
Abstract
Proteinase-activated receptor-2 (PAR-2) was shown to influence immune regulation; however, its role in human macrophage subset development and function has not been addressed. Here, PAR-2 expression and activation was investigated on granulocyte macrophage (GM)-CSF(M1) and macrophage (M)-CSF(M2) macrophages. In both macrophages, the PAR-2-activating peptide, SLIGKV, increased PAR-2 expression and regulated TNF-α and IL-10 secretion in a manner similar to LPS. In addition, HLA-DR on M1 cells also increased. Monocytes matured to an M1 phenotype in the presence of SLIGKV had reduced cell area, and released less TNF-α after LPS challenge compared with vehicle (P < 0.05, n = 3). Cells matured to an M2 phenotype with SLIGKV also had a reduced cell area and made significantly more TNF-α after LPS exposure compared to vehicle (P < 0.05, n = 3) with reduced IL-10 secretion (P < 0.05, n = 3). Thus, PAR-2 activation on macrophage subsets regulates HLA-DR and PAR-2 surface expression, and drives cytokine production. In contrast, PAR-2 activation during M1 or M2 maturation induces altered cell morphology and skewing of phenotype, as evidenced by cytokine secretion. These data suggest a complex role for PAR-2 in macrophage biology and may have implications for macrophage-driven disease in which proteinase-rich environments can influence the immune process directly.
Introduction
Proteinase-activated receptor-2 (PAR-2) is a G protein-coupled receptor that possesses its own cryptic ligand within the receptor N terminus. The activating ligand is unveiled by proteolytic cleavage, resulting in receptor activation. 1 Among the PAR family (PARs 1–4), PAR-2 is the only member insensitive to thrombin and is, instead, activated by a number of serine proteinases. Many of the identified PAR-2 activators are proteinases associated with inflammation and immune cell activity, including endogenous proteinases secreted by activated immune cells, for example mast cell tryptase, trypsin and neutrophil proteinase 3. 2 Recently-described membrane-associated proteinases, such as matriptase, have also been identified as PAR-2 activators. 3 PAR-2 can also be activated directly by a number of exogenous pathogen-derived proteinases, such as gingipains produced by Porphyromonas gingivalis 4 and Der p 3/9 5 proteinases from dust mite pathogens. This suggests PAR-2 may have a direct role in the regulation of innate immune function. Critically, PAR-2 represents a mechanism whereby proteinases can directly orchestrate and influence cellular responses, and alter gene expression within evolving innate immune responses.
Proteinases, including those identified as PAR-2 activators, are found in abundance at sites of inflammation and tissue damage. In particular, the synovial microenvironment in rheumatoid arthritis (RA) contains an abundance of inflammatory cells that secrete PAR-2 activators upon cellular activation and degranulation, including mast cell tryptase 6 and neutrophil-derived proteinase 3. 7 In addition, PAR-2 is expressed in the RA joint, co-localizing with macrophages, fibroblasts and mast cells.8,9 We demonstrated previously a pivotal role for PAR-2 in rodent models of arthritis, suggesting that such expression has functional significance.10–12 Further, we demonstrated the therapeutic potential of PAR-2 inhibition in ex vivo RA primary synovial tissue culture, in which we observed reduced pro-inflammatory cytokine release in the presence of the specific antagonist, ENMD-1068. 8
Macrophages contribute substantially to the cytokines manifested during discrete phases of the inflammatory cascade and play a critical role in development of adaptive immune responses.13,14 Two major subsets of macrophages are postulated, namely the classically activated or inflammatory M1 macrophage, associated with high levels of IL-12 and low IL-10, and the alternatively-activated macrophage (M2 type), associated with a high production of IL-10. 15 It is proposed that human monocytes can be polarised in vitro towards a predominantly M1 or M2 phenotype with the addition of granulocyte macrophage (GM)-CSF or macrophage (M)-CSF respectively. 16 PAR-2 mRNA has been identified previously in human monocytes 17 and PAR-2 protein has been detected at low levels on the cell surface. Monocytes also contain intracellular pools of pre-formed PAR-2 that can be trafficked readily to the cell surface when required. 18 Activation of PAR-2 results in enhanced secretion of pro-inflammatory cytokines and chemokines, including IL-1β, IL-6 and IL-8. 18 Many of these, in turn, have been implicated in the pathology of chronic inflammatory diseases, such as RA. 19 In addition, we have shown recently that RA patients having a disease flare have increased levels of PAR-2 expression on their PBMC than individuals with stable disease, 20 supporting an inflammatory role for this molecule in the disease.
Functional PAR-2 is required for in vitro development of murine bone marrow-derived dendritic cells (DC). DC generated from PAR-2−/− mice have reduced expression of co-stimulatory molecules, CD80 and CD86, than PAR-2 wild type (WT) mice. 21 A similar effect was observed in PAR-2 WT-derived DC treated with soybean proteinase inhibitors. These findings suggest PAR-2 may play a role in the differentiation and maturation of myeloid cells, including monocytes and macrophages. However, detailed analysis of the expression and functional implications of PAR-2 manipulation upon human macrophage subset maturation has not been performed. We hypothesised that PAR-2 manipulation would regulate macrophage differentiation and maturation pathways, and could thereby alter macrophage morphology and phenotype, as well as influencing the effector function of the cell.
Materials and methods
Human PBMC and CD14+ cell isolation
Human buffy coats were obtained from healthy blood donors with ethical approval (Scottish
National Blood Transfusion Service, Glasgow, UK). PBMCs were isolated using density gradient
centrifugation (Histopaque 1077; Sigma-Aldrich Ltd, Dorset, UK). CD14+ cells were
positively selected using MACS beads, magnet and filters (Miltenyibiotec, Surrey, UK) as per the
manufacturer's instructions, with purities routinely found to be 90–95%. Isolated cells were
cultured at 1 × 106/ml (2.5 ml/well in 6-well plates; Corning, Sigma Aldrich) in 10%
FCS/complete RPMI (supplemented with penicillin, streptomysin and
FACS staining of PBMC and monocyte-derived macrophages
Following density centrifugation, an aliquot of PBMC was removed for FACS analysis before application of CD14 isolation beads. Cells were surface stained for CD14 phycoerythrin (PE), CD3 FITC, CD19 APC (all from ebioscience, Hatfield, UK), and PAR-2 FITC or Alexa Fluor 647 (Santa Cruz Biotechnology, Heidelberg, Germany). Appropriate isotype controls were also included. An aliquot of cells was stained for cell surface markers followed by intracellular staining for PAR-2 using the BD Cytofix/Cytoperm™Plus Fixation/Permeabilization kit with BD GolgiPlug™ (BD Bioscience, Abingdon, UK) as per the manufacturer's instructions. Matured macrophages were also stained for PAR-2, CD163 PE (ebioscience) and CD68 PE (Santa Cruz Biotechnology, Hiedelberg, Germany) expression. Following SLIGKV (SLIGKV-NH2; Polypeptide Group, Strasbourg, France) stimulation, matured macrophages were stained for surface expression of TLR2 (CD282) FITC, TLR4 (CD284) PE and HLA-DR FITC (all from ebioscience). At least 10,000 events were collected using a FACSCalibur (BD Bioscience, Oxford, UK) and data were analysed using FlowJo 7.6.1 (Tree Star, Ashland, OR, USA).
Immunohistochemistry and cell area determination
CD14+ cells were isolated as described above and re-suspended at 1 × 106/ml in complete RPMI. Cells were plated into 4-well chamber slides (Fischer UK, Loughborough, UK) in the presence of M-CSF or GM-CSF growth factors alone or growth factor plus SLIGKV for 6 d. Cells were fixed in methanol for 20 min and blocked in 3% hydrogen peroxide (Sigma Aldrich) for 30 min, followed by dehydration through graded alcohols. The cells were then stained using the PAR-2 Ab, SAM-11 or isotype control (Santa Cruz Biotechnology) for 1 h at room temperature in a humidified container and then counterstained with haemotoxylin (Sigma Aldrich) before rehydration through graded alcohols and mounted for microscopy. Once mounted, cell area (µm2) was determined using Zeiss Axiovision LE software (4.8). Fifty cells were measured for each condition (M-CSF + vehicle, M-CSF + SLIGKV, and similarly for GM-CSF) in three donors.
SLIGKV stimulations of human monocyte-derived macrophages
PAR-2 agonist SLIGKV was prepared in complete RPMI as described above. Following culture for 6 d, monocyte-derived macrophages were stimulated with 400 µM SLIGKV or media alone for 24 h. Cells were then removed for FACS analysis of TLR2, TLR4, HLA-DR and PAR-2 surface expression.
Cytokine analysis from GM-CSF and M-CSF matured macrophages
CD14+ cells were differentiated in GM-CSF (M1) or M-CSF (M2) for 6 d. Differentiated cells were then stimulated with LPS (100 ng/ml; Sigma, UK) or SLIGKV (400 µM) for 48 h and culture supernatants collected for cytokine analysis. In a subset of experiments, cells were differentiated in the presence or absence of SLIGKV (400 µM) or an equivalent concentration of a control reverse peptide (RP; TOCRIS Bioscience, Bristol, UK) for 6 d prior to stimulating with LPS (100 ng/ml; Sigma) for 48 h, after which culture supernatants were collected for cytokine analysis.
TNF-α and IL-10 levels were measured in culture supernatants by ELISA (Life Technologies) as per the manufacturer's instructions.
Statistical analysis
Statistics were analysed by Student's t-test or by one- or two-way ANOVA with post hoc Student–Newman–Keuls test or Bonferroni correction. Log10 transformation was performed on non-normally distributed data where necessary to permit parametric analysis. Data are presented as mean ± SEM.
Results
PAR-2 expression in human PBMC
Cells freshly isolated from human buffy coats were analysed for surface membrane and
intracellular PAR-2 expression. Whereas most CD14+, CD3+ and CD19+
cells exhibited only low levels of surface expression (13.8 ± 7.5%, 3.3 ± 2.0% and 4.6 ± 2.8%,
n = 5), commensurate with previous reports,17,18 substantial intracellular PAR-2 expression was observed in a majority of cell
subsets (93.1 ± 2.0%, 84.1 ± 5.3% and 89.5 ± 3.8%, respectively, P < 0.001,
n = 5). No significant difference in PAR-2 expression was observed between
populations of cells (Figure 1A). Characterisation of PBMC PAR-2 and cell surface marker expression in GM-CSF- and M-CSF-matured
macrophage subsets. (A) PAR-2 surface (white bars) and intracellular (black bars) expression was
assessed in CD14+, CD3+ and CD19+ populations in fresh peripheral
blood. CD163 (B) and HLA-DR (C) expression were both significantly higher in M-CSF-matured cells
than in GM-CSF-matured cells. (D) Both GM-CSF- and M-CSF-matured macrophages expressed surface
(white bars) and intracellular (black bars) PAR-2 at d 6, n = 8 donors.
***P < 0.01; *P < 0.05.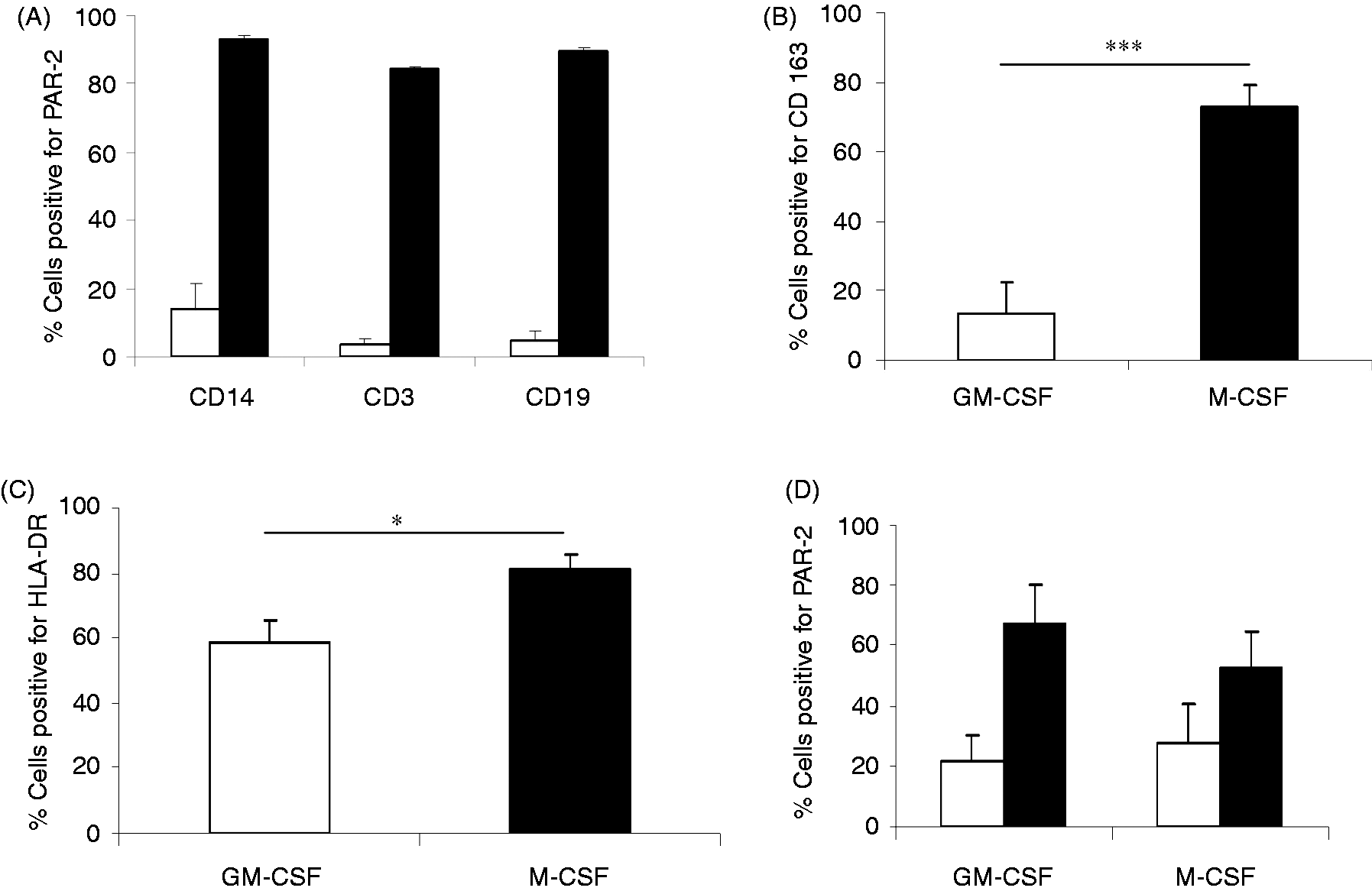
Characterisation of macrophage subsets
After 6 d, the phenotype of M1- and M2-differentiated monocytes was evaluated by FACS analysis and signature cytokine profiles. Cells polarised to an M2 phenotype had significantly higher expression of CD163 than M1 cells, in which expression was minimal (Figure 1B), indicating an M2 phenotype.16,22 HLA-DR expression was also significantly elevated in the M2 matured cells than in M1 cells (Figure 1C). PAR-2 surface expression and intracellular expression was maintained during macrophage differentiation in both M1 and M2 macrophage subsets (Figure 1D). Both M1 and M2 cells expressed CD68, with no significant difference found between the two populations (58 ± 8.4% vs 69.8 ± 5.2%, respectively, n = 11). TLR4 expression was consistent in M1 and M2 cell (58.6 ± 9.7% vs 57.4 ± 12.1%, respectively, n = 8), and while there was a trend toward enhanced TLR2 expression in M1 cells compared with M2 cells (51 ± 11% vs. 41 ± 11, respectively, n = 8), this did not reach significance.
After exposure to LPS, M1 macrophages made significantly more TNF-α and reduced IL-10 compared to
M2 macrophages (Figure 2A, B). These observations suggested the functional
integrity of M1 and M2 phenotypes generated in our cell systems. In addition, this cytokine
signature profile was similarly driven by the PAR-2-activating peptide, SLIGKV, in macrophage
subsets (Figure 2C, D). These data demonstrate direct regulation of M1 or M2 effector
function by a PAR-2 activator that can operate in a similar manner to that mediated via TLR4 agonist
activation. Cytokine signature profiles in GM-CSF- and M-CSF-matured macrophages. (A) GM-CSF-matured
macrophages generated significantly higher levels of TNF-α than M-CSF-matured cells, but
significantly lower levels of IL-10 (B) when stimulated with LPS (100 ng/ml), n = 3
donors. A similar cytokine pattern for TNF-α (C) and IL-10 (D) was observed after activation with
SLIGKV (400 µM), with an example donor from two separate experiments shown.
***P < 0.01; **P < 0.02.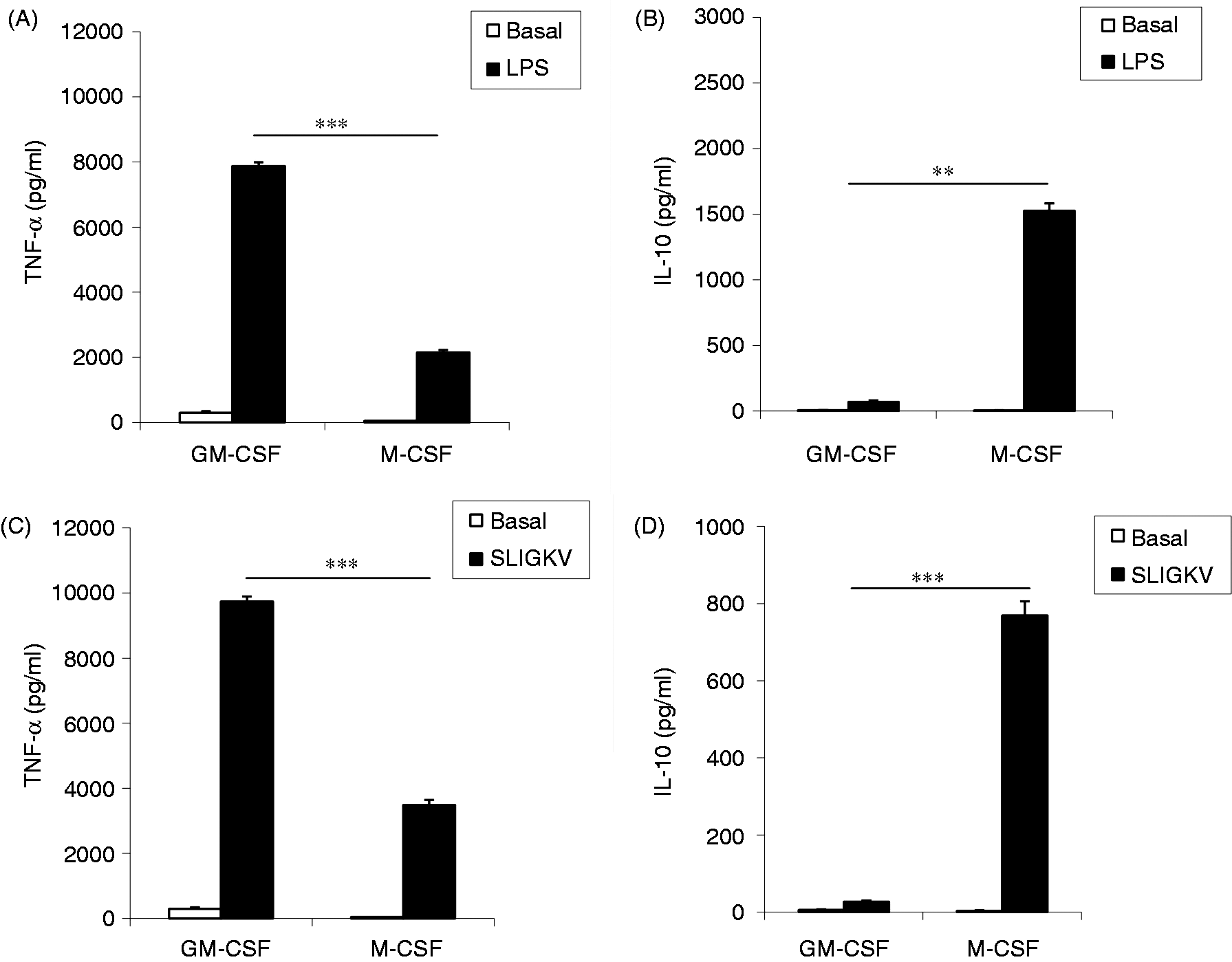
PAR-2 regulation of cell surface markers on M1 macrophages
Exposure of M1 cells to the PAR-2 activator, SLIGKV, for 24 h further enhanced expression of
PAR-2 and HLA-DR (Figure 3A–C). We also measured marginal increased expression
of both TLR4 (59 ± 10% vs 68 ± 7%, M1 vs M1 + SLIGKV, respectively, n = 8) and TLR2
(51 ± 11% vs 58 ± 9%, M1 vs M1 + SLIGKV, respectively, n = 8), suggesting that the
expression of these innate receptors was at least maintained. Regulation of cell surface markers on GM-CSF-matured macrophages by SLIGKV. (A) Histogram plot
from a single donor showing an increase in PAR-2 surface expression after activation of a
GM-CSF-matured macrophage with SLIGKV (400 µM) compared with vehicle (VEH). Isotype staining is also
shown. (B) Summary data for PAR-2 surface expression and (C) HLA-DR expression,
n = 8 donors reported. ***P < 0.01;
*P < 0.05.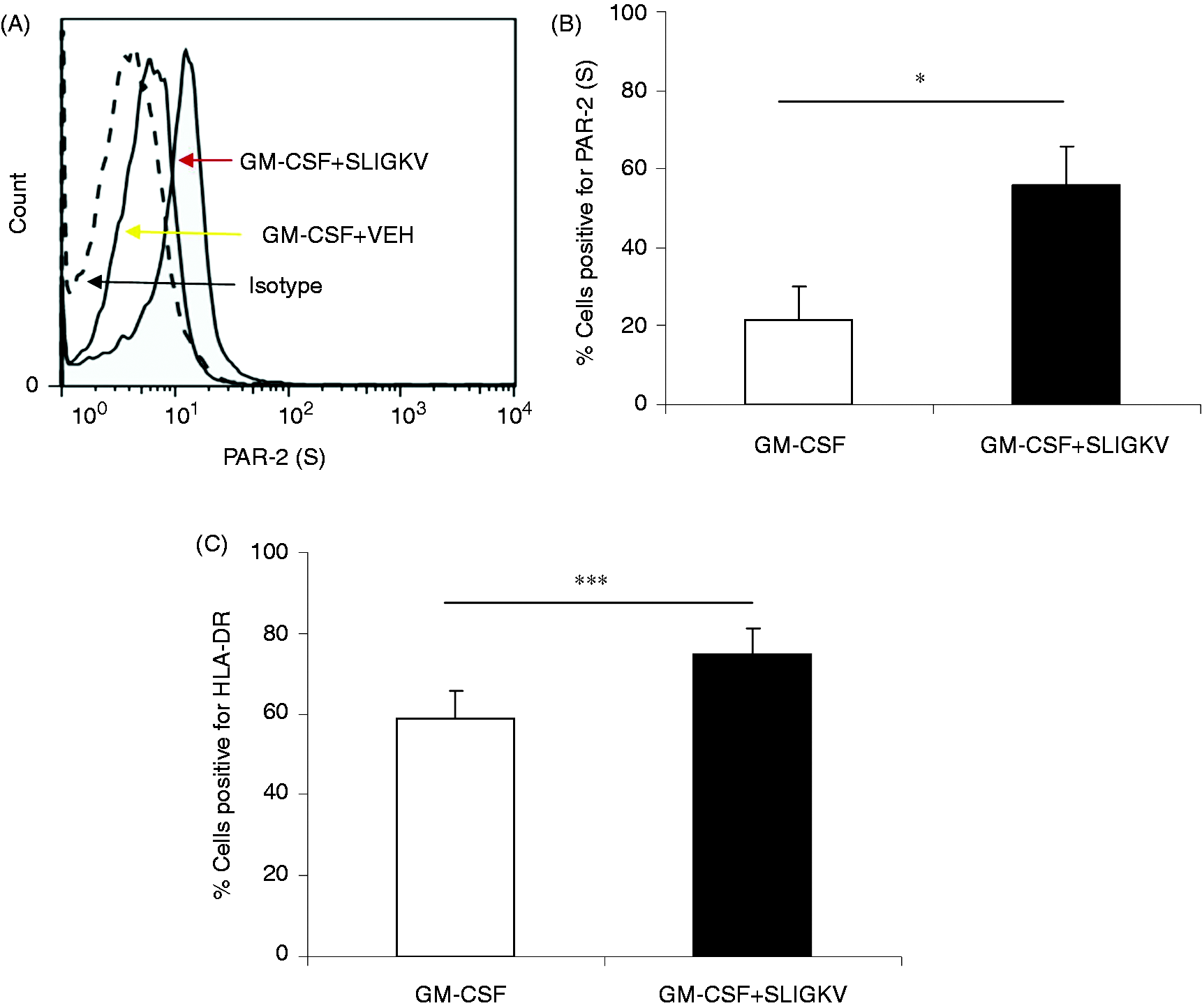
Influence of PAR-2 activation on M1 macrophage differentiation and effector function
Cells polarised to an M1 phenotype in the presence of GM-CSF plus SLIGKV for 6 d stained
abundantly for PAR-2 (Figure 4A). In the
presence of SLIGKV, cells were found to have an altered cellular morphology, appearing spindle-like
and elongated (Figure 4A). This was further
quantified by measurement of cell area. Statistically significant differences in cell area
(P < 0.0001, Bonferroni t-test) were found between vehicle (M1
vehicle) and SLIGKV-treated cells (Figure
4B). Interestingly, M1 macrophages matured in the presence of SLIGKV made reduced levels of
TNF-α than control M1 cells (GM-CSF or GM-CSF + RP) when exposed to LPS (Figure 4C), suggesting a skewing of the cell phenotype. Levels of
IL-10 were not altered significantly (Figure
4D). Maturation studies in the presence of GM-CSF and SLIGKV. (A) Example donor showing changes in
cell morphology and PAR-2 staining when differentiated in the presence of GM-CSF alone or
GM-CSF + SLIGKV (400 µM). Magnification is 200×, with the inset pictures 600×. (B) Summary data
showing differences in the cell area. (C) and (D) show changes in LPS (100 ng/ml)-induced TNF-α and
IL-10 secretion profiles by GM-CSF cells matured in the presence or absence of SLIGKV or RP,
n = 3 donors. **P < 0.02; *P < 0.05.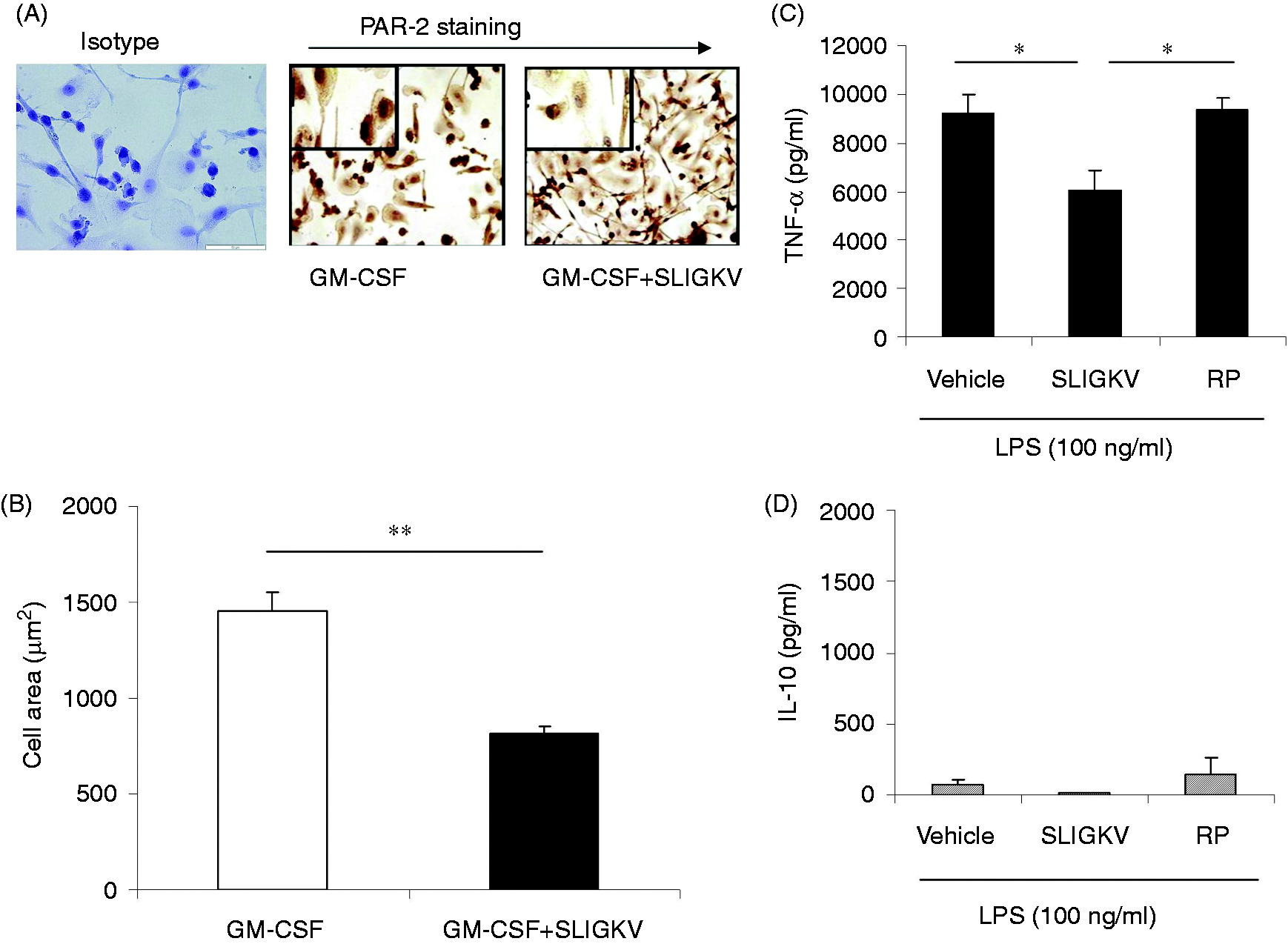
PAR-2 regulation of cell surface markers on M2 macrophages
M2 macrophages exposed to SLIGKV for 24 h exhibited enhanced surface expression of PAR-2 when
compared to vehicle-treated cells, suggesting autocrine regulation (Figure 5A, B), and a modest, but significant, reduction in HLA-DR expression (Figure 5C). No significant change in TLR2 (41 ± 11% vs 46 ± 6.3%,
M2 vs M2 + SLIGKV, respectively, n = 8) or TLR4 (57 ± 12% vs 73 ± 6.4%, M2 vs
M2 + SLIGKV, respectively, n = 8) expression was observed in these cells. Regulation of cell surface markers on M-CSF-matured macrophages by SLIGKV. (A) Example histogram
plot from a single donor showing an increase in PAR-2 surface expression after activation of an
M-CSF-matured macrophage with SLIGKV (400 µM) compared with vehicle (VEH)-treated cells. Isotype
staining is also shown. (B) Summary data for PAR-2 surface expression and (C) HLA-DR expression,
n = 8 donors reported. *P < 0.05.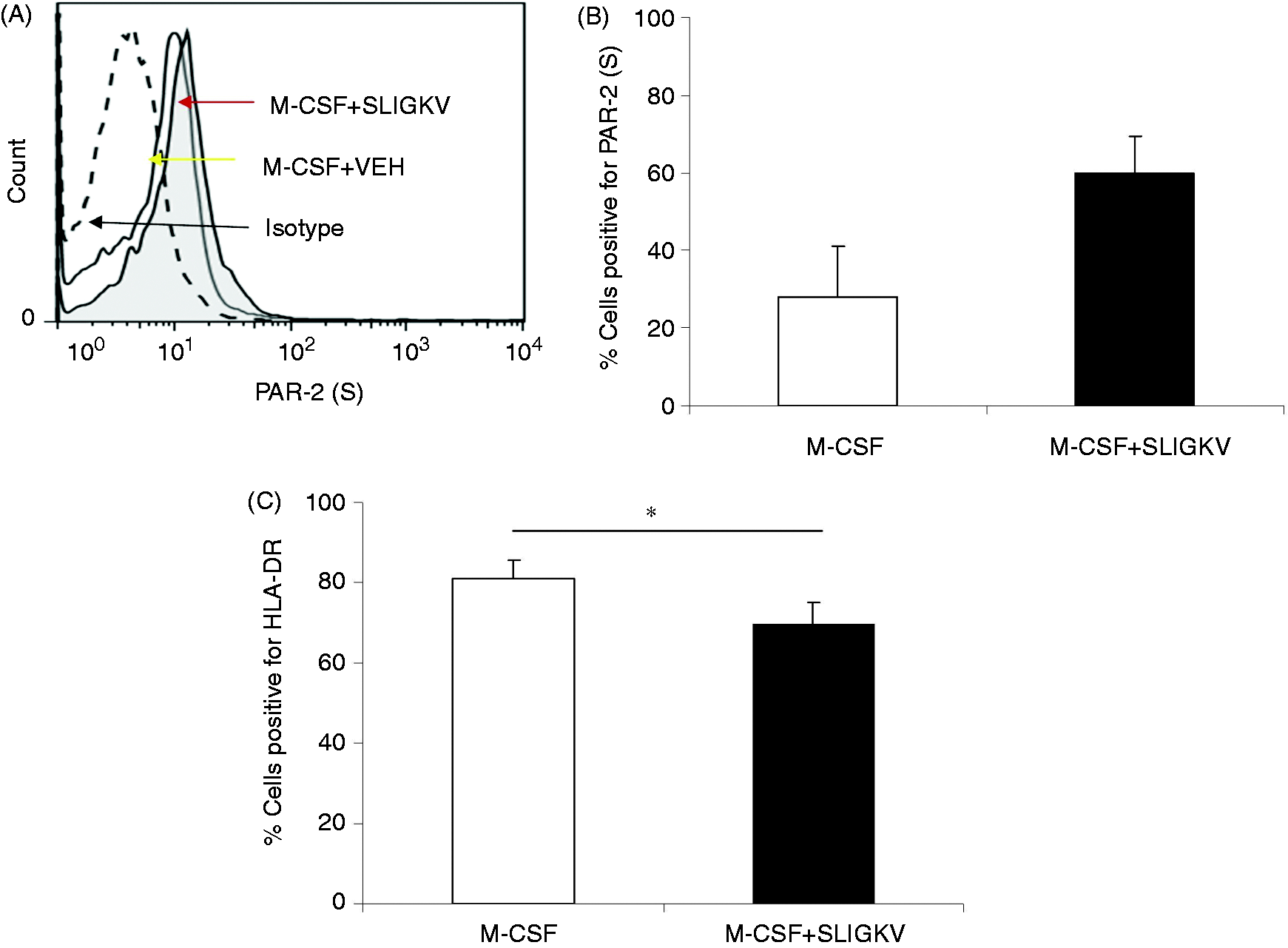
Influence of PAR-2 activation on M2 differentiation and effector function
Cells polarised to an M2 phenotype in the presence of M-CSF ± SLIGKV for 6 d were found to stain
abundantly for PAR-2 (Figure 6A). In the
presence of SLIGKV, cells displayed altered morphology (Figure 6A), with a significant difference in cell area observed
(P < 0.0001, Bonferroni t-test; Figure 6B). In addition, M-CSF + SLIGKV-matured macrophages were
found to make significantly more TNF-α than M2 cells after exposure to LPS (Figure 6C). Altered TNF-α secretion was paralleled by a
significant reduction in IL-10 release with SLIGKV, but not in the presence of control reverse
peptide (Figure 6D). This observation would
suggest that PAR-2 activation in the presence of M-CSF alters the M2 phenotype towards an M1-like
profile. Maturation studies in the presence of M-CSF and SLIGKV. (A) Example donor showing changes in cell
morphology and PAR-2 staining when differentiated in the presence of M-CSF alone or M-CSF + SLIGKV
(400 µM). Magnification is 200×, with inset pictures 600×. (B) Summary data showing differences in
the cell area. (C) and (D) show differences in LPS (100 ng/ml)-induced TNF-α and IL-10 secretion
profiles by M-CSF cells matured in the presence or absence of SLIGKV or RP, n = 3
donors. ***P < 0.01; **P < 0.02;
*P < 0.05.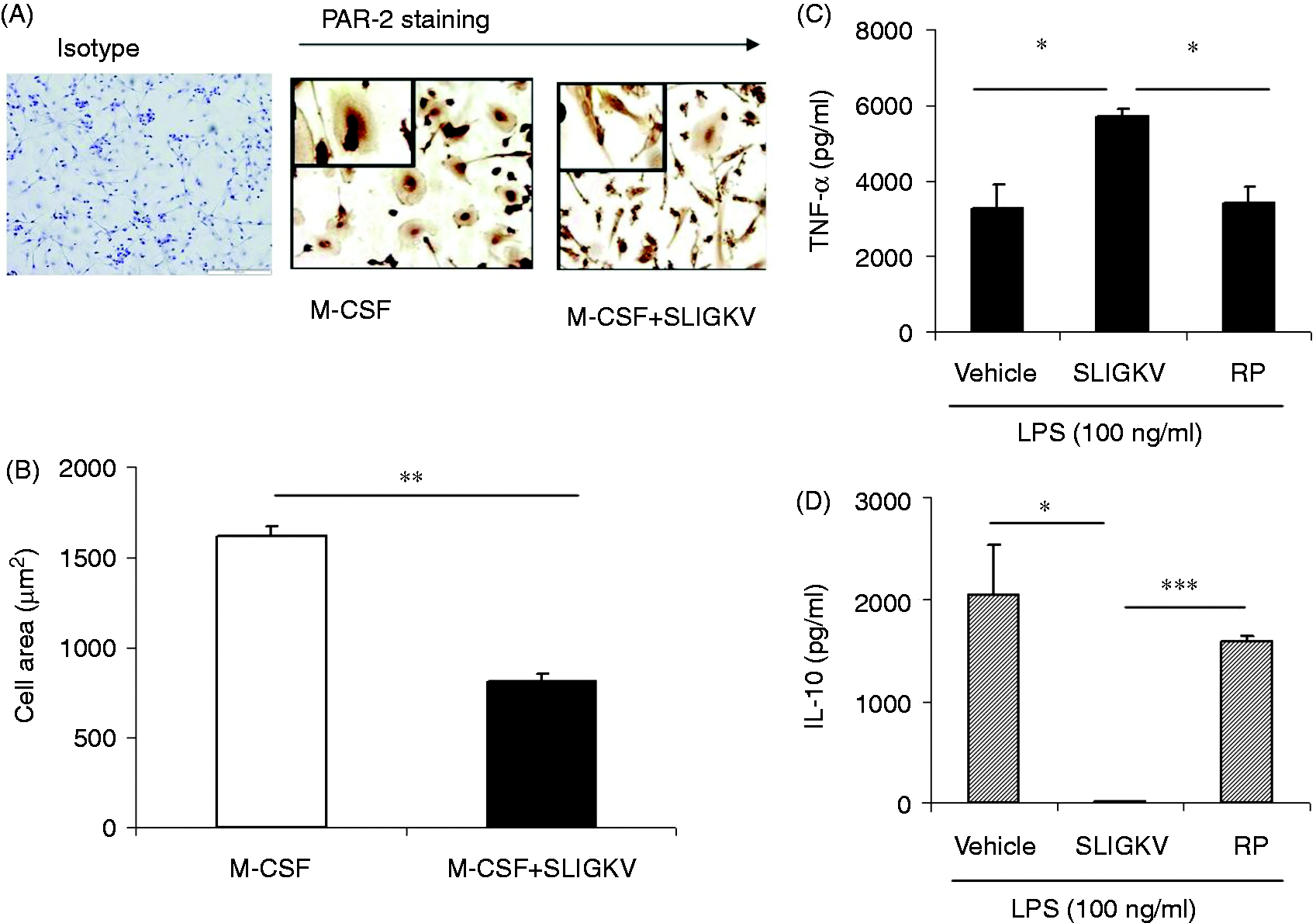
Discussion
Within the context of PAR-2 biology, detailed analysis of this receptor in human macrophage subset development and function has not been undertaken previously. The present study is the first to demonstrate a functionally relevant role for PAR-2 in human macrophage development and subset effector function. While PAR-2 surface expression on M1- and M2-matured macrophages has been reported previously,17,18 the functional consequence of PAR-2 activation on human M1 and M2 subsets has not been explored. To address this directly, we investigated whether PAR-2 activation could regulate expression of surface molecules involved in cellular activation and antigen presentation. Our data suggest that PAR-2 expression on M1 macrophages can be up-regulated using the PAR-2-activating peptide, SLIGKV, possibly through mobilisation of intracellular pools to the cellular surface. PAR-2 cannot be recycled to the cell surface once internalised, and so is targeted to lysosomes for degradation. 23 Re-sensitisation is therefore dependent on trafficking of pre-formed receptor from cytoplasmic stores and our data suggest that once PAR-2 activation has occurred, surface expression is further up-regulated.
PAR-2 activation of M1 macrophages resulted in significant up-regulation of HLA-DR, an antigen-presenting molecule associated with chronic inflammatory disease-associated pathology, exemplified in RA. 24 In contrast, PAR-2 activation in M2 macrophages led to significant down-regulation of HLA-DR expression. Similar observations have been reported in M2 macrophages post-stimulation with LPS. 22 This would suggest that in mature macrophages, in which cellular phenotype has been established, PAR-2 activation will enhance the characteristics of that phenotype—be that inflammatory or anti-inflammatory—and may explain the divergent roles that have been attributed to PAR-2. 2
While not attaining significance, there was a trend to enhanced TLR4 expression in both M1 and M2 macrophages following SLIGKV stimulation. A similar result has been reported for M2 macrophages stimulated with LPS. 22 Physical and functional interactions between PAR-2 and TLR4 have been described previously. 25 It is possible that in innate cell types, such as macrophages, PAR-2 may act as a danger or damage-sensing receptor, similar to pathogen associated molecular patterns, or danger-associated molecular pattern sensors, such as the TLRs. 26 Alternatively, it may act to modulate such responses in the context of tissue damage. Proteinases are generated in response to inflammation and are found abundantly at sites of tissue damage; therefore, it is likely that PAR-2 may act to sustain inflammatory responses under these conditions. A number of endogenous molecules have been identified as TLR activators, including heat shock proteins and nuclear material. 27 These endogenous molecules, along with proteinases, are likely to be in high concentration at sites of tissue pathology, such as in the RA joint. PAR-2-mediated up-regulation of HLA-DR may promote macrophage-mediated antigen presentation to T cells. Although macrophages are generally unable to induce naïve T cell activation, macrophage cellular interactions and mediators are critical in sustaining an inflammatory environment and adaptive cell activation status. Macrophages and T cells are found co-localised within the rheumatoid synovium, and this cellular interaction is thought to be pivotal in RA pathology. 28 It is possible that the pro-inflammatory environment of the RA joint surface enhances PAR-2 expression in T cells, and, in the context of a proteinase-rich environment, this leads to a reduced signalling threshold for T cell activation. Likewise, PAR-2-activating proteinases in the RA joint may lead to amplification of HLA-DR and TLR expression, both of which are known to be enhanced in patients with RA.29,30 This represents a possible mechanism whereby PAR-2 may contribute to activation of innate and adaptive immunity leading to breach of peripheral tolerance, and contributing to autoimmune and chronic inflammatory diseases, such as RA.
Activation of PAR-2 using the activating peptide SLIGKV resulted in the generation of signature cytokines from both M1- (high TNF-α, low IL-10) and M2-matured macrophages (low TNF-α, high IL-10). While similar observations have been reported previously for macrophage subsets activated with LPS, 22 this is the first report of regulation of M1 and M2 signature cytokine secretion via PAR-2 activation. In the mouse it has been reported recently that PAR-2 activation promotes an anti-inflammatory, alternatively-activated (or M2) phenotype in thioglycollate-derived macrophages stimulated with LPS. 31 It is of note, however, that macrophages collected from thioglycollate-induced peritonitis have reduced class II expression, a decreased capacity to support T cell proliferation and make reduced levels of pro-inflammatory cytokines in response to LPS when compared with macrophages derived from an antigen-specific model. 32 These observations would suggest that thioglycollate macrophages may, in fact, have a less inflammatory phenotype to begin with, making them biased or more susceptible to alternative activation. While our data cannot be compared directly to the murine studies of Nhu et al., 31 both provide evidence that PAR-2 is capable of inducing an anti-inflammatory phenotype. In addition to this, our data show that PAR-2 activation can drive the effector function of both M1 and M2 macrophages, not only through regulation of surface molecules, but through cytokine secretion. We would postulate that in M1-matured cells, PAR-2 may have a role in enhancing inflammation, whereas in M2-matured cells PAR-2 may have a more homeostatic role. This dual effect of PAR-2 has been previously reported in murine disease models, with PAR-2 having a protective effect in some disease models, such as allergy and colitis.33–35 Other disease models identify PAR-2 as having a pro-inflammatory effect, including arthritis, encephalitis and dermatitis.11,36–38 Again, this supports the hypothesis PAR-2 has differential effects and functions depending on cell type, environment and polarisation.
It has been documented that human peripheral blood monocytes express low levels of surface PAR-2, 18 which we confirmed in our study. Interestingly, cell subsets expressed substantial intracellular PAR-2. These intracellular stores have been determined previously as a pool of pre-formed receptor, which can be trafficked rapidly to the cell surface, and, once on the cell surface, PAR-2 can mediate release of inflammatory cytokines. 18 Despite this knowledge, the role of PAR-2 during maturation and differentiation to a macrophage subset is not clear for human cells. This is in contrast to murine studies in which a role for PAR-2 in cellular development of murine bone marrow-derived DC has been described previously. 21 Macrophages, DC and osteoclasts are derived from a common precursor, the CD14+ monocyte. PAR-2 has been identified in human osteoclasts, with data suggesting it may be important in cellular regulation. 39 Human DC do not appear to express surface PAR-2 in a non-stimulated state;17,18 however, it is possible that PAR-2 may be up-regulated when DC are cultured in a pro-inflammatory environment. Here, we have investigated the role of PAR-2 in development of monocytes to mature macrophages. Both M1 and M2 macrophages stained abundantly for PAR-2 by immunohistochemistry, with both cell types displaying typical macrophage morphology, with enhanced cytoplasm. However, when cultured in the presence of SLIGKV, morphology appeared altered, with both cell types displaying an elongated and spindle-like appearance. This suggests that PAR-2 may be involved in cytoskeletal rearrangement during macrophage maturation, resulting in the altered and ‘stretched’ appearance of the cells when exposed to a PAR-2 agonist, which was also reflected in altered cell area compared with vehicle treatment. Interestingly, monocytes polarised towards an M1 phenotype in the presence of the PAR-2-activating peptide, SLIGKV, made significantly less TNF-α after challenge with LPS than M1 cells polarised with GM-CSF alone. In addition, these cells made more IL-10 than M1 cells, although this failed to reach significance. Contrastingly, cells polarised towards an M2 phenotype in the presence of SLIGKV made significantly more TNF-α with abrogation of IL-10 secretion when compared with control M2-matured macrophages post-LPS challenge. A role for PAR-2 in mouse macrophage phenotype development has been demonstrated using thioglycollate-derived macrophages from PAR-2−/− mice. 31 While this model is GM-CSF independent, 40 it appears that in the absence of PAR-2 thioglycollate induces a peritonitis characterised by macrophages that have a more inflammatory phenotype, as assessed by their cytokine profile post-stimulation with LPS. While it is difficult to make a direct comparison between our findings and those reported by Nhu et al. 31 in the mouse, we would suggest that both studies highlight a definitive role for PAR-2 in influencing macrophage subset development.
Historically, M2 macrophages have been referred to as inflammation resolving cell. Contrastingly, the M1 macrophage has an inflammatory phenotype with production of inflammatory type cytokines and an ability to support Th1 and Th17 adaptive immunity.16,41 Our data would suggest that PAR-2 activation during macrophage subset differentiation can skew the cell phenotype and, thus, its effector function.
In conclusion, this study identifies a complex role for PAR-2 in innate immunity. Our study highlights PAR-2 as a key regulator of macrophage differentiation and effector function. These observations may be particularly relevant to macrophage-driven diseases in which PARs, acting as molecular sensors in proteinase-rich microenvironments, have the potential to dictate the type of innate response. We would suggest that as a pivotal receptor molecule in innate immune response, PAR-2 may offer an attractive therapeutic target for macrophage-driven disease.
Footnotes
Funding
This work was supported by Arthritis Research UK (grant number 18306) and the Carnegie Trust.
Acknowledgements
We acknowledge the assistance of Ashleigh Rainey in initial PBMC experiments.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.