. pneumoniae.
The presence of surface components on the resistant strain that block polymyxin activity was highlighted by the finding that, following colistin exposure, the wild type strain was, to a large extent, more sensitive to the lytic action of lysozyme and sodium deoxycholate on the periplasmic peptidoglycan and inner membrane respectively (Figure 4). These observations may be attributed to a greater propensity for colistin to disrupt the outer membrane barrier of the wild type strain, thus enhancing the access of these lytic agents to the internal structures that they target.
To examine the role of LPS in providing a barrier function against polymyxin attack of the outer membrane, the binding affinity of polymyxin B and colistin to LPS purified from each strain was measured fluorometrically (Table 2). On average, the Ki values for polymyxin binding to LPS from the resistant strain were 2.5–5.2-fold lower compared with those determined for the wild type strain. In contrast, a 64-fold difference in MIC was observed between the two strains (Table 1). This would indicate that the affinity of polymyxins for LPS measured in vitro is not entirely proportional to their ability to attack the outer membrane and exert a bacterial killing activity. Indeed, the bacterial killing elicited by polymyxins on K. pneumonia must involve a complex series of interactions with various surface components and, possibly, intracellular components.
The bactericidal activity of polymyxin B is known to exhibit a strong temperature dependence.
52
The activity of polymyxin B against Escherichia coli cells was noted to decrease drastically from 98% at 35℃ to merely 5.4% at 18℃.
52
The temperature region at which the change in killing was observed (24–27℃) lies within the phase transition temperature for the E. coli outer membrane, as measured by differential scanning calorimetry.
52
From a mechanistic view, these observations are comprehensible, as self-uptake of polymyxins necessitates penetration of the hydrophobic polymyxin domains (i.e. the N-terminal fatty acyl chain, and
Outer membrane electrostatic variations that potentially affect the electrostatic component of polymyxin activity were assessed by zeta potential measurements. Although, there was no significant difference in the surface charge of stationary phase cells of each strain, the mid-logarithmic phase wild type cells exhibited a greater negative charge than the mid-logarithmic phase-resistant cells. Evidently, the relationship between polymyxin resistance and surface charge variations is very complex and growth phase-dependent.
DiSC3-5 is a cationic cyanine dye that has been widely applied to measure membrane potential in intact bacterial cells.29,59–61 It accumulates in cells on hyperpolarized membranes with its fluorescence self-quenched. Any compound that permeabilizes the cytoplasmic membrane and thus depolarizes membrane potential will result in the release of diSC3-5 dye and an increase in fluorescence. 62 Previous studies showed limited depolarization effects of polymyxin compounds on Pseudomonas aeruginosa. 29 The fluorometric data diSC 3-5 assay indicated that the cytoplasmic membrane of the resistant strain displays similar depolarization characteristics in response to polymyxin treatment as that of the sensitive strain, suggesting that the cytoplasmic membrane may not be crucial for polymyxin resistance in K.pneumoniae.
NPN is a hydrophobic probe whose fluorescence is greatly increased in a hydrophobic environment. 63 The tight packing of the fatty acyl chains of lipid A in the outer membrane leaflet limits the free diffusion of hydrophobic solutes, such as NPN. 11 However, once permeated, intercalation of NPN into the underlying phospholipid inner leaflet and the cytoplasmic membranes produces a resultant increase in fluorescence. 64 In comparison with the wild type strain, the NPN uptake kinetics were greatly enhanced in the resistant strain, even in the absence of polymyxin treatment (Figure 3). This would suggest the resistant strain expresses a more permeable, less densely packed outer membrane structure. Moreover, NPN uptake by the polymyxin-resistant strain was not affected by the chelating agent, EDTA. EDTA is known to destabilize the Gram-negative outer membrane by sequestering divalent cations that bridge adjacent LPS molecules in the outer leaflet. 65 Notably, P. aeruginosa resistant to the cationic surface-acting disinfectant benzalkonium chloride displayed co-resistance to polymyxin B (which also displays cationic quaternary ammonium groups), an also showed a reduction in NPN uptake into the outer membrane with EDTA and polymyxin treatment. 66 It is also noteworthy to mention that a recent study showed that Burkholderia multivorans mutants with a transposon insertion in the open reading frame of their hopanoid biosynthesis gene cluster displayed increased susceptibility to polymyxin B and an increased permeability to NPN, albeit only in the presence of polymyxin B. 67 In bacteria, hopanoids are believed to confer a barrier function by increasing outer membrane stability and reducing molecular permeability in an analogous fashion to eukaryotic sterols. 68 As there was no significant difference in NPN permissibility between the wild type and the hopanoid mutant of B. multivorans to NPN alone (i.e. in the absence of polymyxin) (Dr Rebecca J. Malott, personal communication), the polymyxin resistance mechanism employed by K. pneumoniae ATCC 13883 is unlikely to involve the hopanoid pathway.
Taken together our data suggest that the polymyxin-resistant strain expresses a less negative and more permeable outer membrane phenotype compared with the wild type. Intriguingly, this does not run parallel to the increased polymyxin susceptibility we previously observed with a K. pneumoniae mutant strain B5055ΔlpxM, which expresses an under-acylated lipid A that also results in a phenotype with a more fluid outer membrane. 39 Clearly, in the case of the resistant strain the increased membrane fluidity does not coincide with polymyxin susceptibility; it is more likely a secondary consequence resulting from modifications to another outer membrane component that the bacteria has introduced to attain resistance. In light of the fact that polymyxin resistance in K. pneumoniae has been shown to involve modification of the lipid A phosphates with Arap4N, one possibility is that this modification affects the packing of the lipid A leaflet by abrogating the ability of divalent cations to bridge adjacent LPS molecules. This is certainly plausible given that we observed very little effect of EDTA on the outer membrane permeability of the resistant strain. Intuitively, the lower state of order of the resistant outer membrane should actually make the surface more vulnerable to polymyxin attack by facilitating the self-uptake of the hydrophobic domain of the polymyxin molecule, as per the B5055ΔlpxM mutant. 39 However, we have previously shown that the interaction of polymyxins with LPS and other biomolecular systems involves an initial electrostatic attraction that serves to sufficiently stabilize the complex to allow for initiation of the second stage of the interaction between the hydrophobic domains of each species.31,69 Thus, by employing an outer membrane resistance mechanism that nullifies these crucial primary electrostatic contacts, the increased membrane fluidity that could be seen as a vulnerability in terms of the ability of the polymyxin molecule to attack the surface becomes of little consequence. In summary, our study shows that the polymyxin resistance mechanism(s) employed by K. pneumoniae translates into a complex set of biophysical surface changes that together act to block the ability of the polymyxin molecule to establish crucial electrostatic contacts with the bacterial surface and initiate subsequent surface changes that eventuates in killing activity.
Footnotes
Funding
R.L.N. and J.L. are supported by research grants from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01A1070896 and R01AI079330).T.V, R.L.N, J.L and M.C are also supported by the Australian National Health and Medical Research Council (NHMRC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. J.L. is an Australian NHMRC Senior Research Fellow. T.V. is an Australian NHMRC Industry Career Development Level 1 Research Fellow. MC is a NHMRC Australia Fellow supported by AF511105.
Conflict of interest
The authors have no conflicts of interest to declare.