
Abstract
The impact of under-acylation of lipid A on the interaction between Klebsiella pneumoniae LPS and polymyxins B and E was examined with fluorometric and calorimetric methods, and by 1H NMR, using a paired wild type (WT) and the ΔlpxM mutant strains B5055 and B5055ΔlpxM, which predominantly express LPS with hexa- and penta-acylated lipid A structures respectively. LPS from B5055ΔlpxM displayed a fourfold increased binding affinity for polymyxins B and E compared with the B5055 WT LPS. EC50 values were consistent with polymyxin minimum inhibitory concentration (MIC) values for each strain. Accordingly, polymyxin exposure considerably enhanced the permeability of the B5055ΔlpxM OM. Analysis of the melting profiles of isolated LPS aggregates suggested that bactericidal polymyxin activity may relate to the acyl chains’ phase of the outer membrane (OM). The enhanced polymyxin susceptibility of B5055ΔlpxM may be attributable to the favorable insertion of polymyxins into the more fluid OM compared with B5055. Molecular models of the polymyxin B–lipid A complex illuminate the key role of the lipid A acyl chains for complexation of polymyxin. The data provide important insight into the molecular basis for the increased polymyxin susceptibility of K. pneumoniae strains with under-acylated lipid A. Under-acylation appears to facilitate the integration of the N-terminal fatty-acyl chain of polymyxin into the OM resulting in an increased susceptibility to its antimicrobial activity/activities.
Keywords
Introduction
Klebsiella pneumoniae is an opportunistic pathogen, which has been frequently implicated in serious nosocomial infections 1 and associated with high rates of mortality, 2 particularly in immuno-compromised patients. Concerns have thus been generated in response to the escalation in the incidence of multidrug-resistant (MDR) K. pneumoniae infections,3–5 with the realization that effective therapeutic options are becoming increasingly limited. In view of the waning antibacterial development pipeline, interest in polymyxin E (syn. colistin) and polymyxin B has been renewed and both are increasingly used as a last-line therapy. 6 Indeed, considerable in vitro activity against K. pneumoniae strains has been demonstrated; 98.2% of general clinical isolates of K. pneumoniae were susceptible to polymyxins. 7 Extremely drug-resistant strains, which are also resistant to polymyxins, have emerged.8,9 These findings demand a greater appreciation of the mechanism(s) of polymyxin activity and resistance in K. pneumoniae in order to minimize the development of resistance and assist drug discovery strategies.
The Gram-negative outer membrane (OM) is a permeability barrier to various antimicrobial substances, including numerous antibacterials.10,11 This complex asymmetrical structure comprises an inner phospholipid leaflet, as well as an outer leaflet, which predominantly contains LPS. Structurally, LPS is composed of three domains: the highly variable and serovar-dependent O-antigen chain (encompassing repeated saccharide units) is linked to a more genus-related core oligosaccharide region and these extra-cellular sugar portions are built upon the relatively conserved lipid A moiety (Figure 1). Lipid A is intercalated within the membrane functioning as a hydrophobic anchor.
Silver stained SDS-PAGE analysis of purified LPS from the Klebsiella pneumoniae B5055 strains examined in this study. Increasing sample amounts (0.5, 1, 1.5, 2 µg) were loaded per lane to allow for visualization owing to the differential staining of each sub-structure. The position of each LPS sub-structural component is indicated on the right ordinate. Molecular mass standards are shown on the left ordinate, in lane 1 of the gel. The dashed line indicates the mass shift of the lipid A between the paired strains. The structural organization of LPS is shown schematically.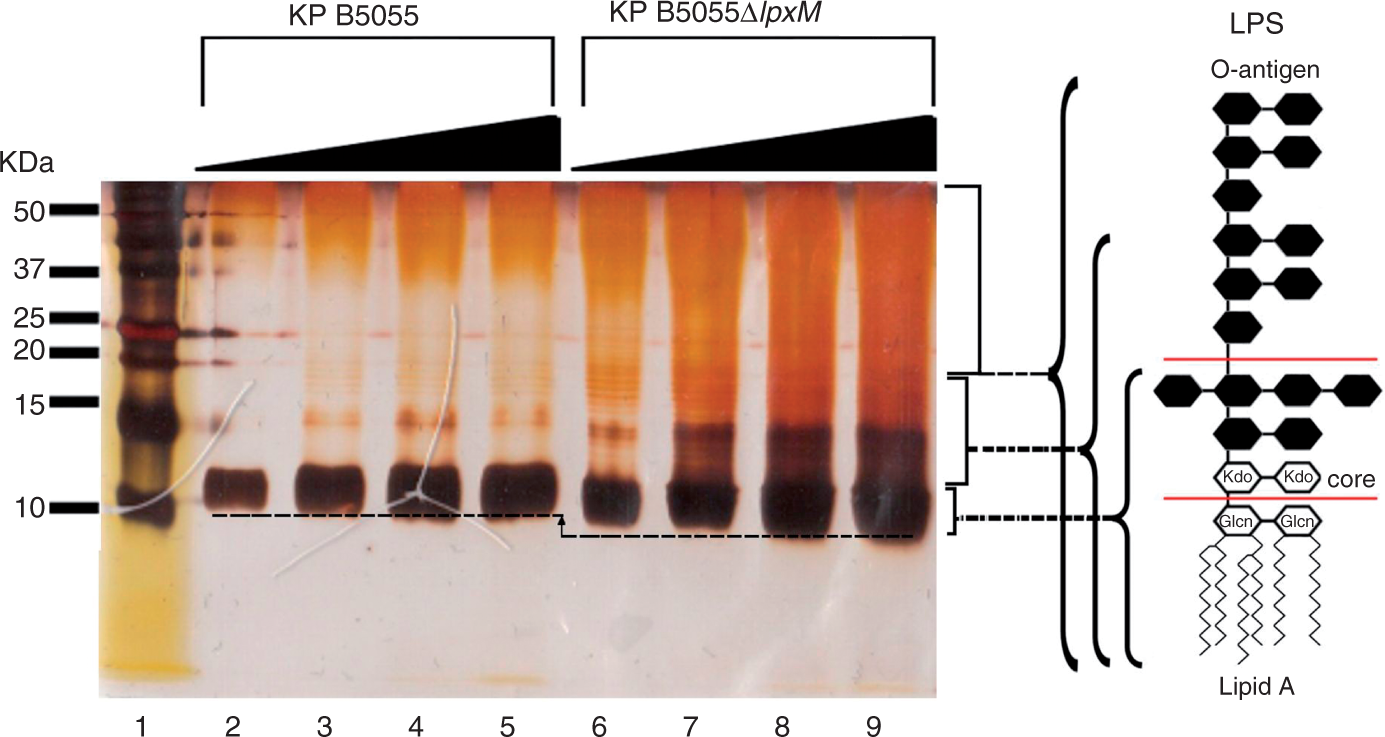
For polymyxins, lipid A represents an important binding target. 12 The cationic amphipathic nature of polymyxins is understood to be crucial to enable ‘self-promoted uptake’ across the OM. 13 The importance of hydrophobic interactions with the lipid A acyl chains for the antibacterial activity of polymyxins against K. pneumoniae has been highlighted in a recent study, which showed that K. pneumoniae strains that express an LPS chemotype with an under-acylated lipid A displayed an increased susceptibility to polymyxins. 14 The relationship between polymyxin susceptibility and lipid A structure has been poorly characterized in K. pneumoniae and warrants further investigation. In the present study we employed a series of biophysical methods to examine the correlation between the increased polymyxin susceptibility of K. pneumoniae and the under-acylated lipid A; to this end, we employed a mutant strain B5055ΔlpxM, 14 harboring a defective lpxM gene (formally msbB or waaN) that encodes the enzyme responsible for the late secondary acylation of the immature lipid A structure. This modification results in the generation of a predominantly penta-acylated lipid A as opposed to the parent B5055 wild type (WT) strain, which predominantly expresses a hexa-acylated lipid A. 14 Furthermore, the effect of polymyxin exposure on the phase transition temperature of the LPS hydrocarbon moieties of each test strain was investigated by fluorometric thermal shift analysis. Finally, on the basis of these data structural correlations are drawn using molecular models of the polymyxin B1–K. pneumoniae lipid A complexes.
Materials and methods
Materials
Polymyxin B (sulfate), polymyxin E (sulfate), 1-N-phenylnaphthylamine (NPN), lysozyme, purpald reagent, 3-deoxy-
Bacterial strains
Klebsiella pneumoniae B5055 is a mouse virulent clinical isolate (serotype K2 : O1) and B5055ΔlpxM is a mutant derivative constructed with a defective lpxM gene. 14 The lipid A structure of both strains has been characterized comprehensively in a previous study. 14
Determination of minimum inhibitory concentrations
Polymyxin minimum inhibitory concentrations (MICs) for each strain were determined by the broth microdilution method. 15 Experiments were performed with cation-adjusted Mueller-Hinton broth (CaMHB) (containing approximately 106 CFU per ml) in 96-well polypropylene microtitre plates. Wells were inoculated with 100 µl of bacterial suspension prepared in CaMHB and 100 µl of CaMHB containing increasing concentrations of polymyxins (0 to 128 µg/ml) over 7 titre steps. The MICs were defined as the lowest concentration at which visible growth was inhibited following 18 h incubation at 37℃. Cell viability was determined by sampling wells at polymyxin concentrations greater than the MIC. These samples were diluted in normal saline and spread plated onto nutrient agar. After incubation at 37℃ for 20 h, viable colonies were counted on these plates. The limit of detection was 10 CFU/ml. European Committee on Antimicrobial Susceptibility Testing (EUCAST; Växjö, Sweden) breakpoints against Enterobacteriaceae were employed. 16
LPS isolation
LPS was extracted by a modified hot phenol/water procedure. 17 The purity of isolated LPS samples was ascertained by SDS-PAGE. LPS samples were re-suspended in SDS-PAGE sample buffer (2.3% SDS, 0.8% Tris, 10% glycerol, 5% dithiothreitol) and boiled for 10 min. SDS-PAGE in 15% polyacrylamide gels and LPS was visualized by silver staining. 18
Kdo purpald assay
The molar concentration of Kdo in the LPS samples was measured following the purpald assay, as described in detail previously. 19 The un-substituted terminal vicinal glycol group of Kdo in LPS is subjected to sodium periodate (NaIO4) oxidation, yielding quantitative formaldehyde, which is measurable by purpald reagent. Briefly, 50 µl of 32 mM NaIO4 was added to 50 µl of LPS sample or Kdo standard (0.05, 0.1, 0.2, 0.3, 0.4 and 0.5 mM Kdo) and incubated for 25 min. Fifty microliters of 136 mM purpald reagent in 2 N NaOH was added to each sample and incubated for 20 min. Then, 50 µl of 64 mM NaIO4 was added to the samples and incubated for another 20 min. The absorbance of each sample was measured at 550 nm. The Kdo molarity in the LPS samples was evaluated from their absorbance and the Kdo standard curve. This value divided by the theoretical number of Kdo residues per LPS molecule gave the molarity of LPS in the sample.
Fluorometric measurement of [Dansyl-Lys]1- polymyxin B3 binding to whole cells and isolated LPS
The fluorometric measurements were performed as described previously using the fully synthetic antibacterial polymyxin probe DPmB3. 20
Deoxycholate sensitivity induced by polymyxins
The protocol for this assay was adapted from Vaara et al. 21 with minor modifications. Mid-logarithmic phase cells (OD500nm = 0.4 to 0.6) were pelleted and re-suspended in 5 ml of 5 mM HEPES buffer (pH 7.2) in the presence of increasing concentrations of polymyxin E, then incubated for 10 min. After the OD500nm of the cell suspensions was measured, cells were again pelleted and re-suspended in 5 ml of the same buffer containing 0.25% (w/v) deoxycholate. These suspensions were incubated for 10 min and the decrease in OD500nm was subsequently measured. All assay operations were performed at 37℃.
Permeability to lysozyme induced by polymyxins
This method was performed as described previously. 21 Lysozyme (5 µg/ml) and polymyxin E (9 µg/ml) were added to a mid-logarithmic phase cell suspension (OD500nm = 0.4 to 0.6), and the decrease in OD500nm was measured.
NPN uptake assay
NPN uptake was measured as described previoulsy.
22
Briefly, mid-logarithmic phase cells were pelleted and washed twice in 5 mM HEPES buffer (pH 7.2) containing 1 mM sodium azide, then re-suspended in the same buffer (OD500nm = 0.5). The cell suspension was allowed to sit at room temperature for 30 min before analysis. NPN fluorescence was measured with the excitation and emission wavelengths set at 350 and 420 nm, respectively, with slit widths of 5 nm. The NPN uptake assay was also employed to measure the OM permeabilizing activity of polymyxins. NPN was dissolved in acetone at a concentration of 500 µM and added to 1 ml of cell suspension to a final concentration of 10 µM. Response-concentration curves were plotted from baseline-corrected values and non-linear regression was used to fit equation (1) to the data
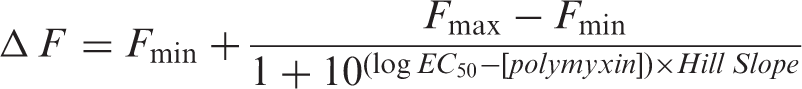
Fluorometric thermal shift assay
The phase transition of the fatty acyl chains of the LPS aggregates was examined in the absence and presence of polymyxin E via a NPN fluorometric thermal shift assay. Fluorescence measurements were performed on a Cary Eclipse spectrophotofluorometer, as detailed earlier. LPS (50 µM) dissolved in 20 mM HEPES buffer (pH 7.4) together with 5 µM NPN was heated from a starting temperature of 5℃ to a target temperature of 95℃ in 1℃ increments. Data-fitting operations were performed as described previously to obtain information on the phase transition range and its mid-point (Tm). 23 The molar concentration of LPS was determined using the purpald Kdo assay. 19
Isothermal titration calorimetry measurement of polymyxin–LPS binding interactions
Microcalorimetric experiments of polymyxin E binding to LPS were performed on a VP-ITC isothermal titration calorimeter (Microcal, Northampton, MA, USA) at 37℃ in 20 mM HEPES buffer (pH 7.0), as described previously. 20 The molar concentration of LPS was determined using the purpald Kdo assay. 19
1H NMR spectroscopy measurements
Polymyxin E was dissolved in 0.5 ml of 2H2O (pH 4.0) to a final concentration of 0.1 mM. In order to examine the binding of polymyxin E to LPS line-broadening experiments were performed by successively adding small aliquots of a solution of LPS to polymyxin E. One-dimensional proton NMR (1H NMR) spectra were acquired on a Varian INOVA 600 MHz spectrometer (Varian, Mulgrave, VIC, Australia) operating at 20℃. The chemical shifts (ppm) were referenced to an internal 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (TMS).
Molecular modeling of the lipid A–polymyxin B complex
The coordinates of the NMR solution structure of polymyxin B1 when bound to Escherichia coli LPS were obtained from Pristovsek and Kidric.24,25 The lipid A structures of K. pneumoniae B5055 WT and B5055 ΔlpxM 14 were modeled using the crystallographic structure of E. coli LPS as a template; the latter was derived from the crystal structure of FhuA, the receptor for ferrichrome-iron in E. coli with bound LPS (PDB code:1QFF).12,26 The polymyxin B1–lipid A complexes were manually docked to reproduce the interactions described by Pristovsek and Kidric24,25 using Accelrys Discovery Studio V2.1. (Accelrys, Inc. San Diego, CA, USA).
Results
Susceptibility testing and LPS characterization
K. pneumoniae susceptibility to polymyxin B and E, and half-maximal effective concentration (EC50) of polymyxins required to produce permealization of the OM to NPN.
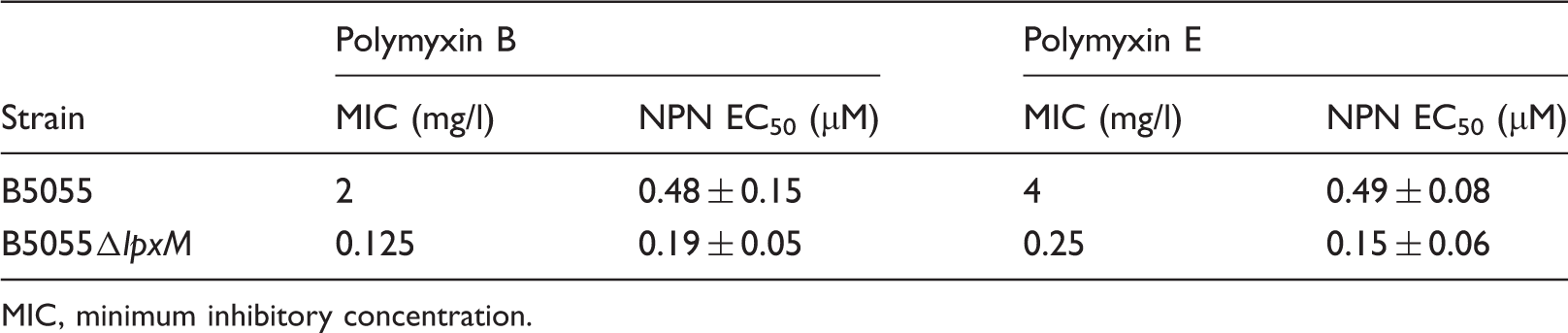
MIC, minimum inhibitory concentration.
Binding of polymyxin B and E to isolated K. pneumoniae LPS
Fluorometric assessment of DPmB3 binding to isolated LPS
Binding affinity of polymyxins for purified LPS from each K. pneumoniae test strain.

Kd, dissociation constant value, a direct measure of the 1 [Dansyl]PmB3 binding affinity.
Ki, inhibition constant value, measured as the ability of the polymyxin to displace 1 [Dansyl]PmB3.
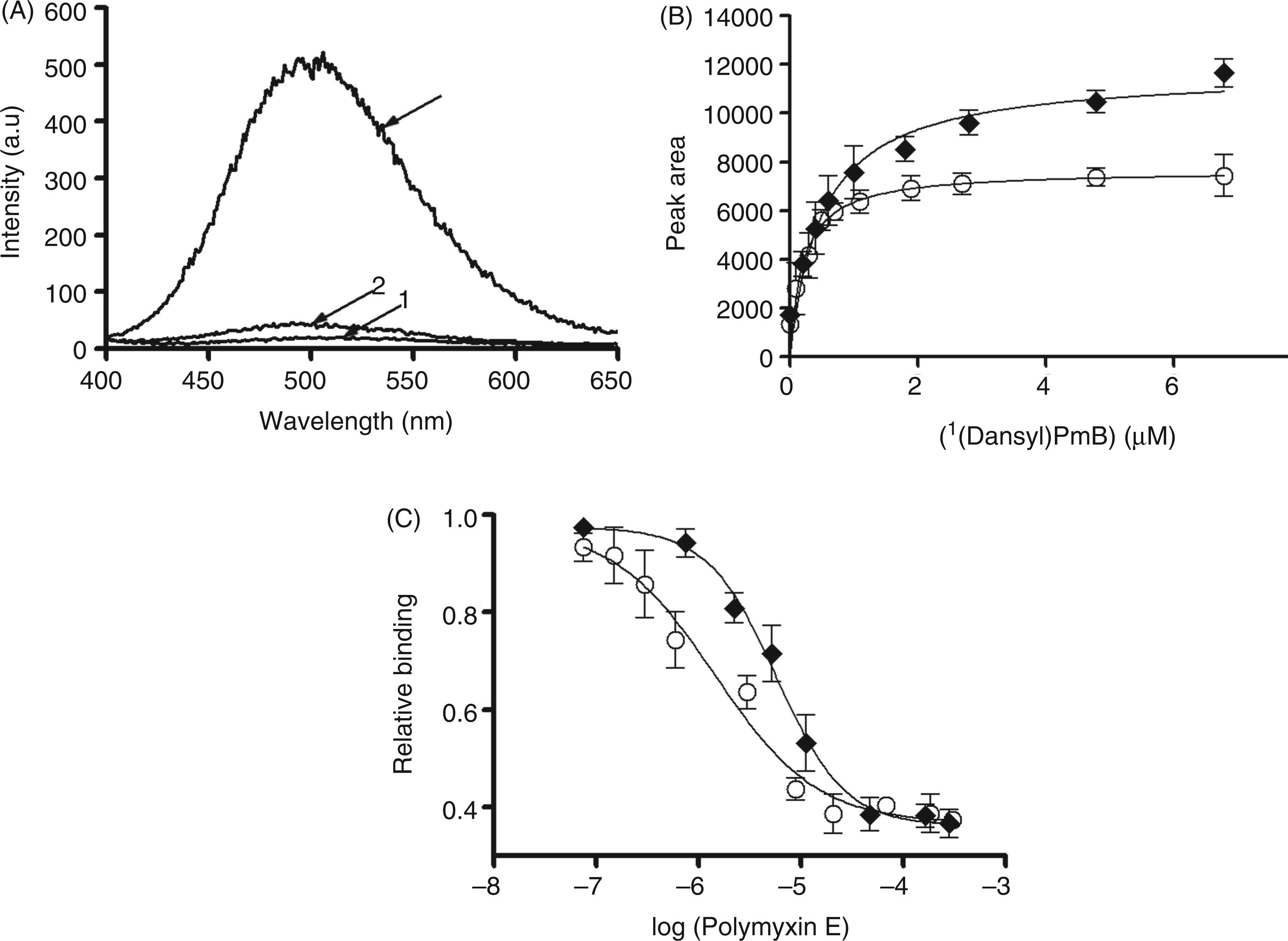
(A) Fluorescence emission spectra of (i) K. pneumoniae B5055 LPS in buffer (3 mg/l); (ii) [Dansyl-Lys]1-polymyxin B3 (12 µM) in buffer; and (iii) [Dansyl-Lys]1-polymyxin B3 (12 µM) bound to B5055 LPS (3 mg/l). (B) Fluorescence assay of the binding of [Dansyl-Lys]1-polymyxin B3 to isolated LPS from K. pneumoniae B5055 (♦) and B5055ΔlpxM (○).The solid lines represent the non-linear least squares regression fit of a one-site binding model as described previously, 20 R2 values for the fits ranged between 0.93 and 0.98. (C) The decrease in fluorescence upon displacement of [Dansyl-Lys]1-polymyxin B3 from LPS K. pneumoniae B5055 (♦) and B5055ΔlpxM (○) by titration with polymyxin E. Solid lines represent non-linear least squares regression fit of a displacement model as detailed previously. 20 All data points are the mean of three independent measurements ± SD.
1H NMR analysis of the binding of polymyxin E with LPS aggregates
Titration of LPS aggregates into a solution of polymyxin E produced a line broadening of all polymyxin resonances; for simplicity, only the amide region is shown in Figure 3. Based on an identical titration regime mass ratio (polymyxin E:LPS), line broadening was observed at a lower mass ratio for titrations with B5055 ΔlpxM LPS compared with LPS from the B5055 WT strain. This suggests that polymyxin E penetrates penta-acylated B5055 ΔlpxM LPS aggregates more readily than it does the hexa-acylated B5055 LPS phenotype (Figure 3A, B).
Six hundred MHz 1H NMR spectrum of the amide region of polymyxin E in 10% D2O pH 4.0 titrated with LPS. The ratio of LPS to polymyxin E is indicated on the right ordinate. (A) KP B5055 LPS (B) KP B5055ΔlpxM LPS.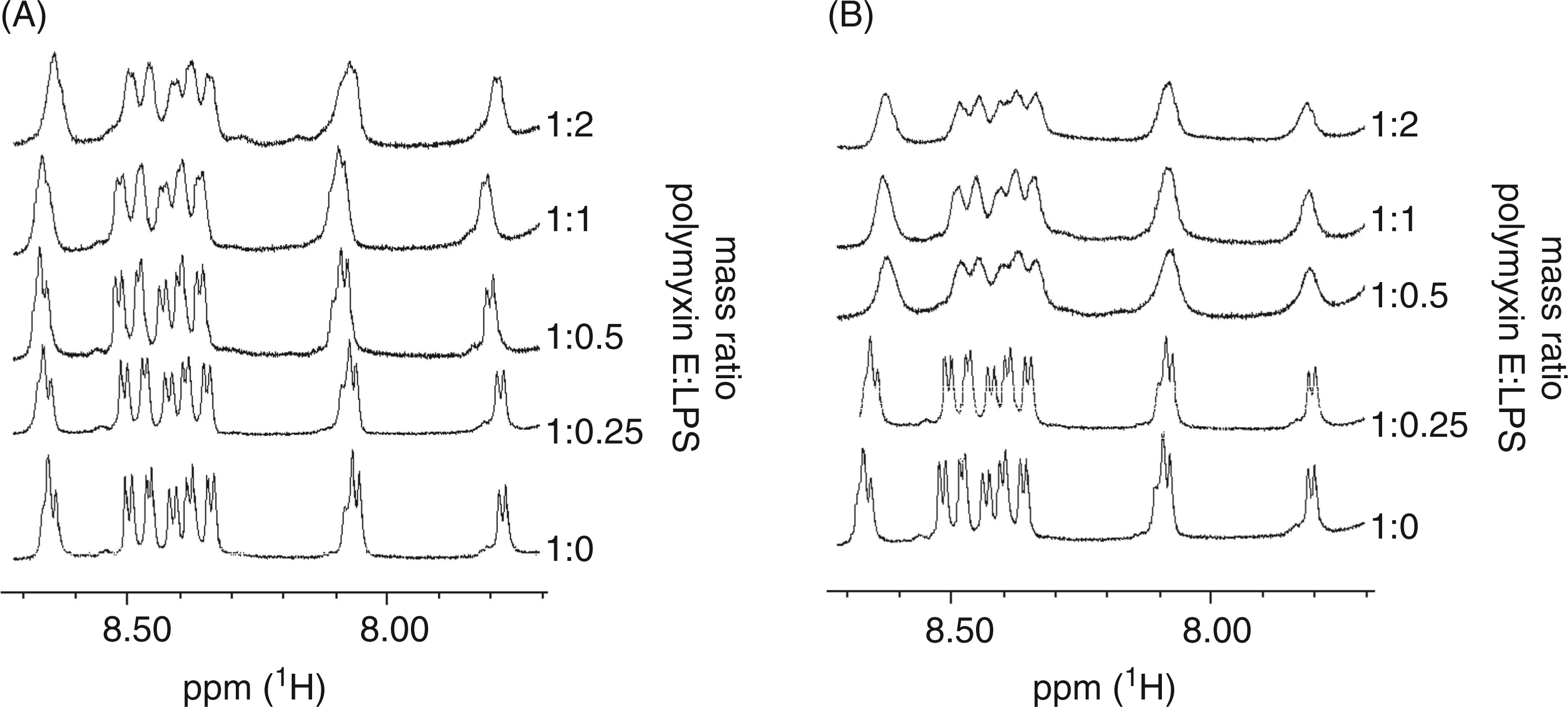
Isothermal titration calorimetry assay of polymyxin E binding to isolated LPS
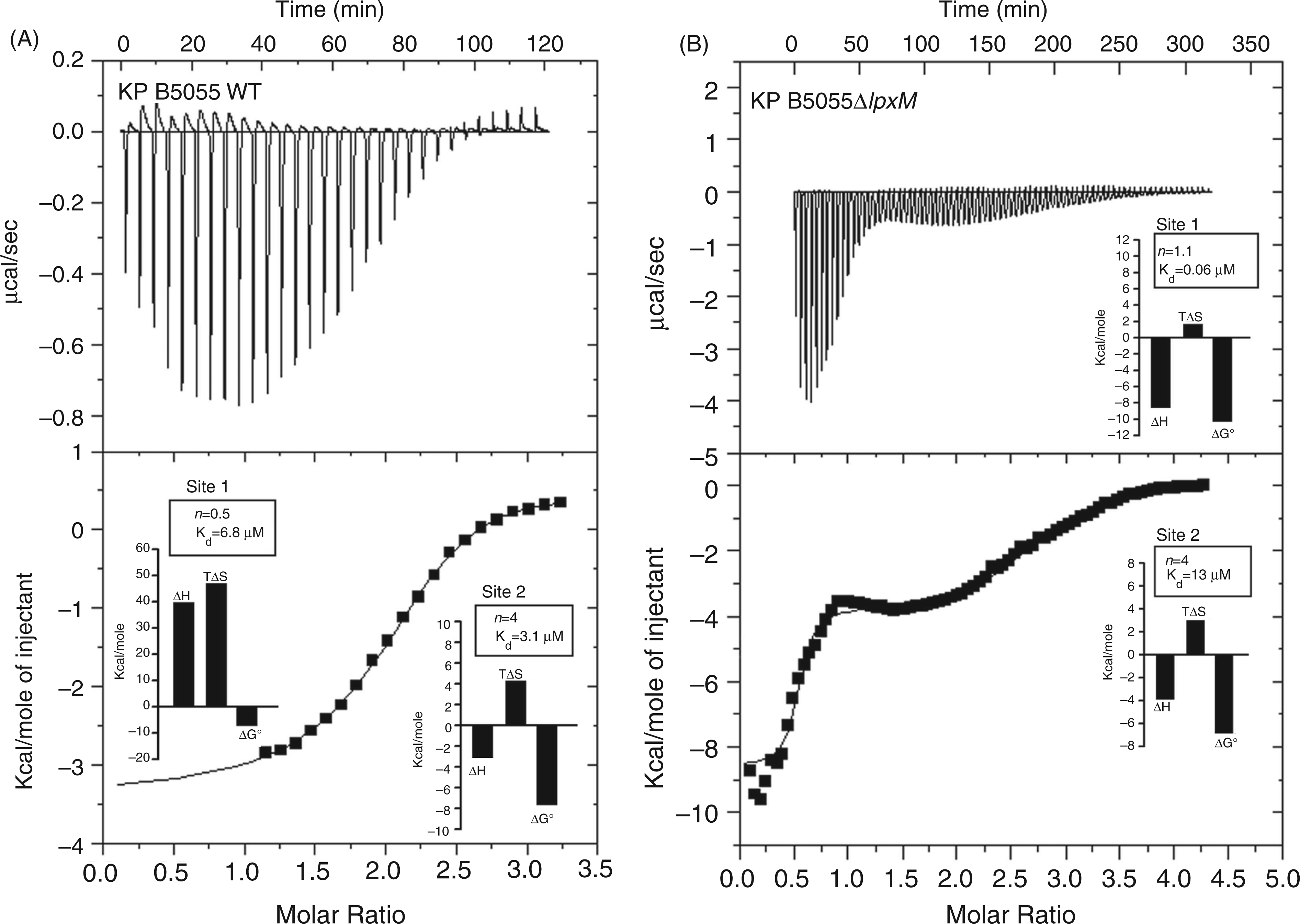
The energetics of the interaction between polymyxin E and LPS from the paired B5055 strains was determined by isothermal titration calorimetry (ITC). The microscopic thermodynamic parameters, the number of binding sites (n), the binding constant (Ka) and binding enthalphy (ΔH) are documented in graphical form as insets in Figure 4. As the nature of the polymyxin B–LPS interaction has been reported to be temperature dependent,
28
all ITC experiments were performed at a physiologically relevant temperature of 37℃. The downward peaks observed for polymyxin E titrations with both B5055 and B5055 ΔlpxM LPS were representative of an exothermic process (Figure 4, top panels). Plots of the enthalpy changes measured calorimetrically versus the polymyxin E:LPS ratio are also presented in Figure 4 (bottom panels). The data were evaluated according to a model which defines two independent binding sites, as described previously.
20
The microscopic thermodynamic parameters for the first set of sites for the polymyxin E–B5055 LPS (hexa-acylated lipid A) interaction revealed an approximately 0.5 : 1 (polymyxin E:LPS) binding stoichiometry, with a moderate binding affinity (6.8 µM). This reaction displayed an unfavorable enthalpic component and a large positive (favorable) entropic component (Figure 4A, insets). The parameters for the second set of sites indicated a 4 : 1 (polymyxin E:LPS) binding stoichiometry with a moderate binding affinity (3.1 µM) and was driven by favorable enthalpic and entropic contributions to binding free energy (Figure 4A, insets). In contrast, the microscopic thermodynamic parameters for the polymyxin E–B5055 ΔlpxM LPS clearly indicated a more thermodynamically favorable binding process; the first set of sites of the interaction revealed a 1 : 1 (polymyxin E:LPS) stoichiometry with a high binding affinity (0.06 µM) (Figure 4B, insets). The reaction was largely driven enthalpically and accompanied by a smaller, albeit favorable, entropic contribution to the binding free energy (Figure 4B, insets). The second phase of the polymyxin E–B5055 ΔlpxM LPS reaction was characterized by a 4 : 1 (polymyxin E:LPS) binding stoichiometry with a moderate binding affinity (13 µM) (Figure 4B, insets). The second phase of the polymyxin E-B5055 ΔlpxM LPS reaction was driven by favorable enthalpic and entropic contributions to binding free energy (Figure 4B, insets).
Isothermal titration calorimetry (ITC) measurement of the polymyxin E–B5055 LPS binding interactions. (A) K. pneumoniae B5055 LPS. (B) K. pneumoniae B5055ΔlpxM, LPS. Top panels show the heat in µcal/sec per injectant. Bottom panels show the enthalpy (kcal/mol) per injectant at 3 µl of 3 mM polymyxin E into a solution of 0.125 mM LPS in buffer 20 mM HEPES pH 7.0 at 37℃. The insets show the stoichiometry (n), binding affinity (Kd) and thermodynamic parameters (ΔH, enthalpy; TΔS, entropy; ΔG°, free energy) for each interaction derived from the fit of a two independent sites model.
20
Assessment of the OM permeabilizing action of polymyxins on K. pneumoniae
NPN uptake by K. pneumoniae
Effect of polymyxin B or E on the uptake of NPN into K. pneumoniae LPS aggregates.

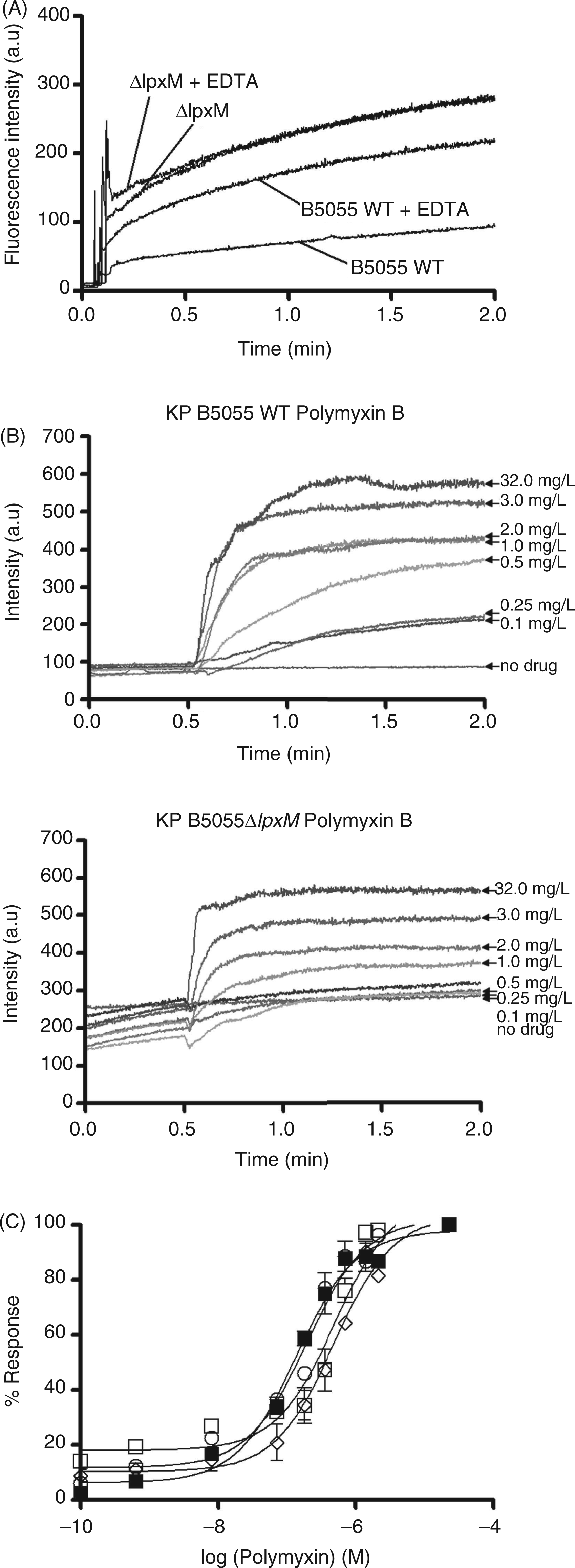
(A) NPN uptake kinetics of a suspension of mid-logarithmic phase K. pneumoniae B5055 WT and B5055ΔlpxM cells in the presence and absence of EDTA. NPN fluorescence emission is shown as a function of time. (B) Effects of polymyxin B on the uptake of NPN into the outer membrane (OM) of K. pneumoniae B5055 WT and B5055ΔlpxM cells. NPN fluorescence emission is shown as a function of time in the presence of increasing concentrations of polymyxin B. (C) The normalized peak NPN fluorescence is plotted as a function of the polymyxin B and E concentration. K. pneumoniae B5055ΔlpxM, polymyxin B (○); K. pneumoniae B5055ΔlpxM, polymyxin E (▪); K. pneumoniae B5055, polymyxin B (□); K. pneumoniae B5055, polymyxin E (◊). Data points are the mean ± SD of three independent measurements.
Lysozyme and deoxycholate permeability assay
Incubation of whole-cell suspensions with deoxycholate, lysozyme or polymyxins alone had a negligible effect on the optical density of the bacterial suspensions (data not shown). However, the addition of polymyxin E to cell suspensions in the presence of lysozyme (Figure 6A) or deoxycholate (Figure 6B) produced a decrease in optical density, which may be interpreted as an increase in deoxycholate- or lysozyme-mediated cell lysis. The lysozyme time course reaction showed that the B5055 ΔlpxM cells were lysed more extensively compared with the B5055 WT cells upon polymyxin E exposure (Figure 6A). Moreover, the B5055 ΔlpxM cells displayed a 20% greater decrease in optical density than the B5055 WT cells at the end of the time course (Figure 6A). Similarly, the deoxycholate permeability assay showed that the B5055 ΔlpxM cells underwent more extensive lysis compared with the B5055 WT cells when exposed to an identical concentration series of polymyxin E (Figure 6B).
(A) Polymyxin E-induced lysozyme sensitivity. The OD500 of a mid-logarithmic phase K. pneumoniae B5055 cell suspensions in the presence of lysozyme following exposure to polymyxin E. (B) Deoxycholate induced cell lysis following exposure to increasing concentrations of polymyxin E. K. pneumoniae B5055 (♦) and B5055ΔlpxM (○). Data are expressed as the percentage of the optical density (OD) in absence of polymyxin E exposure. Data points are the mean ± SD of three independent measurements.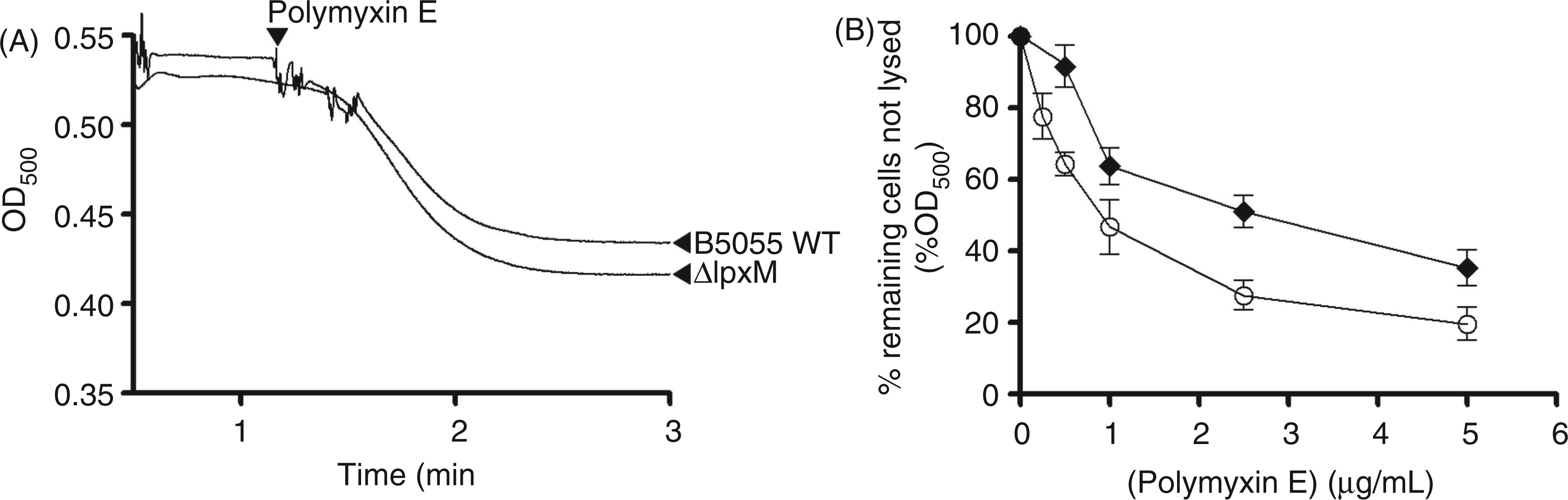
Thermal transition temperature of K. pneumoniae LPS aggregates
Thermal phase transitions in the isolated LPS were monitored using NPN fluorescence as an indicator. Figure 7A shows the phase behavior of LPS from each test strain, the phase-transition temperature and its mid-point (Tm). For the B5055 WT LPS, the gel↔liquid crystalline phase transition of the fatty acyl chains occurred with a Tm of 25℃; in comparison, the B5055ΔlpxM LPS displayed a lower Tm of 23℃ (Figure 7A). In the presence of a fourfold molar excess of polymyxin E to LPS, the Tm of both LPS samples was shifted to lower values (B5055 WT Tm = 23℃; B5055 ΔlpxM Tm = 21℃), indicating increased fluidization of the LPS acyl chains occurred with polymyxin exposure (Figure 7B,C).
Fluorometric thermal shift measurements of K. pneumoniae B5055 LPS melting profiles. The NPN fluorescence intensity is plotted as function of temperature. (A) Comparison of the K. pneumoniae B5055 LPS melting profiles. (B) Comparison of the thermal shift melting profile of K. pneumoniae B5055 WT LPS (50 µM) in the presence and absence of polymyxin E (200 µM). (C) Comparison of the thermal shift melting profile of K. pneumoniae B5055 ΔlpxM LPS (50 µM) in the presence and absence of polymyxin E (200 µM).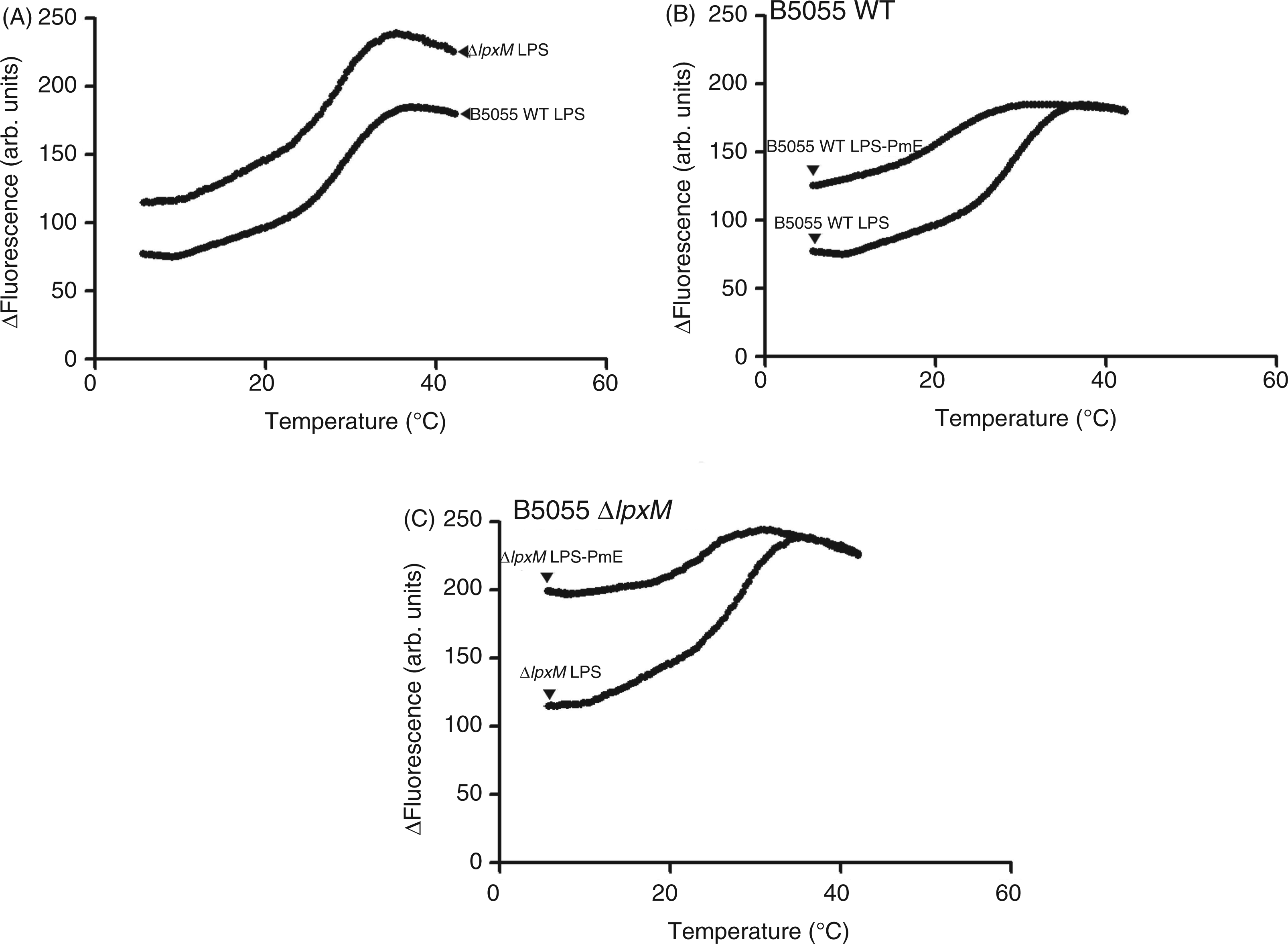
Molecular modeling of the polymyxin B1–lipid A complexes for each K. pneumoniae strain
To demonstrate the effect of lipid A modifications on the polymyxin binding interaction we constructed molecular models of polymyxin B1 in complex with the lipid A structure from each K. pneumoniae test strain (Figure 8). The model of the polymyxin B1–B5055 WT lipid A complexes shows that, in general, the complex is stabilized by a combination of electrostatic and hydrophobic interactions (Figure 8B). The positively-charged side chains of Dab
1
and Dab
5
bond with the negative charge on the 4′-phosphate head group of lipid A, while those of Dab
8
and Dab
9
bond with the 1-phosphate. The buckled configuration of the cyclic peptide portion forces the lipid A binding surface of the polymyxin molecule towards the concave face of the polymyxin molecule. The polymyxin B1–B5055 ΔlpxM lipid A model suggests that the loss of a secondary 3′-myristate fatty-acyl chain leads to a reduced hydrophobic surface area for interaction with the Molecular models of the complex between polymyxin B1 and the lipid A structure from each K. pneumoniae test strain. The lipid A molecule is shown in space-filling representation and polymyxin B1 is shown in stick representation. The chemical structure of each represented molecule is shown in the right hand panels. R2, core oligosaccharide with or without attached O-polysaccharide.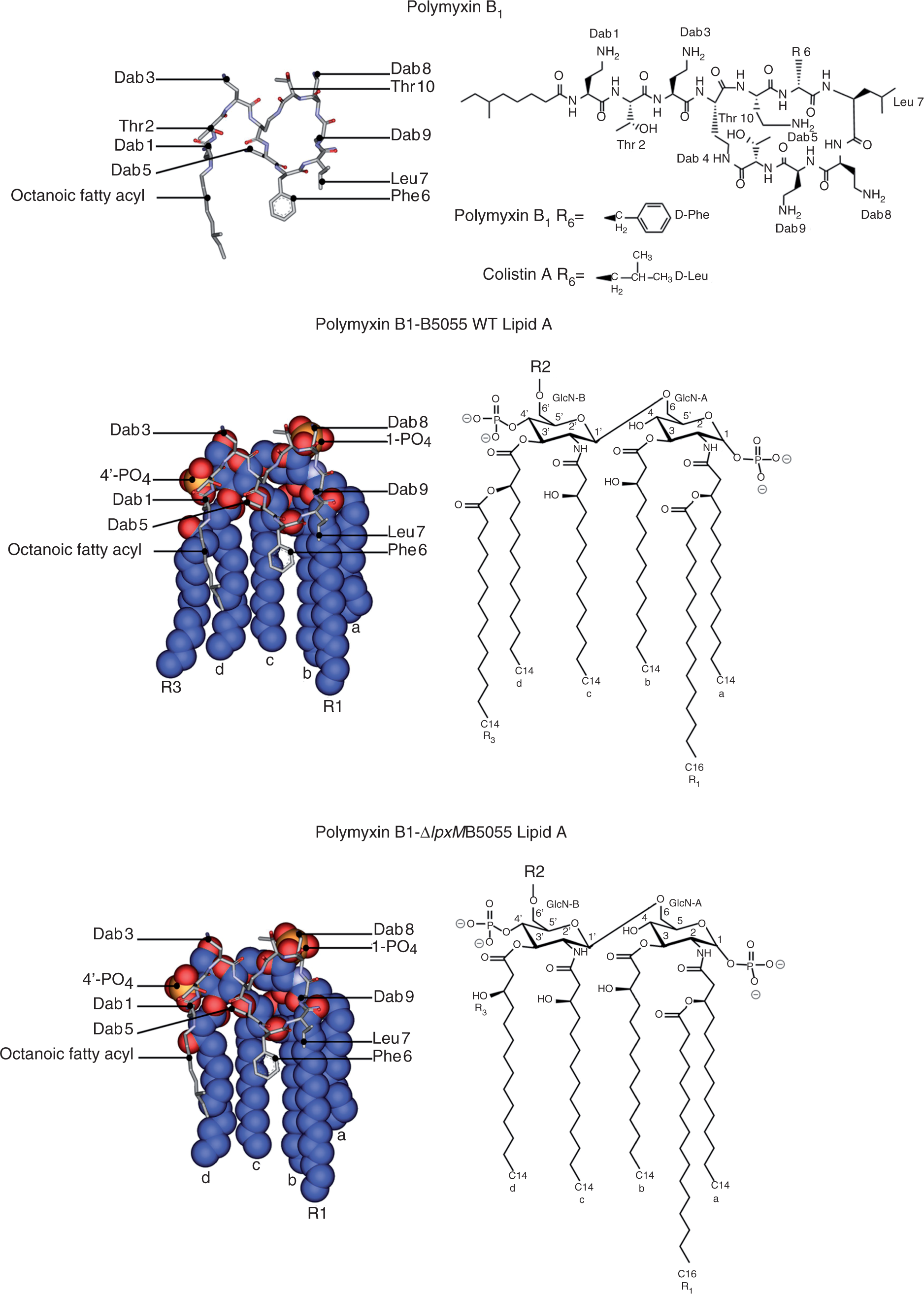
Discussion
Increasing reports of resistance to the last-line therapy polymyxins in K. pneumoniae have provided the impetus to elucidate the molecular mechanism(s) of resistance employed by this ‘superbug’. Although Campos et al.
29
suggested that susceptibility of K. pneumoniae to cationic antimicrobial peptides was dictated by the thickness of capsular polysaccharide material coating the surface, a recent study revealed that polymyxin susceptibility was not altered in a non-encapsulated K. pneumoniae B5055 mutant, possessing approximately one tenth of the WT capsule levels.
14
Alternatively, structural modifications of lipid A are known to confer polymyxin susceptibility in several other Gram-negative species (reviewed in Groisman
30
and Peschel
31
); these encompass changes to the lipid A acylation pattern or covalent modification of lipid A phosphates with cationic moieties, such as 4-amino-4-deoxy-
The mutant K. pneumoniae B5055 ΔlpxM strain was derived from strain B505514 with a defective lpxM gene which encodes for the late acyltransferase LpxM enzyme responsible for the addition of a secondary lipid A acyl chain. Therefore, compared with the hexa-acylated lipid A predominantly expressed by the B5055 WT, the mutant predominantly expresses a penta-acylated lipid A lacking the 3′-myristate chain. 14 K. pneumoniae isolates defective in LpxM acyltransferase have been shown to be less virulent in animal infection models and produce an LPS with reduced endotoxicity. 14
The bactericidal activity of polymyxin B is known to exhibit a strong temperature dependence.
45
For example, the activity (i.e. EC50) of polymyxin B against E. coli cells was noted to decrease drastically from 98% at 35℃ to merely 5.4% at 18℃ following treatment with the same polymyxin B concentration.
45
The temperature region at which the change in killing rate was observed (24–27℃) lies within the phase transition temperature for the E. coli OM, as measured by differential scanning calorimetry.
45
From a mechanistic view, these observations are reasonable as self-uptake of polymyxins necessitates penetration of the hydrophobic polymyxin domains (i.e. the N-terminal fatty-acyl chain, and
Fluorometric and 1H NMR spectroscopic measurements of the binding affinity of polymyxin B and E to isolated LPS from each strain demonstrated polymyxin B and E bound with greater affinity to the LPS from B5055 ΔlpxM compared with the B5055 WT strain. This would imply that the packing of lipid A fatty acyl chains can considerably affect the polymyxin–LPS binding interaction. This was further highlighted by findings that B5055 ΔlpxM was more susceptible to the lytic action of lysozyme and the anionic detergent, sodium deoxycholate, in comparison with B5055 WT, after exposure to polymyxin E. We propose that these observations may be attributed to a greater propensity for polymyxins to bind and disrupt the OM barrier of B5055 ΔlpxM, thus enhancing the action of these lytic agents.
NPN is a hydrophobic probe whose fluorescence is greatly increased in a hydrophobic environment. Tightly-packed lipid A fatty acyl chains limits the free diffusion of hydrophobic solutes, such as NPN, through the intact OM. 10 However, once permeated, intercalation of NPN into the phospholipid inner leaflet of the OM and the inner cytoplasmic phospholipid membrane bilayer produces a resultant increase in fluorescence. Differences in the kinetics and final NPN uptake levels were observed between B5055 and B5055 ΔlpxM. In comparison with B5055, NPN uptake kinetics were enhanced in B5055ΔlpxM, which corresponds with a less densely packed OM of greater permeability likely arising from the loss of a secondary lipid A acyl-chain in this strain. Further evidence in support of a highly permeable OM comes from the observation that NPN uptake by the B5055ΔlpxM isolate was not affected by the chelating agent, EDTA, which is known to release LPS from the Gram-negative OM. 46
Conclusions
Our findings provide a molecular-level understanding of the significant role played by the fatty-acyl chains of lipid A for complexation of polymyxin antibiotics by K. pneumoniae LPS. The increased polymyxin susceptibility of B5055 ΔlpxM was paralleled by a greater binding affinity for polymyxins and an increased fluidity of its LPS in comparison with the B5055 WT strain. This may arise from alterations to the LPS-to-LPS fatty-acyl chain packing, which enhances the fluidity of the OM and can account for an increased permeability of B5055 ΔlpxM to NPN. Therefore, the disruption of the K. pneumoniae OM by polymyxins may be partly dependent upon the ability of its N-terminal fatty-acyl chain to penetrate into the fatty-acyl chain layer of lipid A. This event appears to be more facile in strains which predominantly express an under-acylated lipid A with increased OM fluidity. This study provides important insights into the mechanisms of polymyxin activity in this very problematic pathogen.
Footnotes
Funding
R. L. N. and J. L. are supported by research grants from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01A1070896 and R01AI079330). T. V., R. L. N., J. L., P. E. T. and K. R. are also supported by the Australian National Health and Medical Research Council (NHMRC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Conflicts of interest
J. L. is an Australian NHMRC Senior Research Fellow. T. V. and M. A. B. are Australian NHMRC Career Development Research Fellows. M. C. is a NHMRC Australia Fellow supported by AF511105.