
Abstract
We have performed freeze-fracture replica immunogold labelling of endotoxin preparations (lipid A and deep rough mutant LPS Re from Salmonella enterica sv. Minnesota), i.e. adding the endotoxins to human monocytes, labelling with monoclonal Abs recognizing either lipid A or LPS Re (A6 and A20 respectively), and fixing with immunogold secondary Ab. We have found that the endotoxins intercalated into the cell membranes with subsequent internalization by the cells. Surprisingly, membrane uptake took place only in the inner, plasmic leaflet of the plasma membrane, but there was no uptake of the outer leaflet for both compounds. Remarkable labelling could be also found for the two membranes of the nuclear envelope—in the case of lipid A only at the plasmic leaflet, but in the case of LPS Re on both leaflets. Isothermal calorimetric titration of the AB A20 with LPS and phospholipids showed that the Ab may bind not only to LPS but also to negatively charged phosphatidylserine. These results are discussed in the frame of the published concepts of cell activation induced by the endotoxins, i.e. how they are able to cause a conformational change of signalling proteins, such as the TLR4/MD2 complex.
Keywords
Introduction
Endotoxins (LPS) are some of the strongest immune stimulants known in nature. The stimulation of target immune cells, such as mononuclear cells (MNC; consisting of lymphocytes, monocytes and macrophages), takes place via a complex process by the interaction of LPS with serum and membrane proteins, such as LPS-binding protein (LBP) and soluble and membrane-bound CD14, with a final binding to the TLR4/MD2 complex, leading to cell signalling.1–3 There are numerous articles dealing with the characterization of the physico-chemical unit(s) of endotoxin responsible for the binding and signalling process. Independently of the detailed mechanisms, there seems to be the general opinion that cell signalling is a step which is governed by changes in membrane parameters.4,5 However, various data indicate that the interaction of LPS with some human binding proteins eventually leads to endotoxin monomers, which may fit into a binding pocket of the MD2 molecule and therefore lead to the signalling process. 6 Other data are indicative of endotoxin aggregates responsible for the stimulation process, as the interaction of LPS, in particular, with LBP leads to its intercalation into the cytoplasmic membrane of the target cells forming domains, which leads to conformational changes of the signalling proteins due to the concave curvature of the non-lamellar LPS aggregates representing mechanical stress.7,8
We have applied electron microscopy by performing freeze-fracture replica immunogold-labelling (FRIL) of Ab-labelled lipid A and LPS Re from Salmonella enterica sv. Minnesota strain R595 after they were incubated in vitro with human monocytes. Interestingly, we have found, for the first time and surprisingly, that there is a clear tendency of these endotoxins to intercalate into the inner side of the cytoplasmic membrane, as well as to the nuclear membrane. These data are discussed with respect to the conventional models of cell activation.
Materials and methods
Cells and Abs
Human monocytes from MNC were isolated from peripheral blood up to a final concentration of some 106 cells per ml following methods described previously.9,10 The cells were stored in PBS buffer at 0℃ until further preparation for freeze-fracturing.11.12
The murine mAbs A6
13
required for binding the bisphosphorylated lipid A backbone, and A20 binding to a single α-pyranosidically linked 3-deoxy-
LPS and lipid A
LPS from the rough mutant Re from S. Minnesota (strain R595) was extracted by the phenol/chloroform/light petroleum method from bacteria grown at 37℃, purified and lyophilized. 18 Lipid A was obtained from LPS R595 by acetate buffer treatment and converted to the triethylamine salt form. The results obtained by standard assays on the purified LPS and lipid A (i.e. analyses of the amount of glucosamine, total and organic phosphate, and distribution of the fatty acid residues) were in good agreement with the chemical properties known for this LPS chemotype, whose molecular structure has already been solved. 19
Freeze-fracture preparation
Anti-lipid A
The cells were warmed to 38℃ and lipid A dispersed in HEPES buffer was added to a final concentration of 15 µg/ml. After incubation times of 5 and 20 min the samples were quickly cooled down to 4℃ and cells were sedimented by centrifugation for 5 min at 100 g. Then, the cells were washed from lipid A by removing the supernatant, adding 2 ml cold PBS under shaking and repeating centrifugation. After removing most of the supernatant, glycerol as a cryoprotector was added under shaking to a final concentration of about 17% (v/v).
Freeze-fracturing and freeze-fracture immunogold-labelling were carried out as described by Schlörmann et al. 20 Aliquots of the cell suspension were enclosed between 0.1-mm thick copper profiles, as performed in the sandwich double-replica technique. The sandwiches were physically fixed by rapid plunge-freezing in a liquid (1 : 1) ethane–propane mixture cooled by liquid nitrogen. Freeze-fracturing was performed in a BAF 400 T freeze-fracture unit (BAL-TEC, Balzers, Liechtenstein) at −150℃ using a double replica stage. Carbon, about 20 nm thick was evaporated as the first replica layer perpendicularly by electron evaporation gun, followed by a second 2-nm thick platinum layer evaporated at an angle of 35°. Replication with carbon as a first and platinum as second replica layer is normally not used in freeze-fracturing. It results in more rough fine structures of the replica, but also in a higher frequency of the gold label. 21
Anti-LPS R595
For use of the LPS R595 Ab monocytes were isolated and treated as already described for anti-lipid A. Different to this procedure, the monocytes were incubated with LPS R595 for 5 min only.
FRIL
The replica labelling procedure has been done after the method of Fujimoto. 12 Freeze-fracture replicas were transferred to a digesting solution (2.5% SDS in Tris/HCl 10 mM, pH 8.4 and 30 mM sucrose) and incubated overnight (12 h) under shaking. Then, the replicas were washed in PBS four times to remove SDS and incubated in labelling blocking buffer (LBB; 1% BSA, 0.5% gelatine, 0.005 Tween-20 in PBS, pH 7.2).
For immune labelling the replicas were placed on parafilm on a drop of a solution of anti-lipid A (A6-Ab) or anti-LPS R595 (A20-Ab), respectively (primary mouse Abs), diluted 1 : 50, 1 : 100 and 1 : 1000 in LBB and incubated overnight in the refrigerator. Then, the replicas were washed three times in LBB and transferred to a drop of secondary Ab (10 nm gold-conjugated goat-anti-mouse IgG; British Biocell International, Cardiff, UK) diluted 1 : 50 in LBB and incubated for 1 h.
After the labelling procedure the replicas were rinsed three times in PBS, fixed chemically by 0.5% glutardialdehyde for about 10 min, washed twice with distilled water, and finally picked up onto electron microscopic cupper grids Mesh 300 for examination in an EM 900 transmission electron microscope (Zeiss, Oberkochen, Germany). Beside the human monocytes, the primary dispersion of LPS R595 used for incubating the cells was also freeze-fractured and immunogold-labelled as a control investigation.
Isothermal titration calorimetry
Microcalorimetric experiments of Ab A20 or A6 binding to LPS R595, lipid A or to the phospholipids phosphatidylcholine (PC) or phosphatidylserine (PS) were performed on a MSC isothermal titration calorimeter (Microcal, Northhampton, MA, USA) at 37℃ as described previously. 22 Briefly, after thorough degassing of the samples, the Ab A20 at a 2 mg/ml concentration was titrated in 1.5-µl portions every 5 min into the lipid-containing (0.05 mM) cell under constant stirring, and the heat of interaction after each injection measured by the isothermal titration calorimetry (ITC) instrument was plotted versus time. The enthalpy change during each injection was measured by determining the area underneath each injection peak (Origin software; Microcal), and plotted against the [A20]:[LPS] molar ratio. Titration of the pure Abs into HEPES buffer resulted in a negligible endothermic reaction due to dilution. The experiments were done at least twice.
Results
The results of the labelling procedures are presented in Figures 1 and 2. Because there were no differences in the distribution of 10 nm gold after immune labelling of monocytes incubated 5 or 20 min with lipid A and LPS R595, respectively, here mainly the results of the 5 min incubation time are shown.
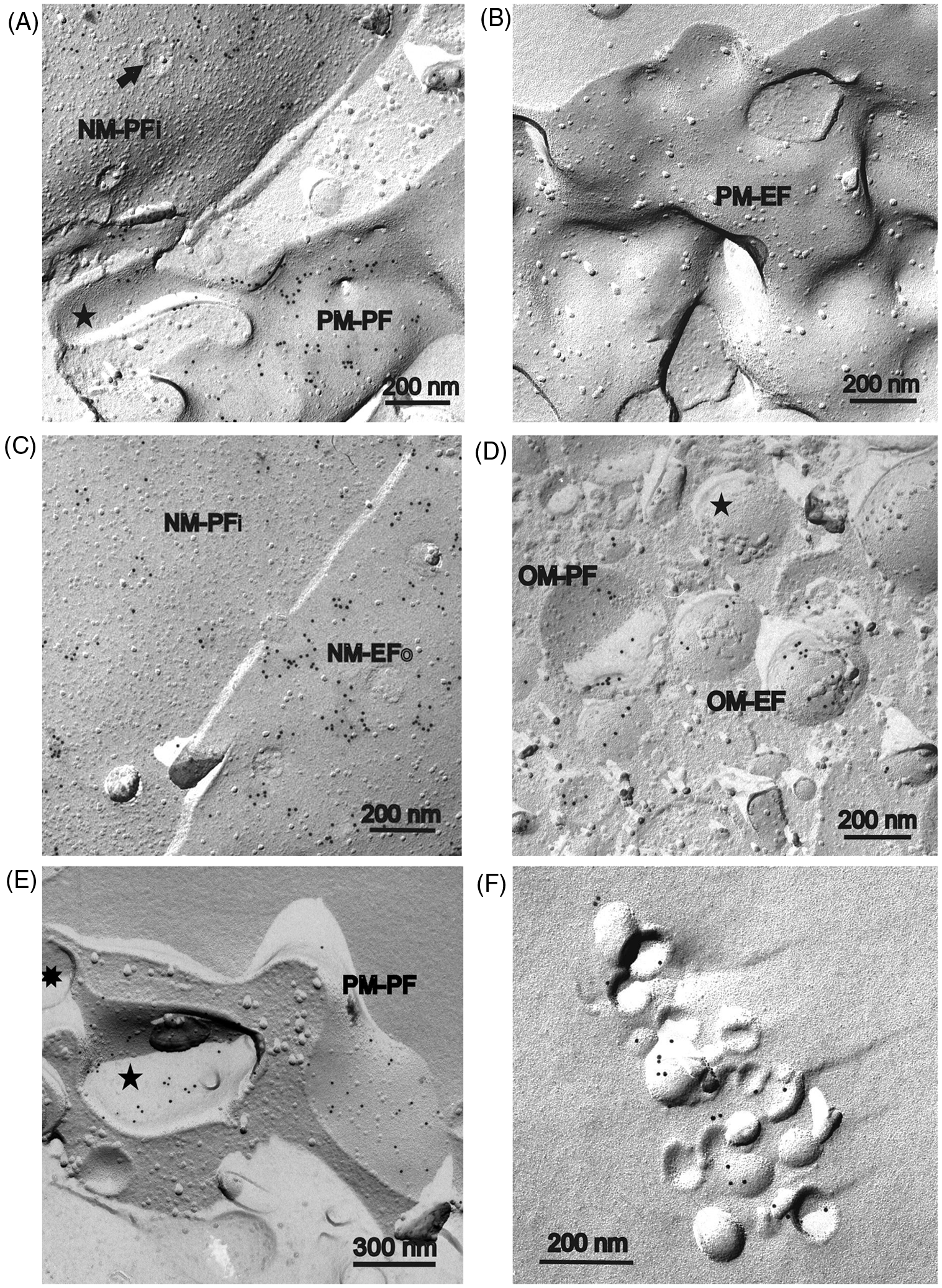
Immunogold-labelled freeze-fracture replicas of human monocytes isolated from peripheral blood after incubation with lipid A. The monocytes have been incubated 5 or 20 min (part B only) with lipid A, freeze-fractured and replicated. Replicas were treated with SDS overnight, incubated in a first step with the primary A6-Ab against lipid A, and after washing in a second step immunogold-labelled by a secondary 10 nm-gold conjugated-Ab. (A) The plasma sided fracture face of the plasma membrane (PM-PF) is covered by small clusters of 10 nm gold grains. The cross-fractured cytoplasm (left side) is free of gold label. (B) Beside the plasma sided (plasmic) inner leaflet of the plasma membrane (PM-PF), the plasma-sided leaflet of a plasma membrane invagination (PMI-PF) also shows gold label, whereas the exoplasmic plasma membrane half (PMI-EF) is free of label. Some gold labelling in the cytoplasm might be artificial owing to the low thinning ratio of 1 : 50 of the primary A6-Ab (only this figure part). (C) A part of the exoplasmic outer leaflet of the plasma membrane (PM-PF) without gold label is shown. (D) Labelled plasma sided leaflet of the outer nuclear envelope (NM-PFo) with clusters of 10 nm gold label. Arrow indicates a nuclear pore. (E) (a) Besides the plasma sided leaflet of the inner nuclear envelope membrane (NM-PFi) the envelopes of mitochondriae (Mt) are also partly labelled. In contrast, the exoplasmic leaflet of the outer nuclear envelope membrane (NM-EFo) is not labelled and bears only a few single gold grains. (b) Part of nuclear membranes of (a) at higher magnification. (c) Mitochondrion from (a) at higher magnification. The exoplasmic half of the outer envelope membrane of the mitochondrion (OM-EF) bears small clusters of gold label. The plasmic half of the inner envelope membrane (IM-PF) seems to be unlabelled. Immunogold-labelled freeze-fracture replicas of human monocytes after incubation with LPS R595 for 5 min and use of A20-Ab against LPS R595. Procedure of treatment as described in Figure 1. (A) The plasma-sided leaflet of the peripheral plasma membrane (PM-PF) and of a plasma membrane invagination (star) is labelled by clustered 10 nm gold grains. Additionally, a part of the plasmic leaflet of the inner nuclear envelope membrane (NM-PFi) is visible and labelled. The nucleus membrane can be identified easily by nuclear pores (arrow). (B) The outer exoplasmic half of the plasma membrane (PM-EF) is nearly free of any labelling. (C) The plasmic half of the inner nuclear envelope membrane (NM-PFi) and the exoplasmic half of the outer nucleus envelope membrane (PM-EFo) are shown. Both inner and outer membrane leaflet are labelled by clusters of gold. (D) A part of the cytoplasm with a number of mitochondria is shown. The plasmic membrane half of the outer envelope membrane (OM-PF), as well as the exoplasmic half (OM-EF), appear to be labelled. But adjacent to labelled mitochondria unlabelled ones have also been found (star). (E) Labelling of the plasmic half of the peripheral plasma membrane (PM-PF) and of invaginated plasma membrane (five-pointed star) could also be found after high thinning of the A20-Ab of 1 : 1.000. Contrary to the thinning ratio of 1 : 100 (Figures 2A–D) clustering of gold label was not so predominant. The invaginated plasma membrane is connected to the peripheral plasma membrane by a pore (eight-pointed star). (F) Control test for immuno-gold labelling of the LPS R595 dispersion used for incubation of the monocytes. Gold label is restricted to convex and concave shells of small vesicles formed by LPS.
Anti-lipid A
Micrographs of monocytes incubated with lipid A and immune labelled by mouse anti-lipid A as primary Ab and 10-nm gold-conjugated goat anti-mouse Ab as second Ab are shown in Figure 1. In Figure 1A a part of the inner plasmic fracture face of the plasma membrane (PM-PF; plasmic leaflet) and an area of cross-fractured cytoplasm (left side) is visible. The plasmic fracture face (PF) of the plasma membrane (PM) is covered by small clusters of 10-nm gold grains, whereas the cross-fractured cytoplasm is nearly free of labelling. In Figure 1B, additionally to the inner leaflet (PM-PF), two plasma membrane invaginations exposing the plasmic fracture face (PMI-PF), as well as the exoplasmic fracture face (PMI-EF) can be seen. Only the plasmic face (PF) of both peripheral and invaginated PM, but not the exoplasmic face (EF), of invaginated PM is covered by clusters of gold label. This is in agreement with the absence of nearly any labelling on the outer leaflet of PM (Figure 1C, PM-EF). Single gold grains in the cytoplasm (Figure 1B) and on PM-EF (Figure 1C) seem to be unspecific labelling because they are absent in most cases.
Concerning intracellular organelles, we found clear labelling of the two membranes of the nuclear envelope (Figure 1D, E) and in a lower frequency of the envelope of mitochondria (Figure 1E). In case of the nuclear envelope only the plasma sided (plasmic) fracture faces [Figure 1D, NM-PFo; Figure 1E(a), NM-PFi, higher magnification in (b)] seemed to be labelled. The exoplasmic fracture faces [Figure 1E(a), NM-EFo; higher magnification in (b)] with the exception of some single gold grains were free of labelling. In the case of mitochondria, in particular, the outer envelope appears to be marked [Figure 1E(a), Mt; higher magnification in Figure 1E(c), OM-EF]. Unfortunately, fracture faces of the inner mitochondrial envelope [Figure 1E(c), IM-PF] were mostly very small and, therefore, labelling could not be characterized.
These data indicate that lipid A incorporates readily into the inner leaflet of the cytoplasmic membrane, as well as into both leaflets of the nuclear cell membranes.
Anti-LPS R595
The results of the incorporation experiments of LPS R595 with monocytes are presented in Figure 2. The primary Ab A20 against LPS R595 seemed to be somewhat more effective than the A6-Ab against lipid A. Thus, we could also reach a sufficient labelling after thinning the A20-Ab at a ratio of 1 : 1.000 (Figure 2E). Nevertheless, for comparison, most results are shown at a thinning ratio of 1 : 100.
Compared with anti-lipid A, nearly the same structures were labelled with some minor variations. In the case of the nuclear envelopes, beside the plasma-sided membrane leaflets (Figure 2C, NM-PFi) the exoplasmic leaflets were also labelled (Figure 2C, NM-EFo). In the case of mitochondria labelled (Figure 2D, OM-PF and OM-EF) and unlabelled outer envelope membranes (Figure 2D, star) could be found located side-by-side in the same cell, and labelling could be even completely absent after use of A20-Ab in a ratio of 1 : 1.000 (data not shown).
In agreement with the results for anti-lipid A, again the plasma sided (inner) leaflet of the peripheral plasma membrane (Figure 2A; Figure 2E, PM-PF) and invaginated plasma membrane (Figure 2A, star; Figure 2E, five-pointed star) were tagged, and, again, the exoplasmic fracture face of the plasma membranes did not show any labelling (Figure 2B, PM-EF).
As a control test the LPS R595 dispersion used for incubation of the monocyte samples was also investigated. Both convex and concave monolayers of the small LPS aggregates (vesicular-like structures) formed were labelled by gold grains (Figure 2F). The frequency of the labelling seemed to be rather low, but labelling was clearly restricted to areas of loosely aggregated vesicles.
ITC
To test the specificity of the Abs for the corresponding endotoxins and their possible interaction with components of cell membranes isothermal calorimetric experiments were performed by titrating the Abs with LPS R595/lipid A, PC and PS, two main components of the eukaryotic cell membrane. The ITC data are presented in Figure 3, showing, in all cases, exothermic reactions (a downward signal at every titration, see the upper half of the top diagram for PC) of the A20 with the three lipids with saturation characteristics. The main information is that LPS und PS both show considerable reactivity with the Ab A20, whereas this reaction is significantly lower in the case of PC. It should be noted that the A20 exhibits also some binding affinity to lipid A (not shown).
ITC of PC (top) with anti-LPS Ab A20, and the enthalpy change versus the weight ratio of [A20]:[PC] (top), and the corresponding data for PS (middle) and LPS R595 (bottom).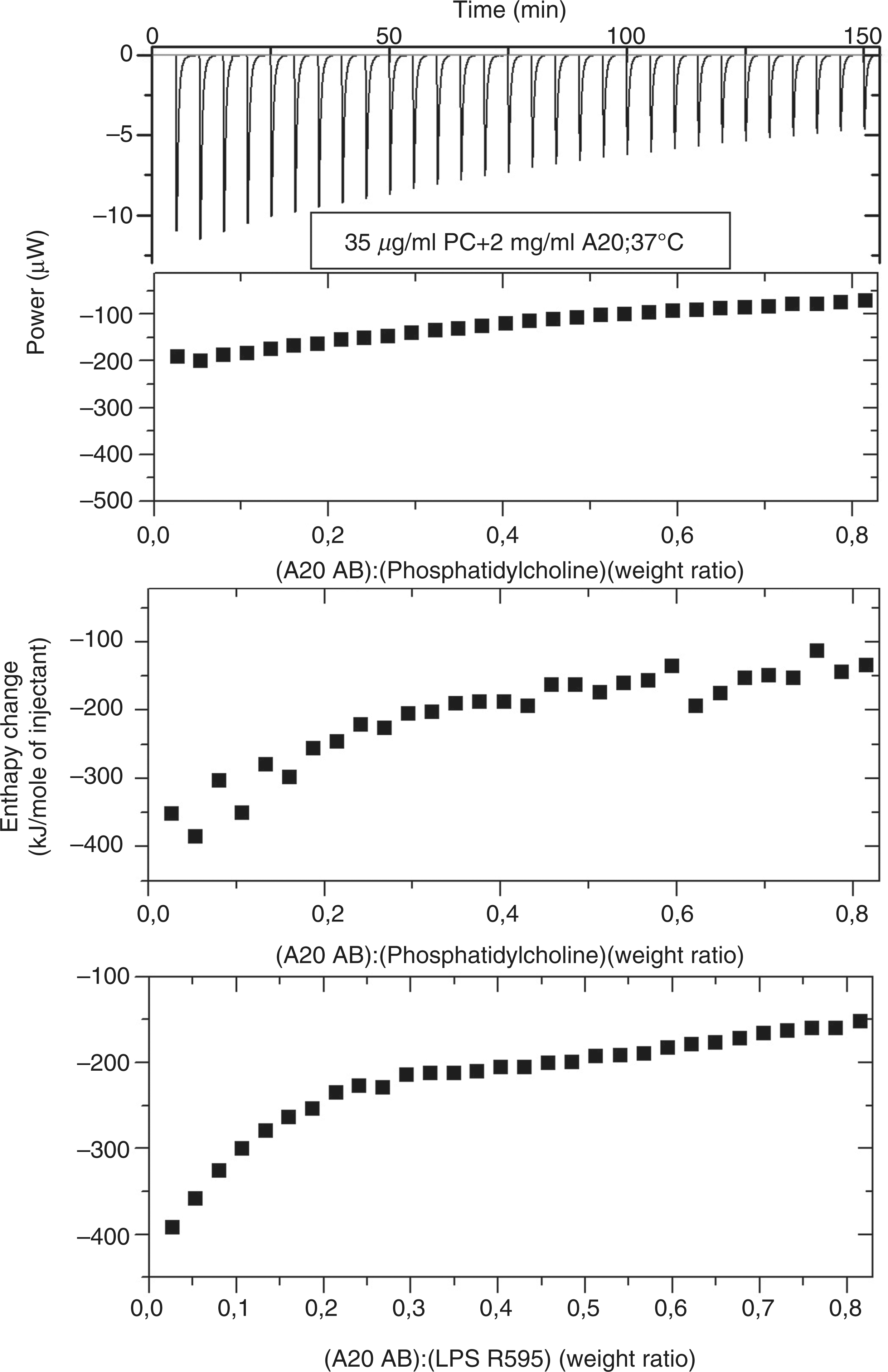
Similar results were found for the system Ab A6 and the lipids. However, the specificity of the A6 to lipid A is more specific and shows only minor reactivity with LPS R595, and also only slight reactivity with PC and PS.
Discussion
The presented data indicate a strong tendency of the investigated endotoxins (lipid A, LPS R595) to incorporate into target cell membranes. Surprisingly, an uptake of the endotoxins into the cytoplasmic side was only observed on the inner side, whereas the nuclear cell membranes were labelled on both sides (Figures 1 and 2). Regarding the mechanisms that lead to a membrane incorporation of endotoxins, it was shown previously that these cannot incorporate solely by themselves by hydrophobic interaction, the presence of a transport protein such as LBP is necessary. 23 In the absence of soluble LBP, this role can be adopted by membrane-bound LBP or by CD14.24,25 The data here show that after the incorporation of the endotoxins into the outer side as first step, a second step must follow consisting of a transfer to the inner side of the cytoplasmic membrane, which must be followed by a further transfer into the cell interior and then into the nuclear membrane. Regarding cell activation, the proposal published previously with model membranes, according to the phospholipid composition of the macrophage membrane that membrane intercalation is a necessary step for cell activation,8,26 is now confirmed for natural cell membranes and extended by the present data. The endotoxins, incorporated into the inner side of the cytoplasmic membrane, represent a strong sterical disturbance of the normal membrane architecture owing to the concave curvature of the endotoxins, resulting from their property to adopt non-lamellar aggregate structures.27,28 Our working hypothesis is now that this membrane-domains form disturbances at the site of signalling proteins such as the TLR4/MD2 complex, which leads to a conformational chance with subsequent cell activation. It will be our aim to analyse this mechanism in detail in further experiments.
The observation that endotoxins do not mix on a molecular scale but form domains has been described. Nomura et al. investigated the miscibility of some phospholipids (PC, PG and PE membranes in different ratios) with LPS and found the phospholipid composition has a critical influence on the distribution of added LPS within the respective phospholipids, and also on the morphology and physicochemical properties of the resulting mixed membranes. 29 Kubiak et al. working with giant unilamellar vesicles made from bacterial lipid extracts arrived at similar results, i.e. that various LPS chemotypes cluster into small domains in the phospholipid matrix, depending on concentration and the particular chemical structure. 30
The observation that there is also an interaction of the A20 Ab with PS from the inner leaflet sounds surprisingly, but such interaction has never been tested before. These findings, however, do not affect our main statements described herein.
The biological role of the incorporation of the endotoxins into the nuclear membrane remains unclear and may be owing only to a pure physico-chemical process without biological consequence, which would be in accordance with earlier data that binding of the endotoxins to the cytoplasmic membrane, but not the observed internalization, is a relevant step in cell activation.4,31 However, there could be also a biological role as found by Cowan et al. 31 for the internalization of LPS into cardiomyocytes, in which the authors found that the transport of LPS to specific intracellular sites in the heart cells are obligatory for activation of LPS-dependent signal transduction.
Our data here and data presented previously 32 do not indicate any role for endotoxin monomers in the cell activation process. This is in accordance with other findings, where a direct comparison of endotoxin aggregates and monomeric solutions derived from them at the same concentration showed only significant induction of TNF-α in human mononuclear cell or macrophages by the aggregates. 33 These data can so far be understood by the physicochemistry of endotoxins. It was found that the critical micellar concentration of lipid A and LPS is in the nanomolar range or even lower, 34 which means that under physiological concentrations of endotoxin activation there are nearly only aggregates present.
In the articles by Weiss and colleagues,9,35 an essential role of endotoxin monomers is described, which was found by a complex interplay of albumin, sCD14, LBP and other proteins. Essential interpretations of these authors resulted from the use of metabolically labelled 3H- or 14C-LPS. Beside the problem that the described interplay of these proteins may be un-physiological, a rough estimate of the radiation damage due to the use of these compounds does not exclude misinterpretations. We are currently investigating differently irradiated LPS preparations for a better description of such radiation-induced decomposition of endotoxins.
Also, the published data of a binding of a monomeric LPS into a pocket of the MD2 molecule 6 may be an artefact due to the cleavage of the membrane anchor of TLR4 necessary for the crystallographic experiments. It is well known that membrane proteins without their membrane anchor lose many of their physiological properties. Moreover, the fact that various non-lipid A structures, such as EISAI 803022, a phospholipid-type structure with 6 acyl chains and a serine-like backbone, are also TLR4-agonists10,27 would imply that such compounds also fit into the MD2 pocket, which seems to be improbable owing to the differing chemical structure.
Funding
The authors are indebted the German ministry BMBF (project 01GUO824) and the Else-Kröner-Fresenius Stiftung (project 2011_A140) for financial help.