
Abstract
Caspase-1 plays a fundamental role in innate immunity and in several important inflammatory diseases as the protease activates the pro-inflammatory cytokines proIL-1β and proIL-18. Caspase-1 itself is activated in different inflammasome complexes, which assemble in response to a variety of exogenous and endogenous stressors. More recently, pyroptosis, a caspase-1-dependent type of programmed cell death, has been identified that is able to support secreted IL-1 and IL-18 in triggering an inflammatory response. Whereas these ‘canonical’ activities are well appreciated, this review also highlights less-known pathways and molecules activated by caspase-1. There is evidence that caspase-1 supports cell survival by activation of NF-κB, induction of membrane repair and regulation of unconventional secretion of certain proteins. The physiologic effects of processing of other downstream targets, such as proteins involved in glycolysis or activation of caspase-7, are less well understood. However, there is increasing evidence that caspase-1 contributes to innate and adaptive immunologic defense mechanisms, repair and pathologic conditions by the regulation of several different and partially opposing pathways.
Introduction
Caspases
Caspases (cysteinyl aspartic proteases) comprise a family of cysteine proteases (caspase-1 to caspase-10, caspase-12 and caspase-14 in humans), which are known mainly for their essential roles in apoptosis.1,2 This form of programmed cell death is characterized by regulated caspase-dependent self-digestion of a cell resulting in membrane-surrounded apoptotic bodies. The dying cells expose ‘find-me’ and ‘eat-me’ signals, thereby mediating their engulfment by phagocytes, which is considered to be immunologically silent. 3
Caspases are expressed as inactive precursors with an amino terminal prosequence. 4 The prosequence may consist of homotypic protein–protein interaction domains, such as the death effector domain (DED) or the caspase activation and recruitment domain (CARD). 1 DED and CARD comprise families of the death domain (DD) fold, characterized by six anti-parallel α-helices, which form a Greek key. Family members can, but do not necessarily have to, allow interactions with members of the same family. However, they never bind to a member of a different family. Interestingly, the DD fold is only found in proteins involved in apoptosis and inflammation, which suggests a special, and most likely evolutionarily conserved, connection between both pathways. 1 Apoptotic caspases with long prodomains (caspase-2, and caspases 8–10) are called upstream or initiator caspases as they are able to react to death signals by DD fold-mediated assembly into macromolecular complexes and subsequent self-activation. Once activated, initiators process and thereby activate the downstream or effector caspases (caspase-3, and caspases 6 and 7), which contain only short prosequences lacking a DD fold. In turn, activated effector caspases execute cell death through cleavage of numerous substrates. In contrast to effector caspases, which require proteolytic processing to be activated, upstream caspases, such as caspase-9, can also be active without proteolytic cleavage.1,2
Whereas apoptotic caspases are expressed ubiquitously, expression of caspase-14 is restricted to stratified epithelia, such as the skin.5,6 Caspase-14 exerts a very specialized function, namely the cleavage of pro-fillagrin, which is needed for the proper formation of the cornified envelope, and thereby protects the cells of the skin from UVB irradiation and water loss.5,6
All other caspases (caspase-1, caspases 4 and 5, and caspase-12 in humans) are called inflammatory caspases. They are transcribed from the same chromosomal locus, contain an amino terminal CARD, and are involved in inflammation through the regulation of activation of the pro-inflammatory cytokines proIL-1β and proIL-18.7,8 Caspase-12 lacks catalytic activity, inhibits caspase-1 and its ablation in mice confers resistance against sepsis,9,10 although there is also evidence for an involvement in endoplasmic reticulum (ER) stress-induced apoptosis. 11
Caspase-1
Caspase-1 was identified in 1989 as the protease able to cleave the pro-inflammatory cytokine proIL-1β.12,13 Three years later its cDNA was cloned as IL-1β-converting enzyme (ICE). 14 It turned out that ICE has 28% similarity to the Caenorhabditis elegans protein CED-3, which is essential for apoptosis. Although overexpressed ICE is able to induce apoptosis, 15 mice lacking caspase-1 do not show a spontaneous phenotype, demonstrating that expression of caspase-1 is dispensable for apoptosis during embryonic development.16,17 Today, it is generally accepted that endogenous caspase-1 is not involved in apoptosis.3,18,19 Nevertheless, some publications claimed a requirement of caspase-1 for apoptosis in neuronal or endothelial cells.20–22 This discrepancy may be, at least partially, explained by the use of different methods for the detection of cell death as caspase-1 activation can induce a lytic form of programmed cell death termed pyroptosis, which can be distinguished clearly from apoptosis (see later).
Besides proIL-1β, caspase-1 can also activate proIL-18, another member of the IL-1 family.23,24 However, there are several reports demonstrating that, in mice, proIL-1β25,26 and proIL-1827–29 can be activated in the absence of caspase-1 by other proteases, particularly in neutrophils.
Interestingly, proIL-1β and proIL-18 are the only substrates of caspase-1 confirmed in vitro and in vivo under physiologic or pathologic conditions. This stands in contrast to more than 600 verified substrates of apoptotic executioner caspases. 30 Although several proteomics approaches led to the identification of novel caspase-1 cleavage products,31–33 the relevance of these cleavage events under physiologic conditions remains to be determined. One reason for the identification of novel substrates in these studies might be the very high, and most likely non-physiologic, concentrations of caspase-1 that have been used. It has been demonstrated recently that caspase-1 has a low substrate specificity at high concentrations, which is drastically increased by lowering its concentration. 30 Most importantly, the half-life of active caspase-1 is very low (about 50 times lower than the half-life of active caspase-3), suggesting that high concentrations, and therefore low specificity, will not be reached under physiologic conditions. 30 Recently, it has further been suggested that substrate binding triggers dimerization and activation of mature caspase-1, but not of caspase-3. This may reflect the fact that caspase-1 cleaves a much smaller number of proteins than caspase-3. 34
IL-1
Inflammation is an essential protective attempt of an organism to restore a homeostatic state after its disturbance by a harmful stimulus. Tissue damage, infection and other immunologic challenges rapidly induce an inflammatory response, which represents the activation of the innate immune system. The pro-inflammatory cytokine IL-1 (IL-1α and IL-1β) plays an important role in these fundamental and beneficial processes. 35 IL-1 induces the expression of adhesion molecules on mesenchymal and endothelial cells, which triggers infiltration of inflammatory and immune cells. 35 In addition, IL-1 causes fever, vasodilation, hypotension and enhances pain sensitivity. Expression and activity of IL-1 are tightly controlled at the transcriptional, as well as at the post-transcriptional, level. 36 Biological responses of IL-1 are mediated by signaling via IL-1 receptor type I (IL-1RI). 36 IL-1α and IL-1β are both expressed initially as pro-peptides with an amino terminal extension. ProIL-1β is not able to bind and activate IL-1RI, whereas proIL-1α possesses biological activity, which is further enhanced by limited proteolysis.35.37 Caspase-1 is described as the principal activator of proIL-1β16,17,35 and, therefore, a central regulator of the inflammatory response. In addition, caspase-1 can activate proIL-18. Interestingly, proIL-1α, proIL-1β and proIL-18 lack a signal peptide for protein secretion and are released from the cell by a poorly understood pathway termed unconventional protein secretion, which occurs independently of the classical ER/Golgi pathway.36,38 IL-1 activity is counteracted by expression of IL-1R antagonist (IL-1Ra), which possesses a signal peptide and is secreted constitutively via the classical pathway. 35 IL-1Ra binds to IL-1RI, thereby preventing binding of IL-1α and IL-1β, and suppressing an inflammatory response in vivo, as IL-1Ra cannot induce signal transduction. 35 Mice lacking IL-1α and IL-1β do not develop a spontaneous phenotype. However, mice lacking expression of IL-1Ra suffer from arthritis, arteritis and skin inflammation. In humans, deficiency in IL-1Ra (DIRA) causes a life-threatening auto-inflammation, affecting mainly the skin and the bones.39,40 This demonstrates that IL-1Ra is required to control the activity of IL-1 also in the human organism. DIRA is treated successfully with a recombinant form of IL-1Ra (Anakinra, Kineret), which was approved initially for the treatment of arthritis.35,41 Anakinra and other IL-1 blockers are effective in a range of important modern diseases, such as type 2 diabetes mellitus or gout, as well as other sterile inflammations now collectively called autoinflammatory diseases, which are all IL-1-mediated.35,41
The NLRP1 inflammasome
In 2002, a protein complex required for the activation of caspase-1 and subsequent processing of proIL-1β was identified and termed inflammasome. 42 This new complex consisted of caspases 1 and 5, the adapter protein apoptosis associated speck-like protein containing a CARD (Asc) and NACHT, leucine-rich repeats (LRR) and pyrin domains-containing protein 1 (NLRP1) or NALP1, the latter most likely acting as backbone protein and as sensor for the assembly. 43 Although, at that time, no ligand for NLRP1 was known, these results suggested that NLRP1 acts like a cytoplasmic Toll-like receptor (TLR) that induces inflammation through activation of caspase-1, proIL-1β and proIL-18 instead of NF-κB. TLRs are transmembrane receptors located at the cell surface or on endosomes, whose LRRs bind to conserved molecules from pathogens [pathogen-associated molecular patterns (PAMPs)], but also to endogenous molecules released by damaged cells [danger or damage-associated molecular patterns (DAMPs)]. This binding induces signal transduction, followed by activation of NF-κB and transcription of pro-inflammatory target genes. 44 NLRP1 belongs to the family of Nod-like receptors (NLRs), which constitutes 22 members in humans and 34 in mice. 43 It binds the CARD of procaspase-5 with its carboxyterminal CARD and the pyrin domain (PYD) of Asc with its amino terminal PYD. In turn, Asc links procaspase-1 to the complex via CARD–CARD interactions.42,43 Therefore, both caspases come into close proximity and are able to activate each other. In vitro, muramyl dipeptide (MDP), a peptidoglycan constituent of bacteria, and ribonucleoside triphosphates induce oligomerization of NLRP1 and caspase-1 followed by activation of the latter, which is enhanced by Asc. 45 Lethal toxin secreted by Bacillus anthracis activates the NLRP1 inflammasome in murine macrophages. 46 Signaling downstream of IL-1RI and pyroptosis (see later) have been suggested to contribute to caspase-1-dependent resistance against B. anthracis.47,48 However, the molecular mechanisms leading to NLRP1 inflammasome activation are not known as there is no evidence that either MDP or lethal factor from B. anthracis bind directly to NLRP1. 45 In human keratinocytes, NLRP1 expression is required for UVB-induced secretion of IL-1β. 49 An important role of NLRP1 in the skin is further suggested by the observation that single-nucleotide polymorphisms in the nlrp1 gene are associated with vitiligo, an autoimmune disorder involving destruction of melanin-producing melanocytes in the skin. 50
The NLRP3 inflammasome
The NLRP3 (also known as NALP3 or cryopyrin) inflammasome is the most important type of inflammasome complex as it is activated by many different PAMPs and DAMPs. 19 Several bacterial pathogens, such as Escherichia coli, Vibrio cholera and Staphylococcus aureus; fungal pathogens, such as Candida albicans and Aspergillus fumigatus; viruses, like influenza A and vesicular stomatitis virus; and parasites, such as Schistosoma mansoni, activate caspase-1 dependent on NLRP3 and Asc expression, which, in turn, induces secretion of active IL-1β and IL-18.51,52 Recently, it has been reported that expression and activity of caspase-4 are required for NLRP3 inflammasome activation in THP-1 cells and human keratinocytes. 53 NLRP3 can sense prokaryotic mRNA, present only in living bacteria, now defined as a special class of viability-associated PAMPs. 54 It has also been suggested that ER stress activates the NLRP3 inflammasome. 55 Most importantly, the NLRP3 complex is also activated by different types of crystals and particles in phagocytic cells.19,51 Examples are monosodium urate crystals, which are the causing agent of gouty inflammations, and islet amyloid polypeptides, which activate the NLRP3 inflammasome in patients with type 2 diabetes mellitus. IL-1 blockers are effective in the treatment of these patients, thus demonstrating a fundamental role of inflammasome-generated IL-1 activity in modern human diseases.19,35 Furthermore, amyloid β aggregates activate the NLRP3 inflammasome in microglia, asbestos and silica particles in alveolar macrophages, or cholesterol crystals in macrophages, which plays an important role in Alzheimer’s disease, pulmonary fibrotic disorders and atherosclerosis, respectively. 19 An involvement of NLRP3 in inflammatory diseases is further demonstrated by the fact that mutations of the nlrp3 gene are the cause of different auto-inflammatory periodic syndromes, which are now collectively called cryopyrin-associated periodic syndromes (CAPS). CAPS patients suffer from rashes, fever and fatigue; in severe cases there can be hearing loss, mental impairment or bone deformities. These patients are free of symptoms as long as Anakinra or other IL-1 blockers are administered.35,56
A heavily discussed question is how all these different PAMPs and DAMPs induce NLRP3 inflammasome activation at the molecular level. It is clear that, in contrast to keratinocytes, expression of proIL-1β and NLRP3 has to be induced in macrophages and dendritic cells prior to inflammasome activation. Therefore, these cells require an activation of NF-κB triggered by stimulation with TNF-α, IFN-γ or TLR agonists.49,57,58 As NLRP3 ligands are highly diverse in their chemical nature and size, a direct binding of DAMPs and PAMPs to NLRP3 is extremely unlikely and could not be demonstrated so far.
19
Therefore, more indirect pathways have been suggested, which integrate the different triggers of inflammasome assembly into ion fluxes, phagosomal destabilization and the generation of reactive oxygen species (ROS).19,51 For example, NLRP3-activating crystals are phagocytosed, which results – due to inefficient clearance – in phagosomal and lysosomal damage, and in cathepsin B-mediated NLRP3 inflammasome assembly.
58
However, particle-induced IL-1β secretion from murine cathepsin B-deficient cells is normal.
59
All NLRP3 activators induce the generation of ROS. Therefore, it has been suggested that ROS-induced binding of thioredoxin-interacting protein (TXNIP) to NLRP3 triggers inflammasome activation.
60
The source of ROS is most likely mitochondria.61–63 However, it is not known whether ROS are always necessary, and particularly if the generation of ROS is always sufficient for the activation of the NLRP3 inflammasome. Other proteins involved in the regulation of NLRP3 inflammasome activation are the protein kinase R (PKR) and guanylate binding protein 5 (GBP5). GBP5 is required for NLRP3 inflammasome responses to pathogenic bacteria and soluble (but not to crystalline) activators.
64
In contrast, ablation of PKR expression prevents not only activation of the NLRP3 inflammasome, but also of the NLRP1, NLRC4 and absent in melanoma 2 (AIM2) inflammasomes (Figure 1).
65
However, a recent report claims that PKR is dispensable for inflammasome activation in macrophages.
66
The different types of inflammasomes. The NLRs NLRP1, NLRC4, NLRP3 and NLRP6, and the HIN200 proteins AIM2 and IFI16 build up inflammasomes upon sensing different PAMPs or DAMPs. Whereas AIM2 and IFI16 assemble through binding of pathogen-derived dsDNA, more indirect mechanisms induce formation of the other types of inflammasomes. NLRC4 is activated by binding to NAIPs, NAIP5 and NAIP6 sense flagellin, and NAIP2 the T3SS rod protein. The NLRP3 inflammasome is activated by many different DAMPs and PAMPs. Here, generation of ROS, efflux of K+ or cathepsin B release upon lysosomal damage may trigger complex formation. The stimulus that results in assembly of the NLRP6 inflammasome remains to be identified. All inflammasomes recruit Asc and caspase-1, what induces activation of the protease, subsequent activation of proIL-1β and proIL-18, and, finally, secretion of the active cytokines.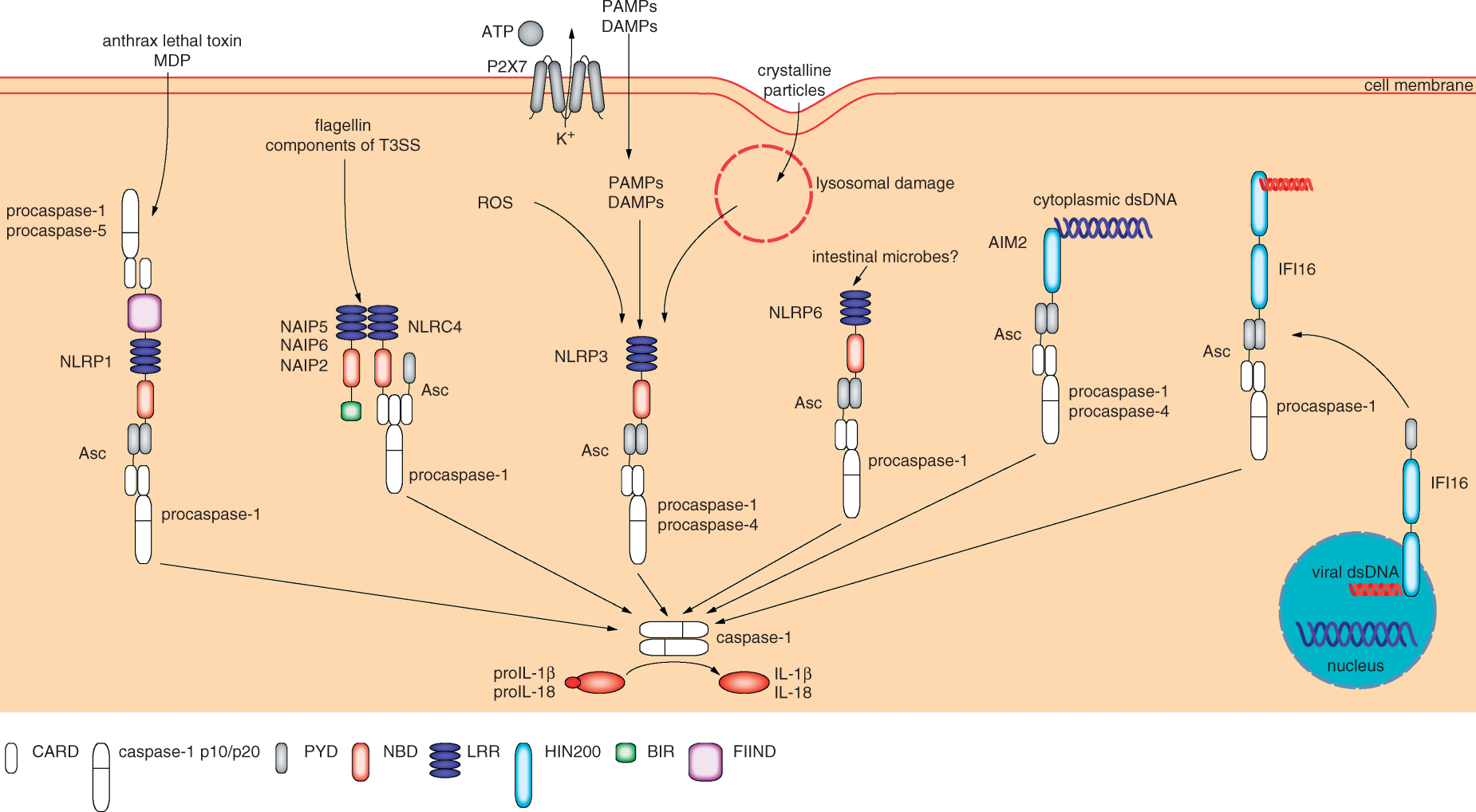
The NLRC4 inflammasome
NLRC4 (NLR family CARD domain-containing protein 4, also known as ICE-protease activating factor) is an NLR required exclusively for bacterial-induced caspase-1 activation and subsequent inflammation.19,67 Asc expression is needed for caspase-1-mediated and NLRC4-dependent activation of proIL-1β and proIL-18, but most likely not for caspase-1-dependent cell death (see later). 52 The NLRC4 inflammasome is activated by flagellin, the principal substituent of the bacterial flagellum, during infection of macrophages with Salmonella Typhimurium, Listeria monocytogenes and Legionella pneumophila, but also by the type III secretion system (T3SS) rod protein from a range of bacteria.67,68 Interestingly, these bacterial products are sensed in murine macrophages by NLR family, apoptosis inhibitory proteins (NAIPs), also known as baculoviral IAP repeat (BIR)-containing 1, which are then recruited to NLRC4, thereby defining the specificity of the NLRC4 inflammasome. 19 NAIP5 and NAIP6 are required for sensing of flagellin, NAIP2 for the T3SS rod protein.69,70 In contrast, there is only one human NAIP, which neither binds to flagellin nor to T3SS rod proteins, but to the T3SS needle subunit from several bacteria. 70 Recently, it has been shown that NLRC4 is phosphorylated – what is required for its ability to activate caspase-1 – thus representing a further level of complexity in NLRC4 inflammasome activation. 71
The AIM2 inflammasome
AIM2 (absent in melanoma 2) is a cytoplasmic member of the hematopoietic expression, interferon-inducible, nuclear localization and a 200 amino acid repeat (HIN200) family that consists of an amino terminal PYD and a carboxyterminal HIN200 motif. The latter can bind to double-stranded (ds) DNA. 72 AIM2 expression is induced by type I and II IFNs, which play an important role in defense against viral infections. AIM2 can bind to transfected or pathogen-derived cytoplasmic dsDNA and subsequently recruits Asc by PYD–PYD interaction, which, in turn, binds caspase-1. This results, most likely, in oligomerization of the complex, in activation of caspase-1 and in secretion of active IL-1β and IL-18. The AIM2 inflammasome is activated by Francisella tularensis, DNA viruses, such as cytomegalovirus and vaccinia virus, as well as by L. monocytogenes, although the latter requires expression of NLRP3 and NLRC4 for maximal activation of caspase-1.73–75 In contrast to AIM2, the HIN200 family member IFN-inducible (IFI) 16 is located mainly in the nucleus, where it is able to detect viral dsDNA and trigger a type I IFN response. 76 During primary and latent infection IFI16 binds to Kaposi’s sarcoma-associated herpes virus DNA. Then, it translocates to the cytoplasm, where it assembles an inflammasome complex with Asc and caspase-1.77,78 An interesting question is how IFI16 can discriminate between self and pathogen-derived DNA. 73
The NLRP6 inflammasome
Recently, it was shown that mice deficient in NLRP6 are more susceptible to dextran sodium sulfate (DSS)-induced colitis, a model for an inflammatory intestinal disease, and subsequent tumorigenesis. 79 The reason for this unexpected phenotype is most likely an aberrant microflora of the gastrointestinal tract.79,80 In addition, not only NLRP6 but also Asc, caspase-1 and proIL-18 expression are required for a ‘healthy’ microflora, suggesting that NLRP6 forms an inflammasome, which protects against colitis and colitis-associated tumorigenesis through IL-18. Interestingly, the phenotype was transferable to wild type mice, which were co-housed with NLRP6-, caspase-1- or Asc-deficient animals. 79 It has been demonstrated that IL-18, rather than IL-1, is the more important cytokine in DSS-induced colitis,81,82 although other publications based on a different mouse strain reported less susceptibility in caspase-1-deficient mice. 83 Apart from using different strains, these conflicting results may also be explained by different gastrointestinal microfloras in different animal facilities. Surprisingly, mice lacking NLRP6 expression are more resistant to infection with certain bacterial pathogens, most likely as NLRP6 expression reduces the levels of NF-κB- and mitogen-activated protein kinase (MAPK)-dependent cytokines and chemokines. 84
Pyroptosis
Overexpression of caspase-1 has been suggested to induce apoptosis. 15 Caspase-1 has also been implicated in neuronal and endothelial programmed cell death.20,21,22 More than 20 years ago, it was shown that macrophages infected with Shigella flexneri or S. Typhimurium undergo apoptosis. 85 The classification of apoptotic cell death was based on morphologic features, DNA fragmentation, chromatin condensation and on the requirement for caspase-1. 3 After generation of caspase-1-deficient mice it turned out that caspase-1 expression is dispensable for cell death induced by known apoptotic agents and for apoptosis during embryonic development.16,17 A more careful characterization of pathogen-induced cell death of macrophages revealed clear differences to apoptosis.3,18 This type of cell death, now termed pyroptosis, is considered to be dependent only on caspase-1, but not on apoptotic caspases. In addition, caspase-1 does not cleave, and thereby activate, caspase-3 during pyroptosis. Most importantly, in contrast to apoptosis, pyroptotic cell death is characterized by an increase in cell size due to osmotic swelling and the subsequent rupture of the cytoplasmic membrane, whereas the mitochondrial membrane remains intact. This results in the release of pro-inflammatory molecules, such as ATP, or proteins, such as proIL-1α and HMGB1, which are able to recruit neutrophils and induce inflammation. In contrast, apoptosis is considered immunologically silent as apoptotic cells attract macrophages to find and phagocytose them without mounting an inflammatory response. Experimentally, apoptotic cell death can be discriminated from pyroptosis by detection of caspase-3 processing, whereas both types of cell death are characterized by DNA fragmentation. 3 Interestingly, caspase-1 does not require processing in order to become active for execution of pyroptosis, in contrast to activity towards proIL-1β and proIL-18, where processing of the protease has to take place. 86 In addition, pyroptosis, if triggered by NLRC4, does not require expression of Asc, 86 whereas Asc is needed for NLRP3 and AIM2 inflammasome-driven pyroptosis.3,86 Reports about pyroptosis in vivo are rare, but an elegant study in mice demonstrated that S. Typhimurium induced NLRC4-dependent pyroptosis in macrophages leads to the subsequent uptake of the released bacteria by neutrophils, which is required for clearance of the infection. 87 Murine neutrophils do not undergo S. Typhimurium-induced pyroptosis as they do not express NLRC4. 87 It is still unclear whether pyroptosis only occurs in macrophages and dendritic cells, or also in other cell types, and whether all inflammasomes can trigger pyroptosis.3,18,51 Interestingly, it has been claimed that pyroptosis depends only on the catalytic activity of caspase-1, but not on caspase-11. 87 However, recently, it turned out that the caspase-1-deficient mice used so far also lack expression of caspase-11 and that caspase-11, rather than caspase-1, confers susceptibility to sepsis and cell death.73,88
Glycolysis and caspase-7
In order to determine the molecular mechanisms of pyroptosis downstream of activation of caspase-1, screens led to the identification of novel caspase-1 substrates.31–33 In contrast to caspase-3, recombinant caspase-1 cleaves several enzymes required for glycolysis, such as aldolase, GAPDH, triose-phosphate isomerase and α-enolase. 33 Glycolysis is essential for the synthesis of ATP, which is required for macrophage survival and activation. Although in vitro the caspase-1 concentration needed for cleavage of GAPDH is higher by a factor of 50 than for cleavage of proIL-1β, this cleavage seems to occur at a consensus site and to inactivate GAPDH. Most importantly, processing of GAPDH is also detected in Salmonella-infected macrophages and in the diaphragm muscle of mice undergoing septic shock, suggesting a novel function of caspase-1 during pyroptosis and septic shock. 33 However, whether inactivation of glycolysis contributes to the protection against pathogens or plays an important role in sepsis is not known.
Caspase-7 is an effector in apoptosis with a similar function as caspase-3. Recently, it has been shown that caspase-7, but not caspase-3, is activated by caspase-1 in vitro and in vivo. 31 This cleavage requires expression of NLRP3, NLRC4, Asc and caspase-1, when macrophages are stimulated with NLRP3 or NLRC4 activators, respectively. 31 Interestingly, restriction of L. pneumophila replication is reduced in caspase-7-deficient macrophages in comparison to wild type cells. Certainly, this is most likely due to a function of caspase-7 in the fusion of bacteria-containing phagosomes with lysosomes, rather than in pyroptosis.31,89 Murine caspase-11 also promotes this fusion event through the modulation of actin polymerization. 90 As S. Typhimurium-induced pyroptosis proceeds normally in cells lacking expression of both caspases 3 and 7, a possible function of caspase-7 in pyroptosis remains to be demonstrated. 91 The induction of expression of NF-κB target genes by inflammasome-induced caspase-7 activation points into a different direction. 92 Interestingly, caspase-1 mediated activation of caspase-7 results in translocation of the latter to the nucleus, where it cleaves PARP1. This induces PARP1 release from chromatin, chromatin decondensation and, eventually, expression of NF-κB target genes. 92
Caspase-1 activates lipid metabolic pathways
Although caspase-1 supports cell death of immune cells infected by certain pathogens (see earlier), it can also support survival and confer resistance to pathogenic bacteria.93,94 Some bacteria express pore-forming toxins as virulence factors, which create holes of various sizes in the plasma membrane of the host cell. Aerolysin secreted by Aeromonas species heptamerizes into a ring, which results in the exposure of hydrophobic surfaces allowing the insertion into the plasma membrane.
93
These pores lead to the efflux of intracellular potassium, which triggers the NLRP3-, NLRC4- and Asc-dependent activation of caspase-1. Interestingly, active caspase-1 is required for activation of sterol regulatory element binding proteins (SREBPs), transcription factors that function predominantly in cholesterol and fatty acid biogenesis. Inactive SREBPs are located in the ER membrane. Upon cholesterol depletion, which induces their activation, SREBPs move to the Golgi apparatus, where they are processed and liberated by two Golgi proteases. Eventually, SREBPs translocate to the nucleus, where they induce the expression of target genes and switch on lipid metabolic pathways (Figure 2).93,94 Aerolysin-induced SREBP activation is blocked by inhibition of caspase-1 activity and by knockdown of expression of caspase-1, NLRP3, NLRC4 and Asc. Most importantly, this increases aerolysin-induced cell death, which most likely does not represent apoptosis, demonstrating an unexpected role of caspase-1 in survival. Most likely, this is due to repair of the damaged plasma membrane through the SREBP-dependent synthesis of lipids. However, the molecular mechanism of how caspase-1 activates SREBPs, which most likely occurs indirectly, is completely unknown. The fact that caspase-1 activity is needed suggests the existence of an unknown substrate of the protease, which, in turn, activates SREBPs. In addition, the role of caspase-1 in survival has been demonstrated only in Chinese hamster ovary and HeLa cells,
93
whereas pyroptosis occurs in macrophages and dendritic cells (see earlier). It would be interesting to know whether caspase-1 is also able to increase survival in macrophages, which are the primary cells involved in defense against pathogens.
94
Pathways targeted by caspase-1. Upon activation, caspase-1 cleaves proIL-1β and proIL-18, and the active cytokines induce inflammation after secretion. Activity of the protease is also needed for unconventional secretion of many other leaderless proteins supporting inflammation, repair and, most likely, survival. In addition, pore-forming toxins induce the activation of SREBPs by active caspase-1, which are required for the repair of the plasma membrane and subsequent survival. Pyroptosis represents a caspase-1 effector mechanism causing a lytic form of cell death with release of pro-inflammatory molecules. Caspase-7 is a substrate of caspase-1 that cleaves PARP, resulting in transcription of NF-κB target genes, which may foster inflammation and survival. Caspase-1 triggers NF-κB activation by two additional mechanisms, requiring either cleavage of MAL by active caspase-1 or RIP2 activation by CARD–CARD interaction.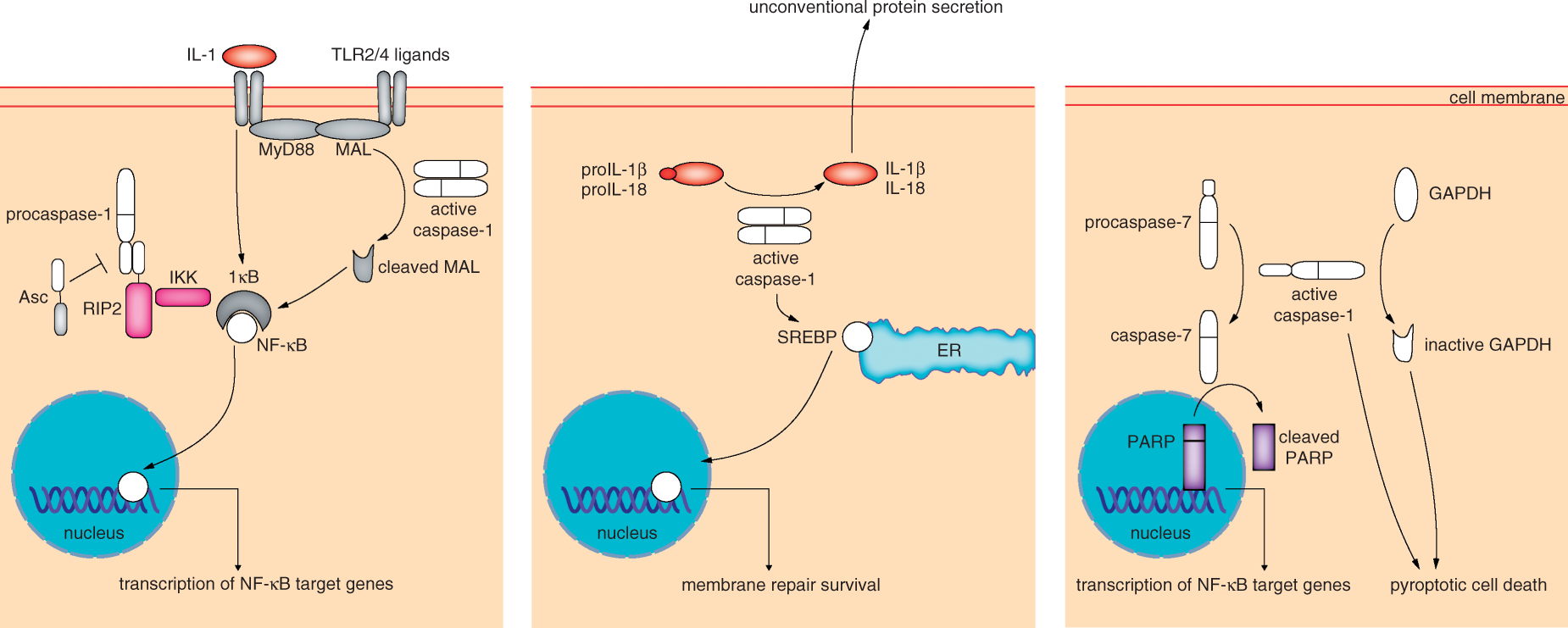
Unconventional protein secretion
Proteins, such as IL-1α, IL-1β, IL-18, or fibroblast growth factor (FGF)-2 clearly fulfill extracellular functions, but lack a signal peptide, which would allow their ER/Golgi-dependent secretion from the cell. 38 Nevertheless, these proteins get secreted through the plasma membrane. However, they use a special and only poorly understood pathway termed unconventional protein secretion. The difficulty of the analysis of this pathway is to discriminate between regulated (unconventional) secretion and passive release owing to cell lysis. This is particularly true because unconventional secretion can be induced by stressors, which have also cytotoxic effects resulting in cell death and lysis. Interestingly, macrophages deficient for caspase-1 are not only defective in IL-1β and IL-18 secretion, but, surprisingly, also in the release of proIL-1α.16,17,95 ProIL-1α secretion requires expression of caspase-1, but also caspase-1 activity, suggesting the existence of an unknown substrate involved in the regulation of unconventional secretion of at least proIL-1α. 96 Consequently, secretion of proIL-1α also requires expression of other inflammasome proteins, which are needed for activation of caspase-1. UVB irradiation activates the NLRP3 and NLRP1 inflammasome in human primary keratinocytes, which triggers secretion of IL-1 and IL-18 and unconventional secretion of inflammasome proteins – all lacking a signal peptide. 49 The advantage of UVB-irradiated keratinocytes over activated macrophages and dendritic cells is the robustness of the former, which allows distinguishing between unconventional secretion and passive release. 49 Interestingly, secretome analysis of UVB-irradiated keratinocytes revealed caspase-1-dependent secretion of several proteins, whose unconventional secretion has already been described, as well as secretion of several other leaderless proteins. 96 These proteins are involved in repair processes, ROS detoxification and apoptosis. Therefore, caspase-1 activity directly links IL-1- and IL-18-dependent inflammation with these processes. It is tempting to speculate that the secretion of pro-apoptotic proteins, such as Bid, enhances the survival of the secreting cells, which has also been suggested for secretion of caspase-1 itself (unpublished results).7,43,96 Unconventional protein secretion is an at least partially evolutionarily conserved process, which is mediated in Dictyostelium by autophagosomes. 97 Recently, it has been demonstrated that unconventional secretion of IL-1β and other proteins by mammalian cells is supported by autophagy. 98 However, these findings are in contrast to reports that suggest that autophagy suppresses inflammasome activation and, therefore, secretion of IL-1β.62,63
Caspase-1 and NF-κB
Caspase-1 has been implicated in the regulation of NF-κB. Transcription factors of this class are involved in inflammation, regulation of immune responses, differentiation and apoptosis. 99 In unstimulated cells, NF-κB is retained in the cytoplasm through binding to the IκB inhibitor. Upon activation, IκB gets phosphorylated by the IκB kinase (IKK) complex, which results in polyubiquitination and proteasomal degradation of IκB. The liberated NF-κB translocates to the nucleus and induces expression of target genes, which are inflammation-related or anti-apoptotic. Pathogen-derived products, which are agonists of TLRs, as well as TNF-α and IL-1, which stimulate their respective receptors, lead to activation of NF-κB through distinct signaling pathways. 100 Therefore, a pathway, by which caspase-1 mediates NF-κB activation, is triggered by inflammasome-activated, caspase-1-dependent IL-1 secretion. 35 However, macrophages isolated from mice lacking expression of caspase-1 have impaired NF-κB activation when stimulated with the TLR4 agonist LPS, which does not result in activation of caspase-1.101,102 NF-κB activation is rescued by overexpression of an enzymatically inactive variant of caspase-1, which demonstrates that caspase-1 enhances NF-κB activation independently of the active site cysteine. 101 In addition, overexpression of inactive caspase-1 enhances NF-κB activity in 293 cells. 100 This effect is CARD-dependent through interaction with the CARD-containing kinase RIP2, resulting in NF-κB activation via IKK recruitment. 100 The inhibitor of caspase-1 CARD only protein (COP) also interacts with RIP2 and thereby mediates NF-κB activation. In contrast, the CARD-containing proteins ICEBERG, and caspases 11 and 12 do not activate the transcription factor, suggesting that caspase-1-dependent activation of NF-κB is a specific event. 100 Asc, which is required for activation of caspase-1 in inflammasome complexes, has an inhibitory effect on NF-κB.101,102 Although the interaction of the CARD of caspase-1 with Asc and RIP2 is mediated by different amino acids, 103 caspase-1 interacts either with Asc or with RIP2, suggesting that Asc may direct caspase-1 away from RIP2, thereby inhibiting direct NF-κB activation, while inflammasome-mediated IL-1 production is induced. 101 This pathway might explain the recent report of patients carrying mutations of caspase-1. 104 Although all these mutations drastically decrease the enzymatic activity of caspase-1, reflected by reduced processing of the enzyme itself and by diminished secretion of mature IL-1β, the symptoms of the carriers are similar to those suffering from IL-1-mediated auto-inflammatory diseases. 104 It is tempting to speculate that a possibly more pronounced activation of NF-κB by these caspase-1 variants underlies the inflammatory phenotype, although additional mutations in other genes cannot be excluded.
In addition, caspase-1 activity can trigger TLR2- and TLR4-mediated NF-κB activation. 105 TLR2 and TLR4 require MyD88 and MyD88 adaptor-like (MAL) for signal transduction. Interestingly, caspase-1 can physically interact with and cleave MAL, which is required for subsequent activation of NF-κB and MAPK. This has been demonstrated by inhibition of caspase-1 activity or by overexpression of a MAL variant lacking the caspase-1 cleavage site, which blocks TLR2- and TLR4-dependent NF-κB activation. 105
Conclusions and perspectives
Inflammation is an essential pathway in host defense. During the last 10 years different inflammasome complexes have been identified, which all activate the protease caspase-1 and thereby the pro-inflammatory cytokines proIL-1β and proIL-18. In addition, many different DAMPs and PAMPs have been described that activate inflammasomes. Inflammasomes are not only involved in acute danger- or pathogen-associated inflammation but also in the development of inflammation-driven diseases such as cancer, atherosclerosis and type 2 diabetes mellitus. 19 In addition, pyroptosis has been identified as a novel form of programmed cell death, which is caspase-1-dependent and supports IL-1- or IL-18-driven inflammation. 18 This is in sharp contrast to apoptosis that does not activate the immune system. Important unsolved questions are how inflammasomes and, particularly, the NLRP3 inflammasome are activated at the molecular level. In addition, as not a single substrate of caspase-1 in pyroptosis has been identified, why is the enzyme's activity needed for this form of cell death? What is the relationship between caspase-1 activation, activity and specificity? Recently, a NLRP7 inflammasome has been identified, which activates caspase-1 upon sensing of microbial lipopeptides. 106 How many more types do exist and how is the assembly of these inflammasome complexes triggered? Is there a crosstalk between different inflammasome complexes? How are caspase-1 activation and activity regulated by post-translational modifications, such as (de)ubiquitination and glutathionylation?107–109 Whereas most reports deal with IL-1β-, IL-18- and pyroptosis-mediated effects of caspase-1, the protease is able to trigger additional pathways. Caspase-1 is capable of supporting the survival of cells through triggering lipid metabolic pathways and most likely repair of the damaged plasma membrane. 93 In addition, activity of the protease is required for unconventional secretion of proteins involved in inflammation, repair, cytoprotection and apoptosis, and it is tempting to speculate that also this pathway supports survival. 96 However, the substrates, which mediate these effects, are not known and it will be a future task to identify them. Caspase-1 inactivates enzymes involved in glycolysis and activates the executioner caspase-7. However, the physiological relevance of these events remains to be demonstrated.31,33 Caspase-1 has also been reported to activate the transcription factor NF-κB independently of its enzymatic activity,100,101 which may result in inflammation in vivo. 104 Clearly, more efforts are required to understand these effects – particularly the generation of novel knock-in mouse models expressing enzymatic inactive caspase-1. A major problem is the fact that a lot of our knowledge about caspase-1 relies on experiments with mice, which lack not only expression of caspase-1 but also of caspase-11. 88 As mice deficient only for caspase-1 are now available, the real contribution of caspase-1 to inflammation can be addressed. It has, however, to be taken into account that in these mice caspase-11 has been reintroduced. More advanced mouse genetics are also needed to address the contribution of caspase-1 in single cell types in vivo in more sophisticated inflammation models and to discriminate between, for example, IL-1-, IL-18- or pyroptosis-mediated effects. Owing to obvious differences between mice and men, particularly at the immunologic level, knock down studies with human (primary) cells are very important for the analysis of the function of caspase-1 in humans. These efforts should result in novel and specific therapies for the numerous and different diseases caspase-1 is involved in.
Footnotes
Funding
Our research is supported by the Swiss National Science Foundation (31003A_132450), the European Science Foundation (31EM30-126141), the Swiss Cancer League (02741), the Center for Clinical Research, University Hospital and University of Zürich, the OPO- and the Hartmann Müller-Stiftung.
Acknowledgements
We thank Professor French and his laboratory members for fruitful discussions. G.E.S., M.G. and J.S. are members of the Zürich graduate program in Molecular Life Sciences.