
Abstract
Defensins are the first endogenous mediators to be characterized as alarmins and play multifunctional roles in immune response. Previous studies reported that human neutrophil peptide (HNP)-1, a member of the α-defensin subfamily, could regulate the IL-1β post-translational process; however, the underlying mechanism remained unknown. Using an LPS-primed THP-1 macrophage model, we found that inhibition of P2X purinoceptor 7 (P2X7) suppressed HNP-1-initiated mature IL-1β release. Confocal microscopy and glutathione S-transferase (GST) pull-down assay demonstrated that HNP-1 bound to P2X7 directly. HNP-1 treatment increased the activated level of caspase-1, and inhibition of caspase-1 abolished IL-1β release. Incubation of LPS-primed macrophages with potassium chloride also prevented HNP-1-induced export of mature IL-1β. Likewise, an ethidium bromide uptake test showed that the P2X7-K+ efflux-caspase-1 signaling pathway triggered by HNP-1 contributed to pyroptotic pore formation. Furthermore, knock down of inflammasome adaptor Nod-like receptor family pyrin domain containing 3 (NLRP3) decreased activated caspase-1 level and reduced pore formation in macrophages, whereas IL-1β release was not significantly impaired. These findings not only illustrated the mechanism for alarmin HNP-1 in enhancing inflammatory response, but also provided therapeutic targets for certain inflammatory diseases in which defensins play important roles.
Introduction
Multicellular animals can not only recognize pathogen-associated molecular patterns from exogenous pathogens to initiate antimicrobial defense, but also sense endogenous dangerous molecules to initiate immune response. 1 Recently, host endogenous mediators that are rapidly released upon microbial invasion or tissue injury/damage, and have the ability both to recruit and to activate APCs have been termed ‘alarmins’.2–4 To date, alarmins include defensins, cathelicidin, high mobility group box-1 protein, eosinophil derived neurotoxin, granulysin and other proteins, which play multifunctional roles in infection, inflammation and adaptive immunity, as well as wound healing.4–7
Defensins, a family of cationic antimicrobial peptides, were the first endogenous mediators to be characterized as alarmins. Based on the organization of the three intramolecular disulfide bonds formed by six conserved cysteine residues, human defensins were classified as α-defensins and β-defensins. The α-defensins, human neutrophil peptides (HNP) 1–3, are constitutively expressed in neutrophils and comprise 30–50% of the granule proteins.8,9 In vitro, their antimicrobial activities depend on environmental conditions such as buffers of low ionic strength and neutral pH. 9 In vivo studies have documented that the levels of HNPs 1–3 in various body fluids (e.g. blood, bronchoalveolar lavage fluid and sputum) are greatly increased in patients with sepsis and pulmonary inflammatory disorders.10–12 Furthermore, high concentrations of HNPs can induce phagocytic defects in patients with cystic fibrosis. 13 Release of HNPs by activated polymorphonuclear leukocytes in HNPs transgenic mice can mediate acute lung injury. 14 These findings suggest that alarmins—HNPs 1–3—may contribute to the pathogenesis of inflammatory diseases.
The pro-inflammatory cytokine IL-1β is a key mediator in host immune and inflammatory responses. Previous studies reported that in LPS-activated monocytes, the alarmin cathelicidin/LL-37 and HNP-1 could regulate IL-1β post-translational processing and promote the release of mature IL-1β.15–17 In addition, LL-37 promoted membrane pore formation. 15 However, when HNP-1 and ATP were simultaneously added to LPS-activated monocytes, HNP-1 blocked ATP-mediated mature IL-1β release. 17 These observations suggest that HNP-1 may serve as a regulator for IL-1β maturation and may share the same signaling pathway with ATP to regulate the processing and export of IL-1β. In monocytes/macrophages, the process of IL-1β secretion occurs via two pivotal steps. A primary pro-inflammatory stimulus, such as LPS, induces high levels of pro-IL-1β synthesis, and a second stimulus (ATP, uric acid crystals, etc.) activates the IL-1β-converting enzyme (also termed caspase-1) through the assembly of the multi-protein complex inflammasome.18–20 Extracellular ATP acting on the purinergic receptor P2X7 induces the assembly NLRP3 inflammasome and activation of caspase-1, leading to IL-1β release and pyroptosis, which is described as an inherently inflammatory process of caspase 1-dependent programmed cell death and featured by rapid plasma membrane rupture and release of pro-inflammatory intracellular contents.21,22 However, the signaling pathway of HNP-1-induced monocyte/macrophage IL-1β release remains unknown.
In the present study, using the differentiated LPS-primed THP-1 macrophage model, we tested the hypothesis that HNP-1 could activate caspase-1 via direct binding to P2X7 receptor, and studied the mechanisms of HNP1-mediated caspase-1 activation.
Materials and methods
Materials
The materials used in this study and their sources were as follows: HNP-1 peptide (Abcam, Cambridge, MA, USA, and also a gift from Dr Wuyuan Lu, University of Maryland School of Medicine, Baltimore, MD, USA); GST protein (Abcam); Phorbol 12-myristate 13-acetate (PMA), Escherichia coli LPS serotype O1101:B4, oxidized ATP (oATP), KN-62, apyrase, potassium chloride (KCl), tetraethylammonium (TEA), gliotoxin, Triton X-100 and ethidium bromide (EtBr) (Sigma-Aldrich, St. Louis, MO, USA); ATP (Roche Applied Science, Indianapolis, IN, USA); YVAD-cmk (Merckmillipore, Darmstadt, Germany); ethidium homodimer-2 (EthD2) and SYTO42 (Life Technologies, Grand Island, NY, USA); anti-caspase-1 p10 rabbit monoclonal Ab (Epitomics, Burlingame, CA, USA); mouse anti-HNP-1 Ab (Hycult Biotech, Uden, the Netherlands); P2X7 recombinant protein with GST tag (Novus Biologicals, Littleton, CO, USA); phycoerythrin-labeled rat anti-mouse IgG1 (eBioscience, San Diego, CA, USA); P2X7 small interfering RNA (siRNA) and scrambled siRNA, NLRP3 small hairpin RNA (shRNA) lentiviral particles, IL-1β Ab and all HRP-conjugated second Abs were from Santa Cruz biotechnology (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The P2X7–EGFP vector cDNA was kindly provided by Professor James Wiley (Florey Neuroscience Institutes, University of Melbourne, Australia).
Cell culture experiments
THP-1 cells, P2X7-knock down (KD) THP-1 cells, and NLRP3-KD THP-1 cells were maintained in RPMI1640 (Thermo Scientific, Rockford, IL, USA) supplemented with 10% heat-inactivated FCS (Thermo Fisher Scientific) and 100 U/ml penicillin, as well as 100 µg/ml streptomycin (Life Technologies). P2X7-KD THP-1 cells were produced using P2X7 siRNA, and scrambled siRNA was used as a negative control. NLRP3-KD THP-1 cells were generated using NLRP3 shRNA lentiviral particles, according to the manufacturer’s instruction. The efficiency of knock down of P2X7 and NLRP3 was verified by a quantitative real-time PCR method using SYBR green with housekeeping gene β-actin as an internal control, as described previously. 23
For experiments, the cells were plated in 24-well plate (1 × 106 cells/well) and differentiated for 12 h with 100 nM PMA in RPMI1640 supplemented with FCS. Then, the culture medium was replaced with complete RPMI1640 medium. After resting for 48 h, the cells were given fresh complete medium with or without 100 ng/ml LPS and incubated for 3 h at 37℃. The media supernatants were subsequently removed and the cells were rinsed twice with RPMI1640. At this point, 5 mM ATP, or 50 µg/ml HNP-1 were added to the culture medium and the cells were incubated at 37℃ as noted. In some experiments, various compounds (100 µM YVAD-cmk, 900 µM oATP, 20 µM KN-62, 20 U/ml Apyrase, or 130 mM KCl) were added to the culture medium and incubated for 30 min at 37℃ before the addition of ATP or HNP-1. After this, the supernatants were collected and centrifuged. The cell-free supernatants were removed and stored at –20℃. The cells were detached with cell scrapers in cold PBS and collected for further analysis.
Western blot analysis
For detection of cellular levels of activated caspase-1 and mature IL-1β, the respectively treated cells were lysed in RIPA Lysis and Extraction Buffer (Beyotime, Shanghai, China) supplemented with 100 µg/ml phenylmethylsulfonyl fluoride (PMSF) and 1:100 protease inhibitor cocktail (Sigma-Aldrich). The cell lysate was incubated on ice for 40 min and centrifuged at 14,000 g for 15 min at 4℃. The total protein concentration of the lysate was measured using a BCA Protein Assay kit (Pierce Biotechnology, Rockford, IL, USA). Then, the cell lysate (20 mg ∼ 40 mg protein) was heated at 70℃ for 10 min with 1 × NuPAGE reducing agent. The proteins were separated on a 12% NuPage Tris-Bis gel and transferred to a nitrocellulose membrane. After blocking with 5% non-fat dry milk in 1 × Tris-buffered saline buffer containing 0.1% Tween-20 for 2 h, the membrane was incubated with the first Ab at 4℃ overnight (12 h), followed by another 2-h incubation with the related second Ab at room temperature (20℃). The membrane was developed using ECL Western Blotting Substrate kit (Pierce Biotechnology) and the blot was visualized using X-ray film.
Activated caspase-1 levels from THP-1 cells or NLRP3-KD THP-1 cells after HNP-1 treatment were detected in the precipitated cell-free supernatant as described previously.24,25 Western blot analysis was performed as described above.
ELISA
The released IL-1β levels in the cell-free supernatant were measure using an ELISA (R& D systems, Minneapolis, MN, USA).
Transfection of P2X7-EGFP recombinant vector
Transfection of P2X7-EGFP vector into HEK-293 cells was performed as previously described, with minor modifications. 26 HEK-293 cells were maintained in DMEM (Thermo Fisher Scientific) supplemented with 10% heat-inactivated FCS (Thermo Fisher Scientific) and 100 U/ml penicillin, as well as 100 µg/ml streptomycin (Life Technologies). Four micrograms of P2X7-EGFP cDNA and 10 µl of Lipofectamine 2000 reagent were diluted in 250 µl serum-free Opti-MEMI medium, respectively, and incubated at room temperature for 5 min. Then, the diluted cDNA and transfection reagent were mixed gently and incubated at room temperature for another 20 min. The mixture was added to a monolayer of HEK-293 cells (1 × 106 in 1.5 ml of Opti-MEMI medium) in a CellView cell culture dish (Greiner Bio-One, Frickenhausen, Germany), and transfection was performed at 37℃ in a CO2 incubator. Six hours later, the medium was changed to DMEM supplemented with 10% heat-inactivated FCS and the cells were kept into the CO2 incubator.
Confocal microscopy
After HEK-293 cells were transfected for 48 h, the culture medium was removed and the cells were incubated in DMEM containing 50 µg/ml HNP-1 or no additive at 37℃ for 1 h. In some experiments, 20 µM KN-62 was added to the culture medium to incubate for 30 min at 37℃ before the addition of HNP-1. Then, the cells were washed three times with PBS and blocked with 5% FBS for 30 min. After washing twice with PBS, the cells were incubated with anti-HNP-1 Ab in 5% FBS containing PBS for 90 min. Following three washes with PBS, the cells were stained with PE-labeled rat anti-mouse IgG1 for 30 min, washed four times with PBS and, finally, placed in 0.2 ml PBS.
Confocal microscopy was performed using a LEICA TCS-SP2 confocal microscope (Leica Microsystems, Wetzlar, Germany), which was equipped with argon lasers and attached to a 63 × oil-immersion objective lens. After standard fluorescence observations, immunofluorescence staining was automatically scanned. The optical sections were acquired and assembled in an image processor and, finally, projected into a single image using PowerScanner physiology software.
GST pull-down assay
GST in vitro pull-down assays were performed using a ProFound pull-down GST protein:protein interaction kit (Pierce Biotechnology), according to the manufacturer’s instructions. GST-tagged P2X7 protein and GST protein were immobilized to glutathione–agarose gel contained column, and the columns were incubated at 4℃ for 2 h with gentle rocking. A non-treated glutathione–agarose gel control was used as a negative control. After five washes with wash solution, the columns were incubated with 8 µg HNP-1 at 4℃ for 6 h with gentle rocking. Then, the glutathione resin was washed with 400 µl wash solution five times and the proteins were eluted using 100 mM glutathione elution buffer. The eluted samples were analyzed by immunoblotting.
The immunoblot procedure was performed as described previously, with minor modifications. Briefly, after the samples were prepared, they were loaded into a Novex 16% Tricine Gel and run at 120 V for 80 min. Then, the proteins were transferred from the Tricine gel to a nitrocellulose membrane using Mini Trans-Blot cell (Bio-Rad, Hercules, CA, USA) at 0.18 amp for 20 mins at 4℃. The blot was treated as described above, with the first Ab mouse-anti-HNP-1 and the second Ab goat-anti-mouse IgG HRP.
Pore formation
To ascertain whether HNP-1 treatment induced morphological change, pore formation was assayed by EtBr stain permeability as described previously. 27 THP-1 cells and/or NLRP3-KD THP-1 cells (5 × 105 cells) were grown on glass coverslips in a 24-well plate. The cells were differentiated by 100 nM PMA and stimulated or not with 100 ng/ml LPS as described above. After treatment with various compounds (100 µM YVAD-cmk, 900 µM oATP, 20 U/ml Apyrase, and 5 mM tetraethylammonium), and 5 mM ATP or 50 µg/ml HNP-1, the coverslips were inverted onto 10 μl PBS containing 10 μM cell-permeant blue fluorescent nucleic acid stain SYTO 42 and either 25 µg/ml membrane impermeable red dyes EtBr or EthD2. Meanwhile, apoptotic cells induced by gliotoxin and cells treated with detergent triton X-100 were included as controls. Images were acquired using an Olympus IX81 fluorescent microscope (Olympus, Tokyo, Japan). Pore formation was assayed by the percentage of the cells stained positive with EtBr, and each sample was counted for at least five fields per coverslip.
Statistical analysis
Data are expressed as the mean ± SD, and were analyzed by Student’s t-test or ANOVA followed by Bonferroni post-test analysis, using GraphPad Prism 5.00 for Windows (GraphPad Software, La Jolla, CA, USA). A two-tailed P-value of less than 0.05 was considered statistically significant.
Results
HNP-1 treatment of LPS-primed macrophages leads to IL-1β release via P2X7
A previous study suggested that HNP-1 and ATP might share a same signaling pathway for processing IL-1β.
17
The effect of ATP on the export of mature IL-1β in LPS-primed macrophages is mediated by P2X7, and this effect can be inhibited by blocking its binding site on P2X7 with inhibitors KN-62 and oATP.15,28 In this study, inhibition of P2X7 with KN-62 and oATP not only reduced mature IL-1β secretion from LPS-primed macrophages stimulated with ATP (Figure 1A) but also suppressed mature IL-1β release from macrophages treated with HNP-1 (Figure 1B). Furthermore, knock-down P2X7 expression in THP-1 with P2X7 specific siRNA obviously inhibited the level of mature IL-1β induced by ATP or HNP-1 (Figure 1C). These findings indicated that the effect of HNP-1 on the processing of mature IL-1β might be a consequence of interaction with P2X7. To test this hypothesis, P2X7-EGFP vector-transfected HEK-293 cells were incubated with HNP-1, and immunofluorescence analysis was performed using anti-HNP-1 Ab and fluorescence-labeled second Ab respectively. Confocal microscope analysis showed that the fluorescent P2X7 and the fluorescent HNP-1 were co-localized on the membrane of HEK-293 cells, while addition of KN-62 reduced HNP-1 mediated red-fluorescence (Figure 1D). Moreover, GST pull-down assay further confirmed the direct interaction of HNP-1 to P2X7 (Figure 1E). Taken together, these results demonstrated that HNP-1 treatment leading to IL-1β release from LPS-primed macrophage was mediated by P2X7.
HNP-1 treatment of LPS-primed macrophages leads to IL-1β release via HNP-1 binding to P2X7. (A, B) The concentration of released IL-1β in the supernatant was measured by ELISA. PMA-differentiated THP-1 cells were stimulated or not with 100 ng/ml LPS for 3 h followed by incubation with 5 mM ATP for 1 h or 50 µg/ml HNP-1 for 3 h. In some wells, 900 µM oATP or 20 µM KN-62 were added 30 min before the addition of ATP or HNP-1. The data were presented as mean ± SD from four independent experiments. *P < 0.001, **P < 0.01 after Bonferroni’s multiple comparison post-test. (C) The cellular levels of mature IL-1β in P2X7 knock-down THP-1 cells were detected using Western blot analysis. THP-1 cells were treated with P2X7 siRNA and scramble siRNA using Lipofectamine 2000 reagent for 3 d. Then the cells were stimulated with 100 ng/ml LPS for 3 h followed by 5 mM ATP for 1 h or 50 µg/ml HNP-1 for 3 h, and cell lysates were analyzed. (D) P2X7 and HNP-1 were co-localized on HEK-293 cell surfaces. P2X7-EGFP transfected cells were incubated with 50 µg/ml HNP-1 or left untreated for 1 h. KN-62 was added 30 min prior to HNP-1. After blocking, the cells were incubated with anti-HNP-1 Ab and PE-labeled rat anti-mouse IgG1. The stained cells were analyzed using a LEICA TCS-SP confocal microscope as described in the ‘Materials and methods’. Representative images were presented from 2–3 independent experiments. (E) HNP binds to P2X7 receptor. GST-tagged P2X7 protein and GST protein were immobilized to the glutathione–agarose gel contained column, followed by incubation with HNP-1. The proteins were eluted and analyzed by immunoblot. A non-treated glutathione–agarose gel control was used as a negative control. Representative results are presented from three independent experiments.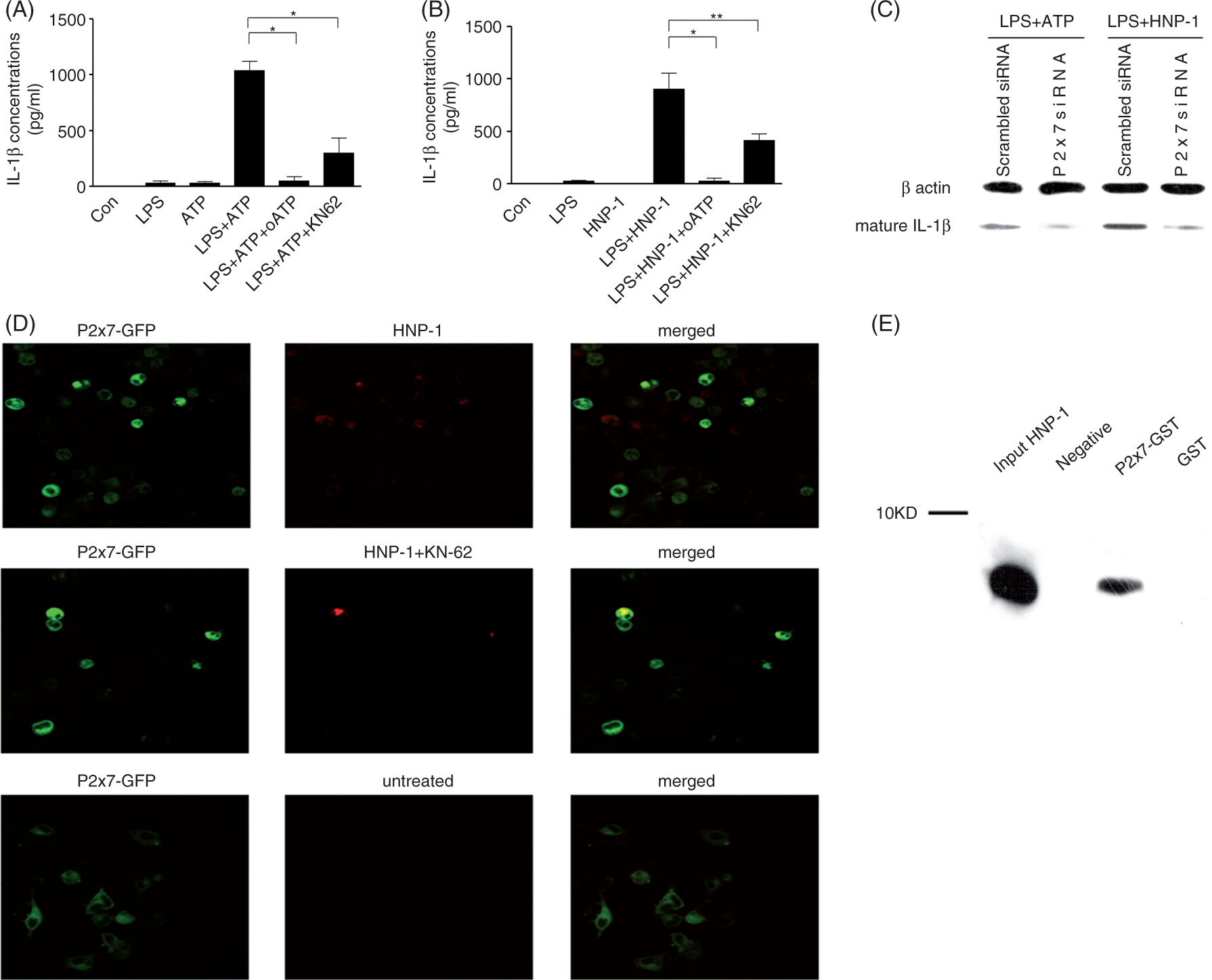
HNP-1 treatment of LPS-primed macrophages leads to IL-1β release via activation of caspase-1
Activation of caspase-1 is a key step for IL-1β regulation and secretion, and inhibition of caspase-1 prevented mature IL-1β from exporting in ATP-treated LPS-primed macrophage.
20
We first investigated whether HNP-1 treatment also activated caspase-1 in LPS-primed macrophages. Based on previous studies and our preliminary experiments, a final concentration of 50 µg/ml HNP-1 was applied in this study. As observed in ATP-treated LPS-primed macrophages, HNP-1 treatment resulted in an obviously enhanced level of caspase-1 p10 component, which is an activated form of caspase-1 critical for IL-1β processing (Figure 2A, B). Furthermore, inhibition of caspase-1 with a specific inhibitor, YVAD-cmk, completely blocked mature IL-1β release from LPS-primed macrophages treated with HNP-1 or ATP (Figure 2C, D).
IL-1β release from HNP-1-treated LPS-primed macrophages is mediated by caspase-1. PMA-differentiated THP-1 cells were stimulated or not with 100 ng/ml LPS for 3 h, followed by incubation with 5 mM ATP for 1 h, or 50 µg/ml HNP-1 for 3 h. Activated caspase-1 levels were detected by Western blot either in the cellular lysate (A) or in the medium supernatant (B). Representative results were presented from 3–4 independent experiments. In some cases, 100 µM YVAD-cmk was added 30 min before the addition of ATP (C), or HNP-1 (D), and the concentration of released IL-1β in the supernatant was measured by ELISA. The data are presented as mean ± SD of 3–4 independent experiments. *P < 0.001 after Bonferroni’s multiple comparison post-test.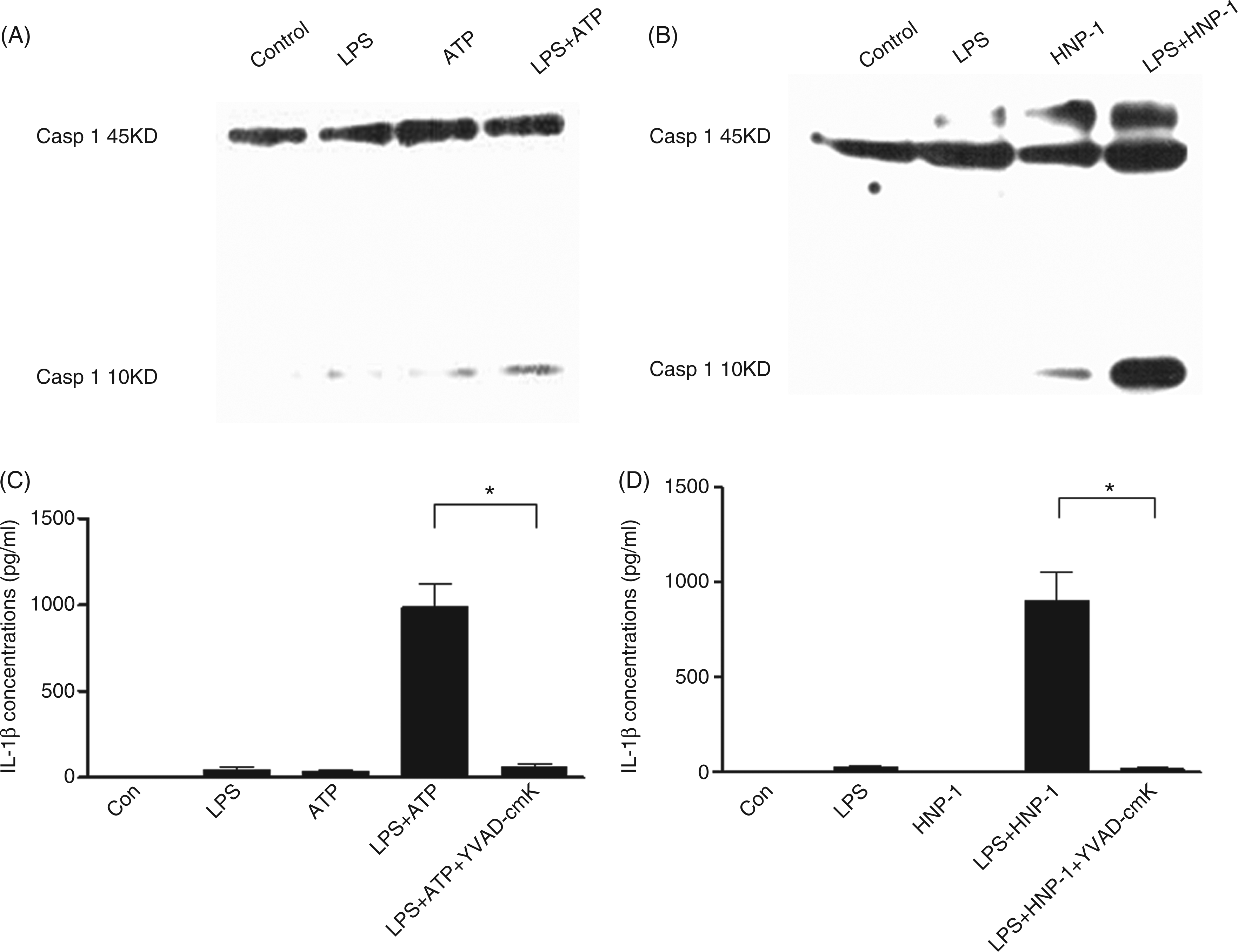
K+ efflux is required for IL-1β release in HNP-1 treatment of LPS-primed macrophages
A previous study reported that treatment of LPS-primed monocytes with alarmin LL-37 lead to a transient release of endogenous ATP, the concentration of which was far lower than the dose of exogenously added ATP required for IL-1β processing.
15
Defensins have been demonstrated to promote ATP efflux from bacterial cells.
29
We used an enzyme known to inhibit autocrine ATP, apyrase, to exclude a possible role of endogenous ATP in IL-1β release in HNP-1-treated LPS-primed macrophages. The presence of apyrase at a concentration of 20 U/ml did not influence the effect of HNP-1 on IL-1β release in the LPS-primed macrophages, while the effect of exogenous ATP was abolished (Figure 3A, B).
K+ efflux is required for IL-1β release from HNP-1-treated LPS-primed macrophages. PMA-differentiated THP-1 cells were stimulated with 100 ng/ml LPS for 3 h, followed by incubation with 5 mM ATP for 1 h, or 50 µg/ml HNP-1 for 3 h. In some cases, 20 U/ml apyrase or 130 mM KCl were added 30 min before the addition of ATP (A, C), or HNP-1 (B, D), and the concentration of released IL-1β in the supernatant was measured by ELISA. The data are presented as mean ± SD of three independent experiments. *P < 0.01, **P > 0.05, ***P < 0.001, ****P < 0.05 after Bonferroni’s multiple comparison post-test.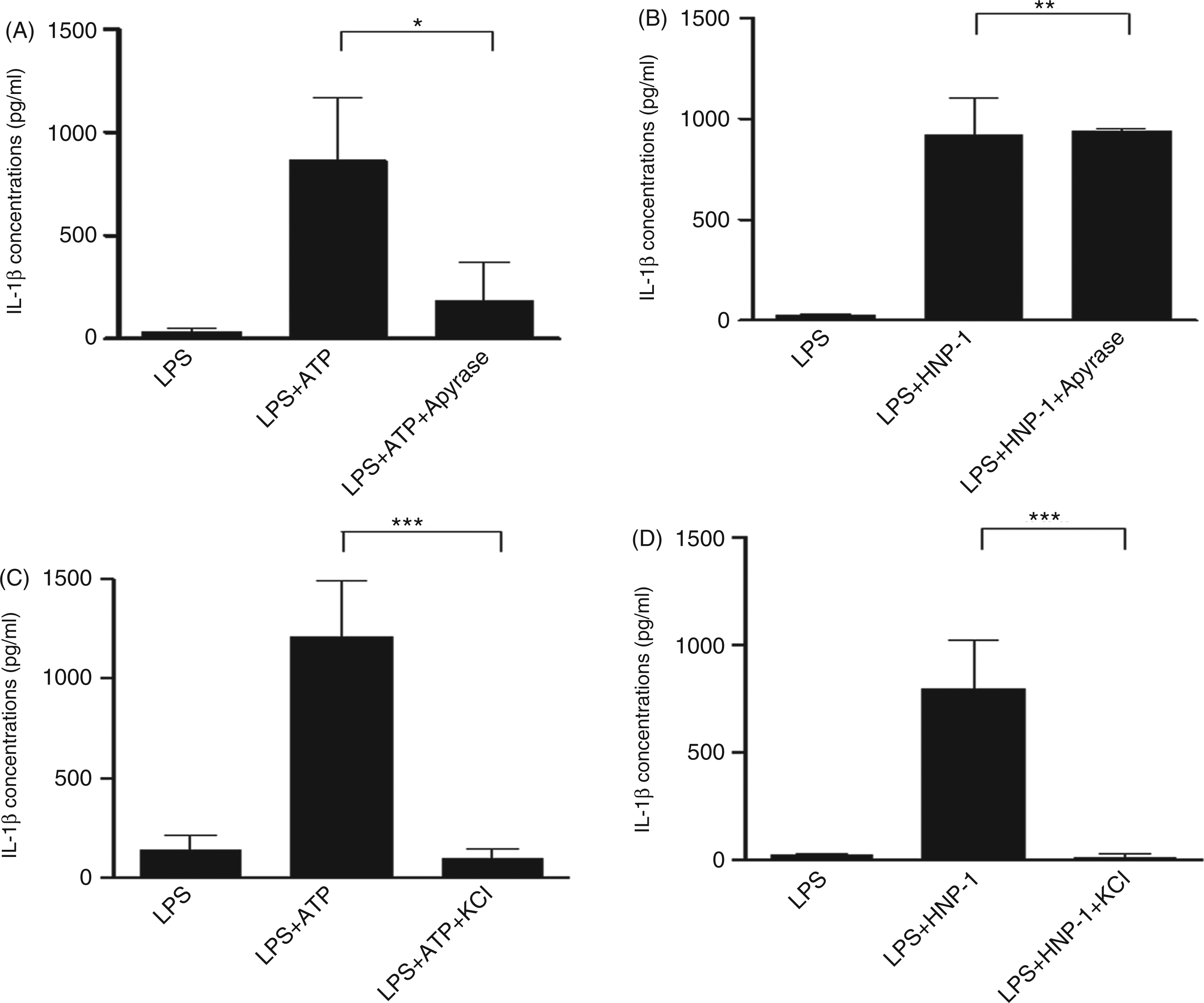
Activation of P2X7 by extracellular ATP can rapidly induce the efflux of cytosolic K+, which is a necessary signal for caspase-1 activation and IL-1β post-transcriptional processing. 30 To address whether K+ efflux is involved in the mechanism for IL-1β release after HNP-1 treatment, LPS-primed macrophages were incubated in medium with a K+ concentration of 130 mM. As expected, ATP-induced IL-1β maturation was blocked by KCl (Figure 3C). Figure 3D illustrates that prevention of cytosolic K+ efflux by high extracellular K+ medium nearly abrogates HNP-1-induced IL-1β processing.
P2X7-K+ efflux-caspase-1 signaling pathway contributes to pore formation in HNP-1 treatment of LPS-primed macrophages
Stimulation of P2X7 with exogenous ATP in LPS-primed macrophages induces membrane pore formation and pyroptosis by a caspase-1-dependent mechanism.
31
Pyroptotic pores with a functional diameter of 1–2 nm rapidly form in the plasma membrane of macrophages, allowing the passage of small molecules, such as EtBr, but excluding the larger EthD2.
27
HNP-1 treatment significantly enhanced EtBr uptake by LPS-primed macrophages, whereas inhibition of either caspase-1 or P2X7, or blocking of K+ current by TEA, prevented the increase in EtBr permeability caused by either HNP-1 or ATP (Figure 4A, B). EthD2 was not permeable in any experimental group (data not shown). In contrast, apoptotic cells retained an intact plasma membrane, which excludes both EtBr and EthD2, while cells treated with detergent triton X-100 allowed the influx of both EtBr and EthD2 (Figure 4A). Therefore, the P2X7-K+ efflux-caspase-1 signaling pathway also contributed to pyroptotic pore formation in HNP-1 treatment of LPS-primed macrophages.
P2X7-K+ efflux-caspase-1 signaling pathway contributes to pore formation in HNP-1-treated LPS-primed macrophages. THP-1 cells were grown on glass coverslips, differentiated by 100 nM PMA and stimulated or not with 100 ng/ml LPS. After treatment with various inhibitors (100 µM YVAD-cmk, 900 µM oATP or 5 mM TEA) for 30 min followed by incubation with 5 mM ATP for 1 h (A), or 50 µg/ml HNP-1 for 1 h (B), the coverslips were stained with blue fluorescent nucleic acid stain SYTO 42 and either membrane impermeable red dyes EtBr or EthD2. Cells treated with gliotoxin and Triton X-100 were included as controls. Images were acquired using Olympus IX81 fluorescent microscope with 10 × objective. Pore formation was assayed by the percentage of the cells stained with EtBr. The data are shown as mean ± SD of three independent experiments, and representative images are presented. *P < 0.001 after Bonferroni’s multiple comparison post-test.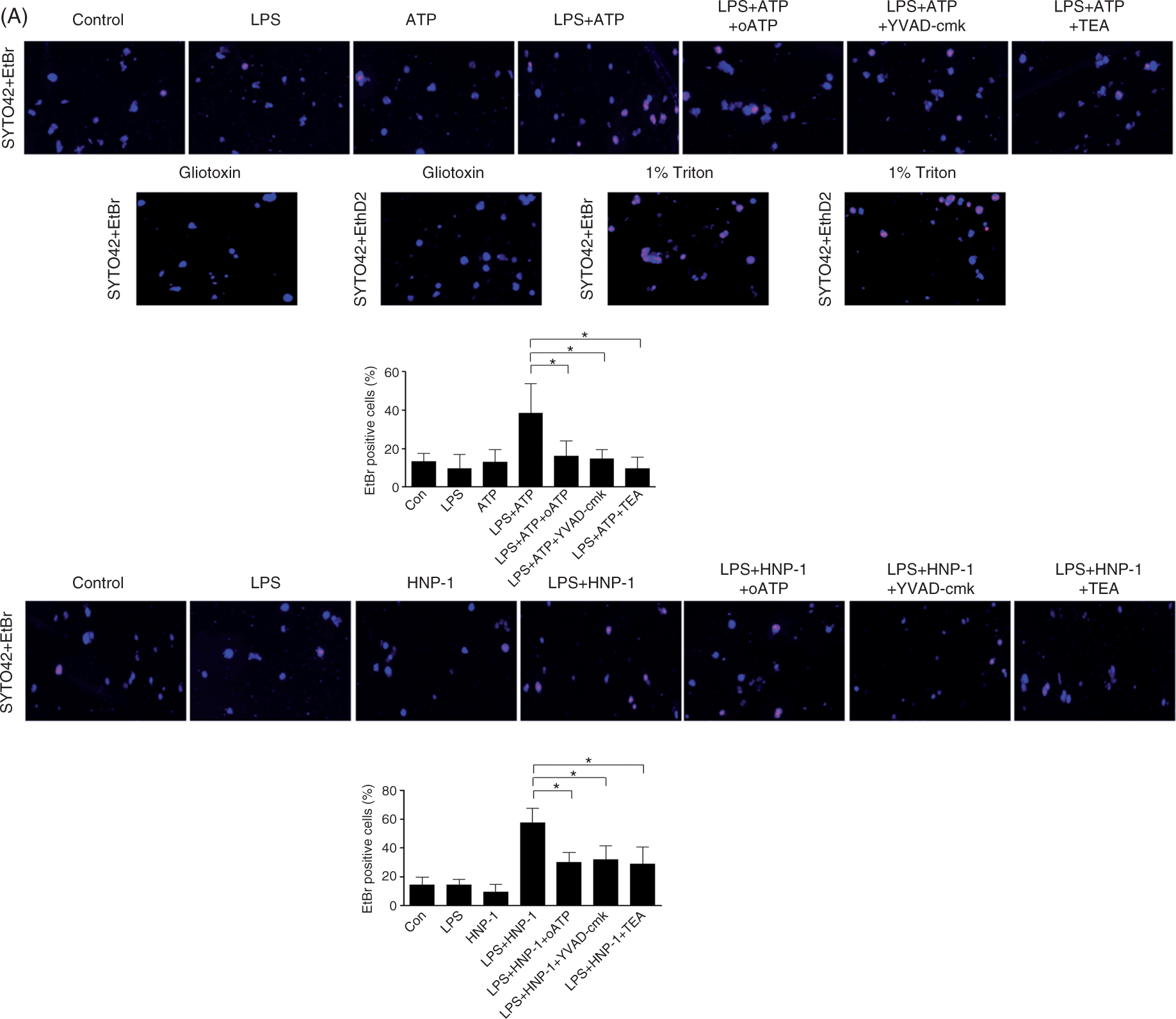
Knock down of NLRP3 significantly affected pyroptosis, but did not influence on IL-1β release in HNP-1 treatment of LPS-primed macrophages
The inflammasome adaptor NLRP3 plays an important role in P2X7-mediated caspase-1 activation by ATP in LPS-primed macrophages, and the assembly of the inflammasome is a crucial step in activating caspase-1 to process IL-1β.18–20 Using NLRP3 shRNA lentiviral particles, the mRNA level of NLRP3 in the NLRP3-KD THP-1 cells was reduced to around 50% of that in the wild type (WT) THP-1 cells (Figure 5A). Furthermore, knock down of NLRP3 diminished the activation of caspase 1 induced by ATP or HNP1, as evidenced by the reduction in caspase-1 p10 levels in LPS-primed NLRP3-KD macrophages in comparison with LPS-primed WT THP-1 macrophages (Figure 5B). However, IL-1β release in response to ATP or HNP1 was not affected by NLRP3 KD (Figure 5C). On the contrary, EtBr uptake was significantly suppressed in LPS-primed NLRP3-KD macrophages treated with the stimuli (Figure 5D).
NLRP3 is essential for pyroptosis, but not IL-1β release, in HNP-1-treated LPS-primed macrophages. (A) Real-time quantitative PCR detected the mRNA levels of NLRP3 in WT and NLRP3-KD THP-1 cells. *P < 0.001, t-test. (B) Differentiated WT and NLRP3-KD THP-1 cells were stimulated with 100 ng/ml LPS for 3 h followed by incubation with 5 mM ATP for 1 h, or 50 µg/ml HNP-1 for 3 h. Activated caspase-1 levels were detected using a Western blot either in the cellular lysate (ATP) or in the medium supernatant (HNP-1). Representative results are presented from three independent experiments. (C) The concentration of released IL-1β in the supernatant from the above-treated cells was measured by ELISA. The data are presented as mean ± SD from three independent experiments. (D) WT and NLRP3-KD THP-1 cells were grown on glass coverslips, differentiated by 100 nM PMA and stimulated or not with 100 ng/ml LPS. After treatment with various inhibitors (100 µM YVAD-cmk, 900 µM oATP or 5 mM TEA) for 30 min followed by incubation with 5 mM ATP for 1 h, or 50 µg/ml HNP-1 for 1 h, the coverslips were stained with 10 μM SYTO 42 and either 25 µg/ml EtBr or EthD2. Images were acquired using Olympus IX81 fluorescent microscope with 10 × objective. Pore formation was assayed by the percentage of the cells stained with EtBr. The data are shown as mean ± SD from three independent experiments, and representative images are presented. *P > 0.05, **P < 0.0001, t-test.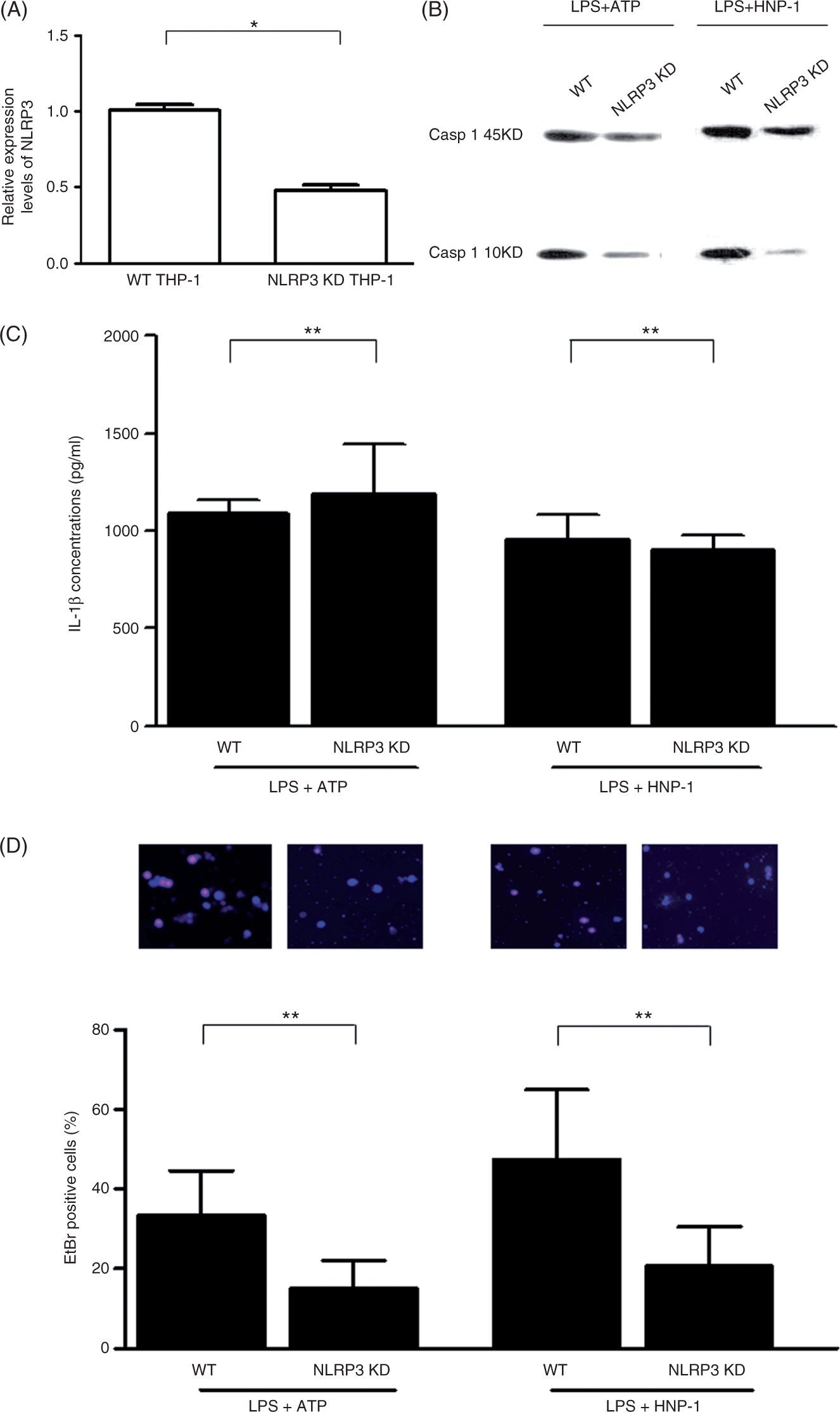
Discussion
Our study found that the alarmin HNP-1 induced caspase-1 activation via P2X7 receptor and stimulating cytosolic K+ efflux, leading to mature IL-1β release and pyroptosis in LPS-primed macrophages. Furthermore, partial knock down of the inflammasome adaptor NLRP3 did affect HNP1-induced caspase-1 activation and pyroptotic pore formation, whereas HNP1-induced IL-1β release was not impaired.
Alarmins are endogenous molecules that can attract and activate APCs, such as dendritic cells and monocytes/macrophages; consequently they are capable of promoting innate and adaptive immune responses.2–4 The chemotactic activity of defensins, the first characterized alarmins, was demonstrated to be mediated by Gαi protein-coupled receptors, such as CC chemokine receptor (CCR) 2 and CCR6.32,33 However, data concerning the activating receptor(s) for defensins are limited. Biragyn et al. 34 observed that murine β-defensin 2 activated dendritic cells in a TLR4-dependent manner. Funderburg et al. 35 reported that human β-defensin 3 (hBD-3) activated monocytes and dendritic cells via TLR1/TLR2 heterodimers. Using inhibition experiments, confocal microscopy and GST pull-down assays, we found that HNP-1 could bind to P2X7 to activate macrophages, leading to the release of proinflammatory cytokine IL-1b and pyroptosis. These findings not only identify P2X7 as another activating receptor for HNP-1, but also demonstrate defensins’ capacity to boost host inflammatory immune responses by promoting macrophage IL-1β production. As the concentration of HNP-1 applied in this study can be achieved in local inflammatory environments, such as pneumonia and cystic fibrosis, these findings may also suggest an important role of HNP-1 in the pathogenesis of inflammatory diseases. Recently, Khine et al. 36 reported that HNP1-3 induced IL-8 production in lung epithelial cells through the purinergic receptor P2Y6. Combined with the current findings, it is likely that alarmin defensins may interact with different receptors in different cells to play multifunctional roles in the immune system.
The export of caspase-1-processed IL-1β from monocyte/macrophages in response to extracellular ATP has been proposed to be mediated by activation of P2X7, K+ efflux and assembly of the NLRP3 inflammasome.21,30 In the present study, incubation of LPS-primed macrophages in a medium with a high concentration of K+ prevented IL-1β release after ATP or HNP-1 stimulation, indicating that ATP or HNP-1 treatment promoted K+ efflux, which is critical for inflammasome assembly and subsequent activation of caspase-1. However, partial knock down of the inflammasome adaptor NLRP3 did not impair IL-1β secretion. Indeed, a low amount of active caspase-1 p10 was observed in LPS-primed NLRP3-KD macrophages after challenge with ATP and HNP-1. Combined with the important role of NLRP3 inflammasome in ATP mediated caspase-1 activation and IL-1β release, the present results indicate that the low levels of caspase-1 from partial knock down of NLRP3 might be sufficient for complete processing of IL-1β. However, previous studies have shown that another inflammasome component, apoptosis-associated speck-like protein containing a C-terminal caspase-activating recruiting domain (ASC), is essential for activation of caspase-1 and generation of mature IL-1β upon ATP-triggered P2X7 activation.30,37,38 ASC is an adaptor protein not only linking NLRP3 to procaspase-1 to assemble the NLRP3 inflammasome, but also required to form other inflammasomes, such as the NALP2 inflammasome. ASC can even oligomerize and activate caspase-1 independently of NOD-like receptors or other proteins at sufficiently low K+ concentrations.18–20 Thus, it is possible that inflammasomes other than NLRP3 inflammasome may also contribute to caspase-1 activation and IL-1β release in LPS-primed macrophages after treatment with ATP or HNP-1.
Activation of caspase-1 is not only critical for IL-1β secretion, but also crucial for pyroptosis. 31 Treatment of LPS-primed macrophages with HNP-1 or ATP caused significant pore formation, which was mediated by the P2X7-K+ efflux-caspase-1 signaling pathway. In contrast to the mechanism in IL-1β release, partial knock down of NLRP3 significantly affected pyroptotic pore formation, which was consistent with previous studies showing that P2X7 activation by ATP leads to pore formation by a mechanism independent of IL-1β processing and release.39,40 This finding suggests that HNP-1 utilizes distinct intracellular signaling pathways for the induction of IL-1β maturation and pyroptosis, both of which promote host inflammatory and immune responses.
Several issues need to be studied in the future. First, defensins are cationic antimicrobial proteins with six cysteine residues and three intramolecular disulfide bonds that play important roles in innate and adaptive immunity. Recently, the residues responsible for the biological activities of defensins have been investigated. In hBD-3, the cysteine residues, rather than the intramolecular disulfide bonds, appear to be important for its chemoattractant function. 41 Trp-26 residue in HNP-1 is a crucial molecular determinant of HNP-1 contributing to its functional versatility. 42 The present study indicates that HNP-1 shares the same binding sites on P2X7 with ATP. However, the residues in HNP-1 important for its binding to P2X7 have not been identified. Second, our data indicate that NLRP3 inflammasome is not required for the caspase-1-mediated IL-1β release in LPS-primed macrophages in response to HNP-1; however, it remains to be determined which inflammasome is responsible. Third, several mechanistically distinct models of non-classical release of caspase-1-processed IL-1β from LPS-primed monocyte/macrophage after activation of P2X7 have been proposed, including exocytosis of secretory lysosomes, formation of multi-vesicular bodies containing exosomes, direct outflow through membrane protein transporters and so on. 43 Although HNP-1 triggered IL-1β secretion via activation of P2X7, the detailed model needs to be identified. Further studies aimed at elucidating the aforementioned issues would help gain additional insight into the mechanism by which these alarmins modulate immune responses.
In summary, the present study demonstrates that HNP-1 via binding to P2X7 can induce macrophage pyroptosis and IL-1β release by NLRP3 inflammasome-dependent and inflammasome-independent pathways respectively. This study not only illustrates the mechanism for the alarmin HNP-1 in enhancing the inflammatory response, but also provides therapeutic targets for certain inflammatory diseases in which defensins play important roles.
Footnotes
Funding
This work was supported by National Science Fund for Distinguished Young Scholars (No. 30825037), and a Key Program from National Natural Science Foundation of China (No. 81130036), Changjiang Scholar Program of China, and Zhejiang Provincial Program for the Cultivation of High-level Innovative Health talents.
Acknowledgements
We thank Professor Tomas Ganz (David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA) for proofreading and editing the manuscript. The authors also thank Professor James Wiley (Florey Neuroscience Institutes, University of Melbourne, Australia) for the P2X7-EGFP vector and Dr Wuyuan Lu (Institute of Human Virology, University of Maryland School of Medicine, Baltimore, MD, USA) for the HNP-1 peptide.