
Abstract
Leucine-rich repeats (LRRs) are versatile motifs present in more than 6000 proteins throughout the phylogenetic kingdom. Tandem LRRs generate a characteristic horseshoe with a diverse range of functions. Fulfilling a key role in the innate immune system, LRRs form the TLR and NOD-like receptor (NLR) pathogen-recognition domain. Host–pathogen interactions mediated by LRRs drive those involved in ligand recognition to become distinct from their consensus motif. Most LRRs range between 21 and 30 residues; however, large insertions in certain TLRs can generate repeats of over 60 amino acids. LRR variability makes them ideal for species-specific mediation of host-pathogen interactions. Teleost TLRs show large insertions, making cross-species alignments difficult without prior demarcation of their LRR motifs. We present LRRfinder2.0, a webserver for LRR prediction. LRRfinder2.0 utilizes scoring matrices comprising more than 60,000 LRR motifs from more than 200 species. The underlying TLR database tLRRdb contains more than 3500 manually annotated sequences, augmenting identification of irregular LRR motifs.
Introduction
Leucine-rich repeats (LRRs) have been identified in more than 6000 proteins, belonging to a more general class of solenoid structures. 1 Structurally stable LRR motifs provide a solution to the conservative nature of evolution, facilitating modifications to perform a diverse range of functions. LRR-containing proteins have been identified in many biologically important processes in plants, invertebrates and vertebrates, including extracellular matrix assembly, cell signalling and adhesion, neuronal development and host–pathogen interactions.2–6
LRR domains are comprised of between 2 and 45 tandemly arranged LRR motifs for which 7 classes have been proposed, characterized by differing lengths and motif consensus: ‘Cysteine-containing’, ‘RI-like’, ‘SDS22-like’, ‘Bacterial’, ‘Plant-specific’, ‘Treponema pallidum’ and ‘Typical’. 7 Each 20–30 amino acid-containing motif can be separated into a highly conserved segment (LRRhs) and a variable segment (LRRvs), with a LRRhs sharing consensus of LxxLxLxxN/C(x)xL where L represents Leu, Ile, Val or Phe, N stands for Asp, Thr, Ser or Cys, and x is any amino acid. 1
The structure and tandem arrangement of LRRs in stretches of variable length provides a highly evolvable and versatile framework for binding myriad ligands.4,8 Throughout the phylogenetic kingdom, LRRs are involved in pathogen recognition, as demonstrated by the vastly expanded repertoire of LRR-containing proteins in the sea urchin that respond to microbial-associated molecular patterns (MAMPs) in the absence of an adaptive immune system. 2 In mammals, MAMPs are recognized primarily by pattern recognition receptors (PRRs), distinguishing between self and microbial structures to initiate an innate immune response. 9 In this process, TLRs, as well as NOD-like receptors (NLRs), are key components of the innate immune system. TLRs and NODs are comprised of an ‘extracellular’ LRR domain, mediating pathogen recognition and an ‘intracellular’ signal domain initiating cytokine production.9,10 In TLRs, the extracellular LRR domain has been shown to be under positive selection, generating a diverse collection of motifs as an ideal basis for an LRR predictor.11–15 As advances in high-throughput technologies generate sequences at a rapid pace, we need to have improved tools allowing for the fast and precise identification of LRRs in innate immune receptors, to allow for detailed ligand-binding analysis, as well as comparative studies.
Here, we present LRRfinder2.0, a webserver for the prediction of LRRs. Based upon TLR sequences, the LRRfinder prediction method has been applied previously to LRR identification in both plant and mammalian immune receptors. 16 A searchable database of domain and motif annotated TLR-sequences has been extended to include more than 60,000 sequences from more than 200 species. The latest release offers improved prediction with more than 14,000 unique LRR motifs and incorporates a post-translational modification site, surface accessibility and structural predictions. LRRfinder2.0 has a broad range of applications, including LRR demarcation for improved alignments in comparative modelling, identification of functionally important residues and scanning of novel genomes for immune-related proteins.
Material and methods
TLR database: tLRRdb
The LRRfinder2.0 prediction model is based upon a sequence-generated position-specific scoring matrix (PSSM), as described in Offord et al. 16 The current release includes more than 3000 full-length and partial nucleotide sequences translated from the NCBI and Ensembl databases.17,18 LRRhs motif positions were annotated manually using multiple Clustal 19 alignments of known LRR-containing structures from the Protein Data Bank (PDB). 20
Most TLRs are comprised of up to six key regions. Signal peptide cleavage sites were predicted using SignalP and Phobius.21,22 The spanning region between the cleavage site and the first LRRhs was determined as the LRR N-terminus (LRRNT) if it contained at least one cysteine residue. An annotated LRRCT domain includes the last LRR, containing the consensus CxC region, and ends at the transmembrane barrier. The exceptions to this rule are the insect Toll proteins, which can include several LRR C-terminus (LRRCT) regions between LRR-containing domains. The transmembrane helix was predicted using a combination of TMHMM, MEMSAT and Phobius.22–24 The downstream signalling Toll/interleukin-1 (TIR) domain was annotated as any region following a transmembrane helix. Partial sequences consisting of only a TIR domain were annotated manually from multiple sequence alignments.
The annotated sequences are stored in the TLR database tLRRdb and can be searched via several options. These include: accession, keyword, phylogeny, TLR, LRRhs motif, LRR length and sequence. BLAST 25 and Clustal have been implemented to allow users to identify and align the most related sequences to their input query. In addition, known LRR-containing PDB sequences have been provided, allowing for several alignment colour schemes to be available. These include Clustal colours, similarity, secondary structure and domain annotation.
LRRfinder2.0 predictor: Features and description
The LRRfinder2.0 server accepts protein queries via four input methods: user-defined protein sequence, NCBI protein accession, NCBI nucleotide accession or user-defined nucleotide sequence. Nucleotide sequences are automatically translated prior to analysis.
Protein sequences are analysed using the protocol described in Figure 1. Several PSSM options are available to the user in this release. TLR- and taxon-specific matrices are provided to allow for the observed variation in amino acid usage between phylogenetic groups (data not shown). LRR-containing proteins are not limited to TLRs therefore we have included a PSSM option based upon all LRR-containing PDB sequences and their related NCBI counterparts.
Schematic layout of LRRfinder2.0 webserver protocol for LRR prediction. The LRRfinder2.0 sequence processing can be divided into six stages. Following LRR identification, the submitted sequence is passed to several applications for domain, post-translational modification, structural property and surface accessibility prediction. The information is then compiled and presented in subsections for user-friendly viewing.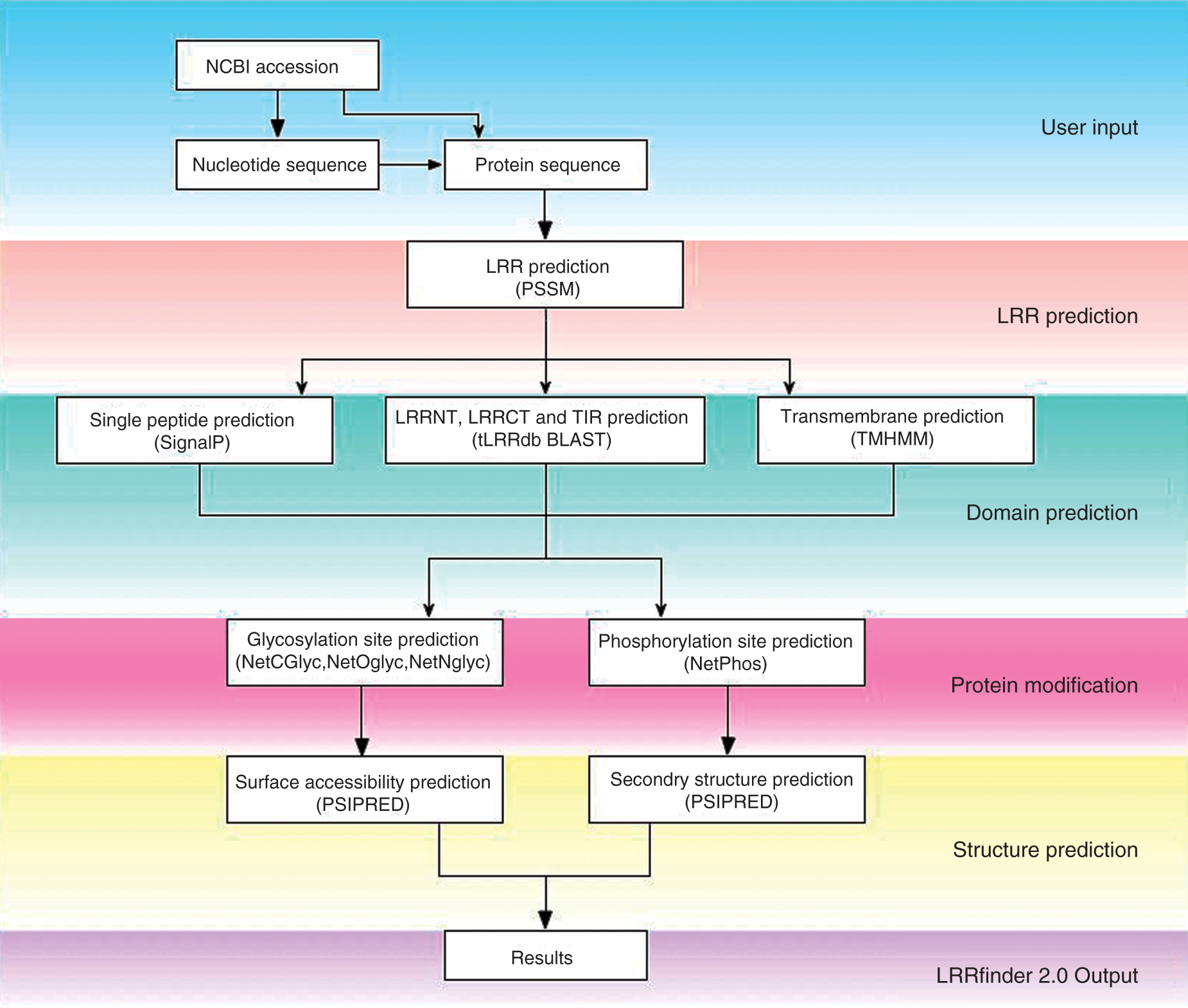
LRRfinder2.0 prediction is applied using an 11-residue sliding window, a user-defined PSSM and annotated LRRhs motifs from tLRRdb. Known motifs have precedence over predictions due to the irregularity of ligand-binding repeats and the tabular output provides details on all tLRRdb matches within the sequence. User-defined e-value thresholds categorize predicted LRRhs regions as either ‘significant’ or ‘insignificant’, and list overlapping predictions.
TLRs are comprised of several domains, including signal peptide and transmembrane regions that are predicted by SignalP and TMHMM respectively. LRR-domain capping regions and TIR domain are identified via a BLAST comparison to tLRRdb, listing the top hits and similarity scores. Post-translational modifications, such as glycosylation and phosphorylation sites, are predicted using the CBS tools NetCGlyc, NetOGlyc, NetNGlyc and NetPhos.26–28
Comparative protein modelling relies upon accurate alignments, assisted by the demarcation of domains and LRR identification. Secondary structure and surface accessibility predictions via PSIPRED 29 have been included to allow users to validate LRRhs predictions, identify functionally important residues and improve alignment of the LRRvs, which may vary in sequence, but maintain structural conservation.
Results and discussion
The evolution of innate immune receptors has been studied by many researchers. Phylogenetic analyses have shown that the vertebrate TLR and NLRs evolved independently by gene duplication prior to the divergence of protostomes and deuterostomes,30,31 with the occurrence of both gene loss and gene conversions. 13 Although vertebrate TLRs have been cited as an example of evolutionary conservation and strong functional constraint, 30 recent studies have suggested positive selection in some TLRs in an extremely broad range of organisms, including primates, teleosts and birds.11–15,32–34 Within innate immune receptors the solenoid LRR domains often show higher rates of evolution than their intracellular counterparts. 35
Taking this evidence into consideration, the exact identification of LRRs is imperative. The relevance of LRRfinder2.0 in computational modelling of LRR-containing proteins has been shown for bovine TLR2, improving MODELLER
36
sequence alignments by influencing manual adjustments (Willcocks et al., submitted). In addition, the LRRfinder methodology has also been applied to the identification of LRR motifs in echinoderm, plant and mammalian immune-related proteins.14,15,37–42 The results generated provide insight into the functional diversification of LRRs between innate immune receptors of different species. Indeed, using LRRFinder2.0 with sequences from urochordates, we were able to detect a variety of LRR-containing proteins. Urochordates are model organisms in comparative and evolutionary immunology. Genome-wide analysis of Ciona intestinalis has previously identified two TLR candidates capable of recognizing myriad MAMPs and inducing cytokine production.
43
A standalone version of the LRRfinder2.0 PSSM predictor was used to scan the C. intestinalis and C. savignyi Ensembl protein libraries finding more than 500 and 800 LRR-containing proteins respectively. LRRfinder2.0 was able to identify more LRRs within the two TLR candidates, improving on previous motif predictions by SMART,
44
as shown in Figure 2. The success of the current system for the identification of LRR-containing proteins via genome scanning strongly suggests that the process can be applied to a much greater sample size, spanning all LRR protein classes and may therefore provide a better insight into LRR evolution and the ancestral immune repertoire.
Comparison of LRR prediction methods for Ci-TLR1 and Ci-TLR2. Sasaki et al.43 identified 7 and 10 LRRs in the Ciona intestinalis receptors Ci-TLR1 (NP_001159599.1) and Ci-TLR2 (NP_001159600.1), respectively, using SMART. LRRfinder2.0 shows a significant increase in the number of predicted LRRs by comparison, identifying 18/19 and 17/18 motifs in Ci-TLR1 and Ci-TLR2 respectively.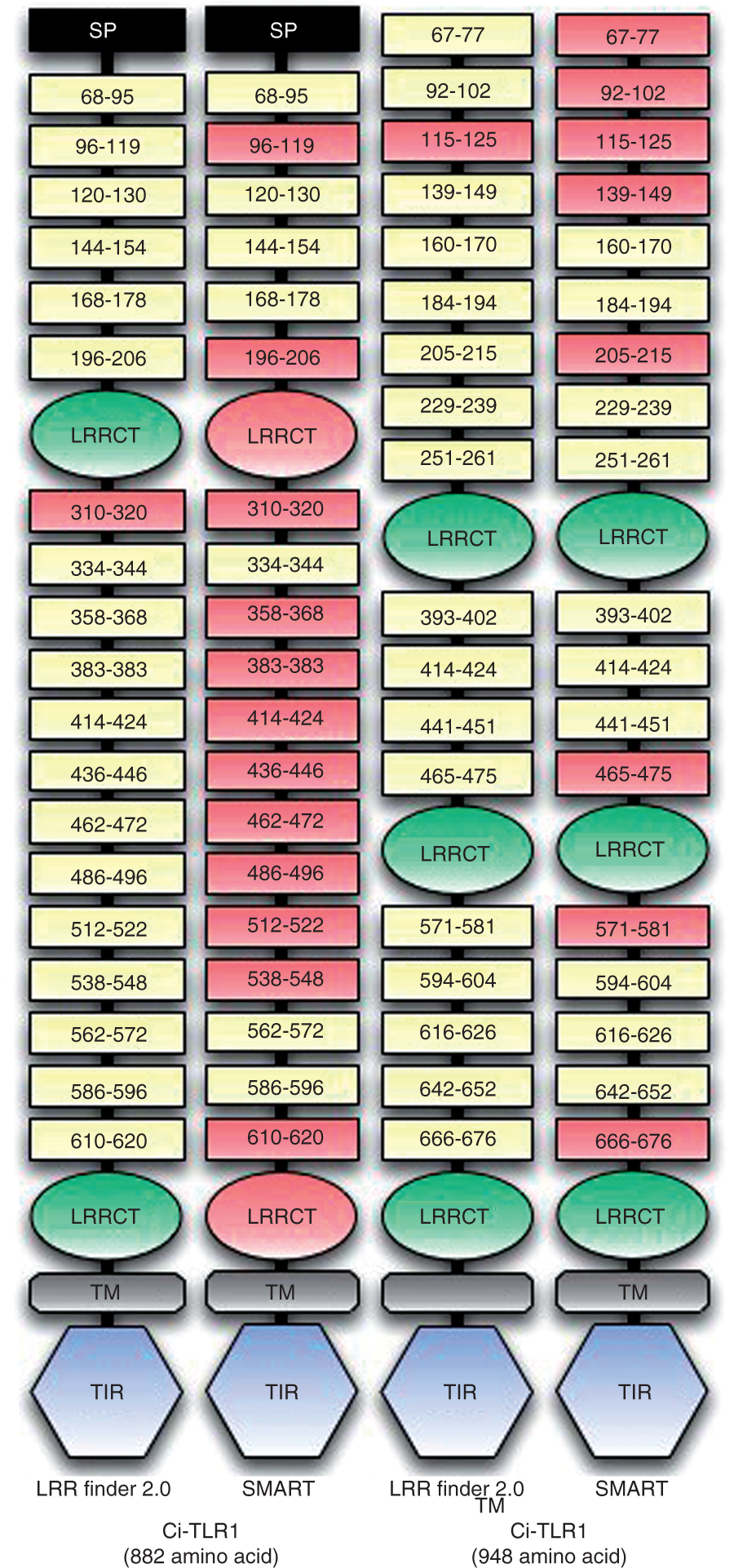
The LRRfinder2.0 webserver provides a user-friendly application for the identification of LRRs, post-translation modification sites, surface accessibility and secondary structure prediction in LRR-containing proteins. LRRfinder2.0 has broad applications, including improving alignments for computational modelling, identifying functionally important residues and scanning novel genomes for LRR-containing proteins which have the potential to be involved in immune-related processes. Further investigation using our dataset could be used to provide insight into the structural variations caused by naturally-occurring indels and, together with available crystal structures of LRR-containing proteins, may provide explanations for the differences in protein-ligand interactions identified in the TLRs of different species.
Availability and requirements
Project home page: www.lrrfinder.com. Operating system(s): Platform independent (web server). Programming language(s): Perl, PHP, JavaScript, Ajax, CSS and HTML. License: No license required. For information about a standalone version, please contact: vofford@rvc.ac.uk
Footnotes
Acknowledgements
We would like to thank Mr S. Thompson for infrastructure support during the development of LRRfinder2.0.
Funding
This work was supported by grant R.9VPR.OFFV of the RVC to DW. The manuscript represents publication number PID_00447 of the RVC.