
Abstract
Organ failure is a severe complication in sepsis for which the pathophysiology remains incompletely understood. Recently, the matri-cellular cysteine-rich, angiogenic induced, 61 (Cyr61/CCN1); connective tissue growth factor (Ctgf/CCN2); and nephroblastoma overexpressed gene (Nov/CCN3) (CCN)-protein family have been attributed organ-protective properties. Their expression is sensitive to mediators of sepsis pathophysiology but a potential role in sepsis remains elusive. To provide an initial assessment, 50 rats were subjected to 18 h of cecal-ligation and puncture or sham operation. Hepatic and pulmonary CCN1 mRNA displayed an average 7.4- and 3.3-fold induction, while its cardiac expression was unchanged. The changes coincided with excessive hepatic and pulmonary inflammatory gene activation and a restricted cardiac inflammation. Furthermore, hepatocytes displayed a dosage-dependent CCN1 mRNA response in vitro, supporting a cytokine-mediated CCN1 regulation in sepsis. CCN2 mRNA was 2.2-fold induced in the liver, while 2.0-fold and 1.4-fold repressed in the heart and lung. Meanwhile, it did not respond to TNF-α exposure in vitro, which indicates different means of regulation than for CCN1. Taken together, this study provides the first evidence for multi-organ regulation of CCN1 and CCN2 in early stages of sepsis, and implies the eruption of inflammatory mediators as a potential mechanism behind the observed CCN1 regulation.
Keywords
Introduction
Sepsis is a complex disease that results from a harmful host response to severe infection. 1 Despite improvements in our understanding of its pathophysiology, mortality remains high. The presence of concurrent organ failure, in particular, is a strong predictor of sepsis-related death. 2 The pathophysiologic mechanisms behind sepsis-induced multiple organ failure (MOF) are, however, still controversial. Current knowledge implies a role of microvascular, and mitochondrial dysfunction, as well as leukocyte-mediated parenchyma disturbances.3–7
The extra cellular matrix (ECM) has received little attention in sepsis research. However, the identification and investigation of a new family of matri-cellular proteins, termed the CCN-family, suggests a potential role in sepsis-induced MOF.8,9 The CCN-family is the acronym derived for the first three family members: cysteine-rich, angiogenic inducer, 61 (Cyr61/CCN1); connective tissue growth factor (Ctgf/CCN2); and nephroblastoma overexpressed gene (Nov/CCN3). The family was later expanded to include three additional members (WNT1 inducible signaling pathway protein 1-3; Wisp1-3/CCN4-6); the proteins are now denoted CCN1-CCN6. 8
The biologic functions of the CCNs contrast with those of classical ECM proteins, as they serve to modulate cellular responses to environmental disturbances rather than sustaining structural roles.8,9 Functioning as autocrine and paracrine multi-functional signal integrators, they support the adhesion, migration and proliferation of endothelial cells, as well as the proliferation and adhesion of vascular smooth muscle cells in vitro and drive angiogenesis in vivo.10–16 The proteins play a potential role also on the endothelial surface, as they act as adhesion substrates for activated platelets and macrophages.17,18 Moreover, they regulate macrophage migration and pro-inflammatory signaling through receptors widely distributed on innate immune cells and promote cytokine and chemokine expression.19,20 Lastly, the CCNs engage in regulation of reactive oxygen species (ROS) accumulation, thereby affecting another pivotal element of inflammation and potentially interfering with vascular tone control.21–25
Furthermore, the expression of the CCN proteins is highly susceptible to cytokine and growth factor expression, hypoxia, hyperoxia, ROS and infections. 23 These features are essential parts of the septic environment, as well as involved in the pathophysiologic background of endothelial, mitochondrial and leukocyte dysfunction which is observed in sepsis-induced MOF.3–5,8,26,27 This provides compelling evidence that the CCN expression might be affected in sepsis and, hence, hints to the potential involvement of CCNs in sepsis and MOF pathophysiology. In fact, renal CCN1 induction has been documented in a mouse model of poly-microbial sepsis. 28 However, the study presented here is the first multi-organ investigation of CCN expression in experimentally-induced sepsis in vivo.
Materials and methods
Animal model
Fifty male Wistar rats (Taconic, Hudson, NY, USA), weighing between 250 and 300 g, were kept under a 12/12 h light/dark cycle with unlimited access to food and water. The animals were randomized to cecal-ligation and puncture (CLP) or sham operation as previously described. 29 Briefly, the cecum was assessed through a midline laparotomy, ligated with 3-0 silk suture and punctured in two places with an 18-G needle. With this injury, approximately 10% mortality was observed within 18 h. The animals received saline (3 ml/100 g) and analgesia (Temgesic 0.05 mg/kg) subcutaneously following surgery, as well as 9 h postoperative. Eighteen hours after surgery a re-laparotomy was preformed and the animals were sacrificed by terminal bleeding from the abdominal aorta, using a 23-G needle and 10-ml syringe. Blood was collected for biochemical analysis and organs (lungs, livers and hearts) were harvested. All animal experiments were approved by the local animal care committee and conducted in accordance with the national animal welfare guidelines.
Biochemical markers
Levels of alanine transaminase, aspartate transaminase and troponin-T were measured in heparin anti-coagulated blood by the Department of Clinical Biochemistry (Oslo University Hospital, Rikshospitalet, Norway), using validated assays for routine patient analysis. Total leukocyte and platelet counts were done on EDTA anti-coagulated blood with assays validated for routine patient analysis. Arterial blood gas analysis was performed on heparin anti-coagulated blood in capillary tubes using an ABL 800 Flex analyzer (Radiometer Medical Aps, Copenhagen, Denmark).
Gene expression analysis
Total RNA was isolated from snap-frozen tissues by means of RNeasy mini kit (Qiagen, Düsseldorf, Germany) according to the manufacturer's instructions. Samples were treated with an RNase free DNase set (Qiagen) to remove genomic contamination. RNA quality was controlled at random using the Experion™ automated electrophoresis system (BioRad Laboratories Inc., Hercules, CA, USA). Reverse transcription was done using a TaqMan RT kit (Applied Biosystems, Foster City, CA, USA).
RT-PCR was run in triplicates using a relative standard curve approach. CCN1-CCN6 and RN18S1 (ribosomal protein S18, internal control) were analyzed using pre-inventoried TaqMan assays according to the manufacturer's instructions (Applied Biosystems). Cytokine and chemokine expressions were analyzed by SYBR Green technology, using 2× Universal master mix (Applied Biosystems), cDNA template, and 300 mM sense and anti-sense primers. SYBR Green primers were designed to span intron junctions. Specificity of the SYBR Green primers was assessed by melting point analysis. All assays were optimized to CT-value between 20 and 30 cycles. The primers utilized to amplify target genes were: TNF-α, forward 5′- ATG GCC CAG ACC CTC ACA CTC A, reverse 5′- CCG CTT GGT GGT TTG CTA CGC; IL-1β, forward 5′- GAC CTG TTC TTT GAG GCT GAC A, reverse 5′- CTC ATC TGG ACA GCC CAA GTC; IL-6, forward 5′- TAG TCC TTC CTA CCC CAA CTT C, reverse 5′- TTG GTC CTT AGC CAC TCC TTC; CXCL1 forward 5′- CAA TGA GCT GCG CTG TCA GT, reverse 5′- TTG AAG TGA ATC CCT GCC ACT; IL-10 forward 5′- AGC TGC GAC GCT GTC ATC GAT, reverse 5′- CAC CTG CTC CAC TGC CTT GCT T.
Western blotting
Snap-frozen tissue samples were gently homogenized in RIPA buffer with protease and phosphatase inhibitors (Thermo Fisher Scientific Inc., Waltham, MA, USA) prior to 30 min incubation on ice. The lysates were centrifuged twice at 10000 g for 10 min to remove insoluble material and the total protein concentration was determined by Bradford assay (BioRad). Proteins from randomly selected animals, with mRNA expression close to the average mean, were separated by 10% SDS-PAGE and transferred to a Hybond-N membrane (Amersham Bioscience, Amersham, UK). Blocking was carried out by incubating membranes with 5% non-fat milk in Tris-buffered saline with 0.05% Tween-20 (TBST) for 1 h at room temperature (20–22°C). Membranes were incubated with primary Abs against mouse-CCN1, mouse-CCN3 (R&D systems, McKinley Place NE, MN, USA), human-CCN2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and β-actin (Cell Signaling technology Inc., Beverly, MA, USA) 18–20 h at 4°C. After washing three times with TBST, the membranes were incubated with a secondary anti-sheep, anti-goat or anti-rabbit Ab (Santa Cruz) for 1 h at room temperature and proteins visualized by LumiGlo (KPL, Inc., Gaithersburg, MD, USA). Displayed images were developed using a Kodak Image station 4000 mm Pro (Kodak, New Haven, CT, USA).
Immunohistochemistry
Paraffin-embedded pulmonary sections (6 µm) were rehydrated and blocked 1 h prior to incubation with purified anti-CCN2, raised as previously described. 30 Subsequently, CCN2 staining was performed with an avidin-biotin-peroxidase system, Vectastain Elite kit (Vector Laboratories, Burlingame, CA, USA), according to the manufacturer's instructions. Visualization was done by 3,3′-diaminobenzine (DAP)-staining (Thermo Fisher) and a hematoxylin counter-stain.
Cell culture
The human hepatocyte carcinoma cell line HepG2 was obtained from the European Collection of Cell Cultures (ECACC, Salisbury, UK). The cells were maintained in EMEM (M2279) (Sigma Aldrich, St. Louis, MO, USA) with 10% FCS,
Statistical analysis
Data distribution was evaluated in inverse normal plots and by means of the Kolmogorov-Smirnov and Shapiro-Wilk tests. Normally-distributed data were analyzed by one-way ANOVA with Bonferroni or Thamhane's T2 post-test or by Students t-test. When normal distributions were violated, analyses were done by Mann-Whitney U test or Kruskal-Wallis multiple comparison test, followed by Mann-Whitney U post-test with Bonferroni adjustment. Differences were considered significant at the level of P < 0.05.
Results
CLP animals suffer from early sepsis with inflammatory organ impairment
Animal characteristics.
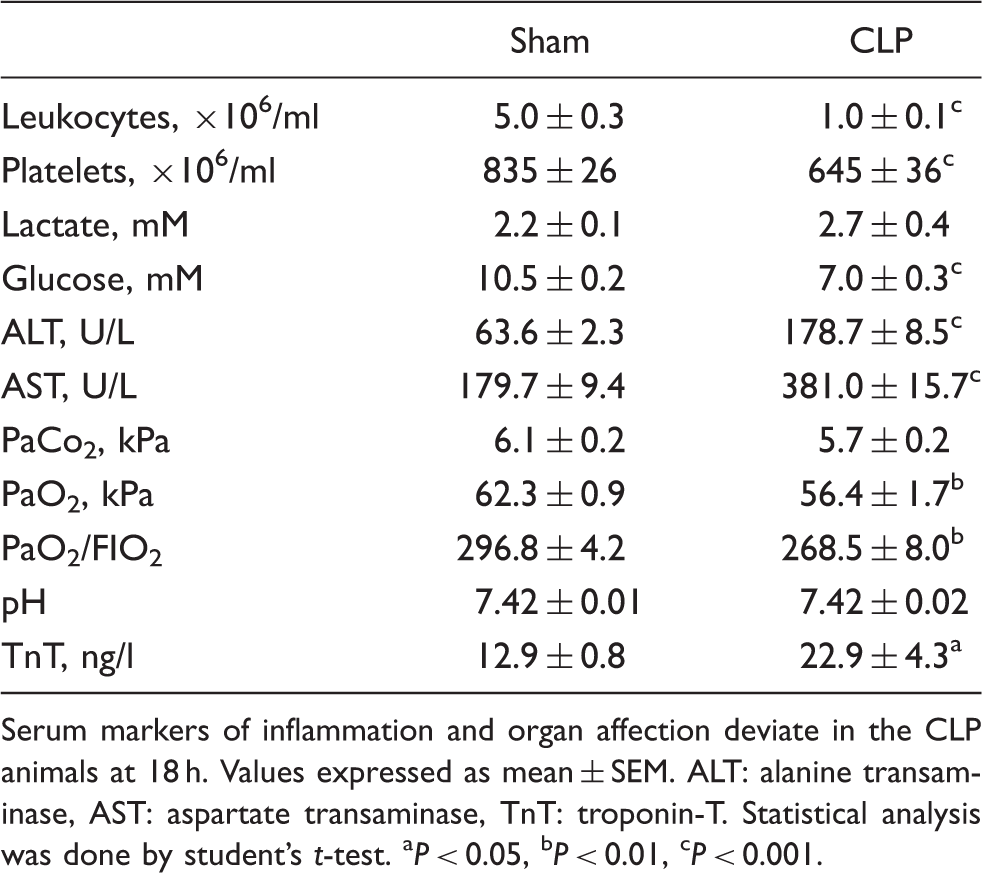
Serum markers of inflammation and organ affection deviate in the CLP animals at 18 h. Values expressed as mean ± SEM. ALT: alanine transaminase, AST: aspartate transaminase, TnT: troponin-T. Statistical analysis was done by student's t-test. aP < 0.05, bP < 0.01, cP < 0.001.
Hepatic alanine transaminase and aspartate transaminase were significantly elevated, consistent with inflammatory liver injury. Severely elevated troponines is indicative of sepsis-induced cardiac failure;
31
however, this marker displayed only a minor increase among the CLP animals. The blood gas analysis revealed an unchanged pH value and PaCo2 among the CLP animals, while PaO2 and, hence, the PaO2/FIO2 ratio were significantly decreased (Table 1). To further address this pulmonary inflammatory impairment, selected tissue sections were evaluated by means of light microscopy. No visible signs of inflammatory cell infiltration or histological damage were, however, observed (Figure 1).
Lung morphology and CCN2 protein expression. Hematoxylin and eosin stained lung sections from cecal ligation and puncture (CLP) and sham operated animals at 18 h (A). CCN2 immunohistochemistry (B–D) were done to identify involved cell types. The alveolar septa (B) showed no protein expression, while intense signals were observed in the endothelial lining of the larger vessels (C, arrow 1), and in the extra cellular matrix (D, arrow 3), as well as around the minor airways (D, arrow 2) in the central lung parts.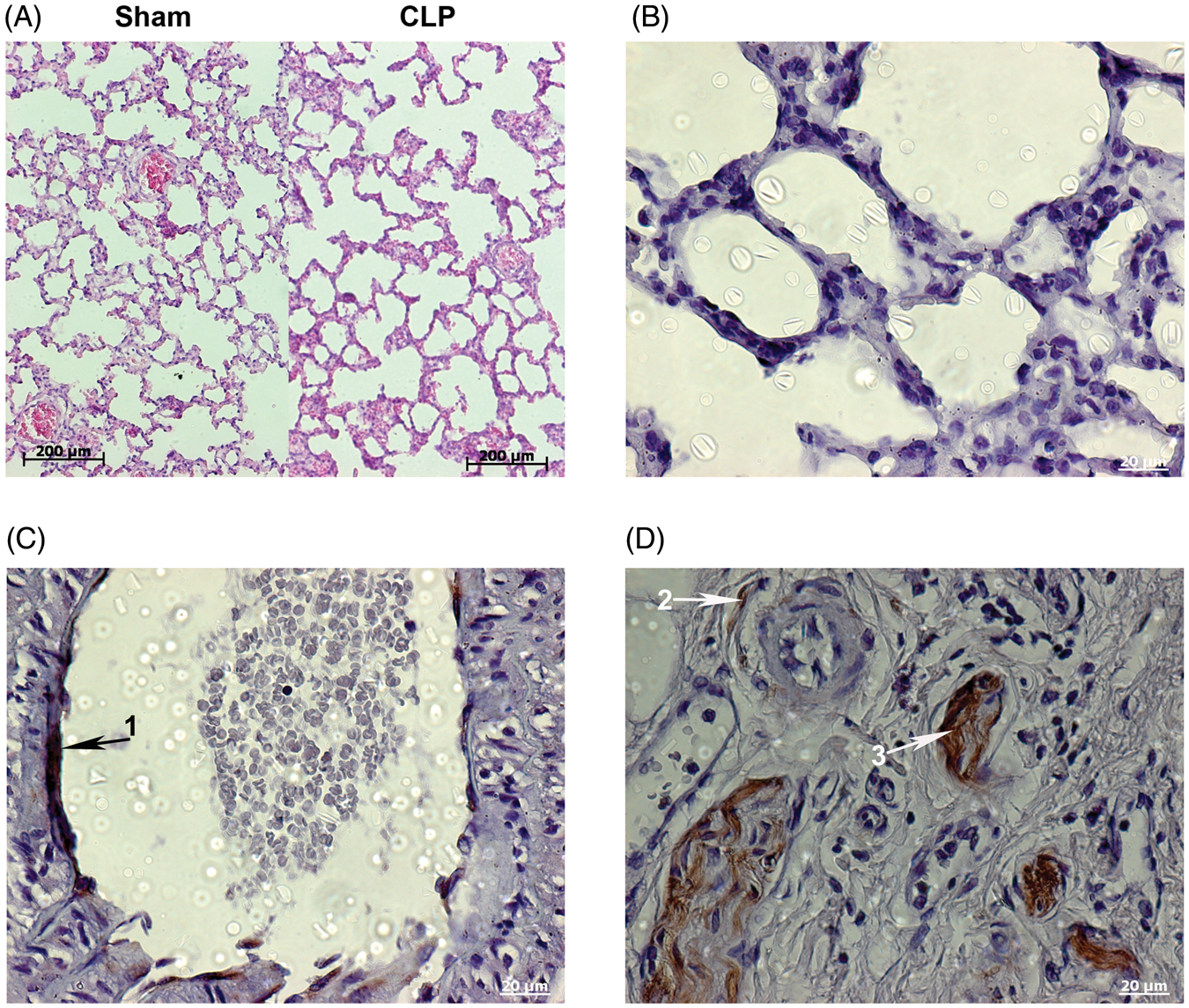
CLP animals display tissue-specific patterns of inflammation
Inflammatory gene expression.
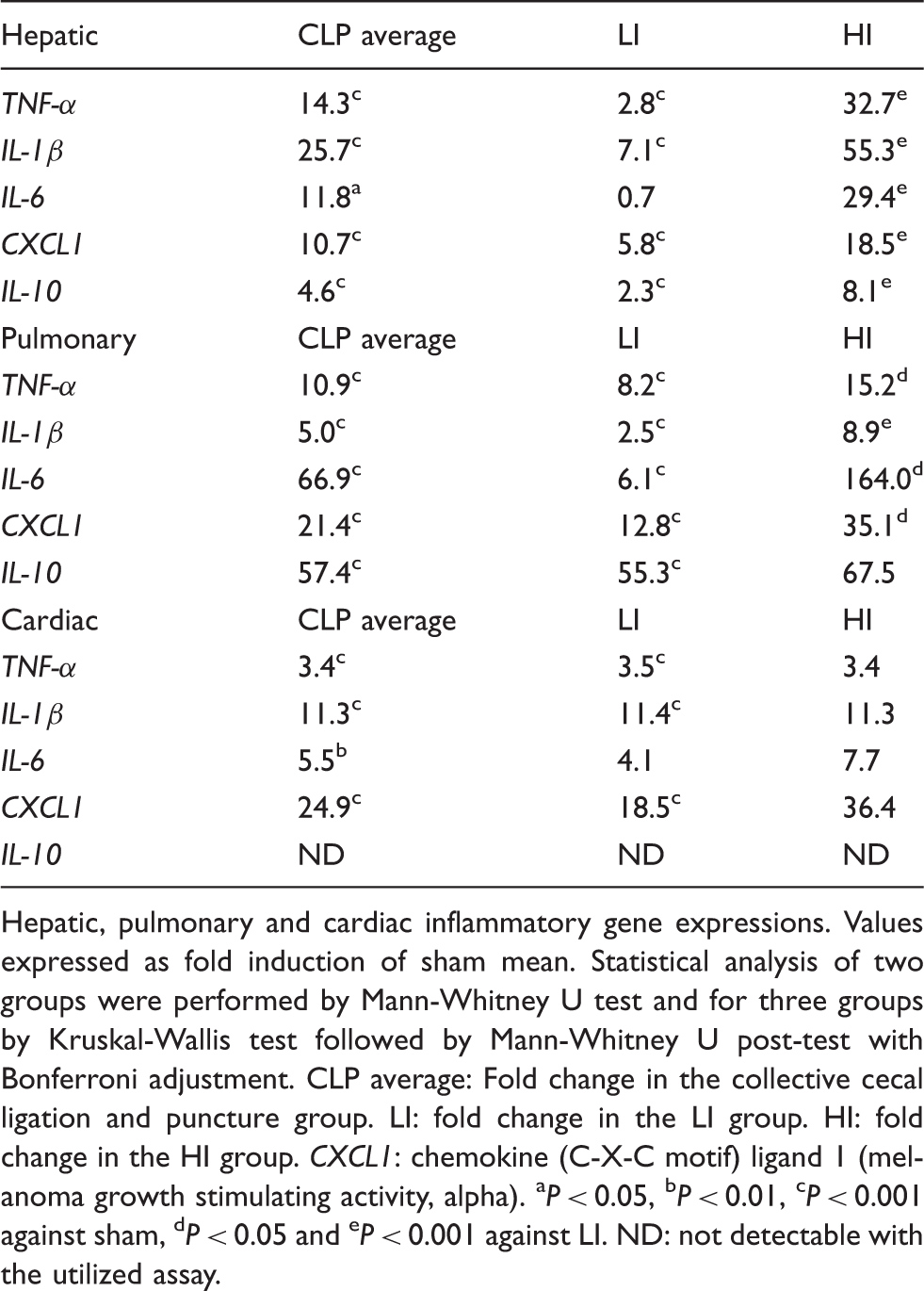
Hepatic, pulmonary and cardiac inflammatory gene expressions. Values expressed as fold induction of sham mean. Statistical analysis of two groups were performed by Mann-Whitney U test and for three groups by Kruskal-Wallis test followed by Mann-Whitney U post-test with Bonferroni adjustment. CLP average: Fold change in the collective cecal ligation and puncture group. LI: fold change in the LI group. HI: fold change in the HI group. CXCL1: chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating activity, alpha). aP < 0.05, bP < 0.01, cP < 0.001 against sham, dP < 0.05 and eP < 0.001 against LI. ND: not detectable with the utilized assay.
Average expression of cardiac and hepatic pro-inflammatory genes revealed similar patterns. In both tissues, IL-1β was the most induced gene, while the TNF-α and IL-6 increases were alike. Meanwhile, average hepatic induction of these genes was excessive and cardiac expression only limited, clearly showing differences in the extent of pro-inflammatory activation. Indicative of differences in the pro-inflammatory process, the average pulmonary mRNA expression manifested a vastly induced IL-6, followed by medium TNF-α, and minimum IL-1β expression.
Peak expression of CXCL1 was observed in the heart, closely followed by lungs, while being less in the liver. Furthermore, pulmonary IL-10 expression was highly induced, while hepatic expression was restricted and cardiac expression non-detectable. This picture clearly demonstrates tissue specific patterns of inflammatory activation.
The CLP animals divide in high and low inflammation responders
Based on the inflammatory gene activation, the CLP animals were divided into two sub-groups (division criterion was a TNF-α fold-induction of <5 or >8). One, termed ‘high inflammation’ (HI), presented with excessive mRNA induction while the other, termed ‘low inflammation’ (LI) had only modest gene activation. This division was evident for all hepatic and pulmonary inflammatory genes tested, except for pulmonary IL-10, but not observed for any of the genes in the heart (Table 2).
There was no correlation between the HI/LI groups and changes in measured markers of organ function or hematologic parameters (data not shown).
CCNs are regulated in early stages of sepsis-induced organ impairment
CCN1-CCN6 mRNA expressions in early sepsis.
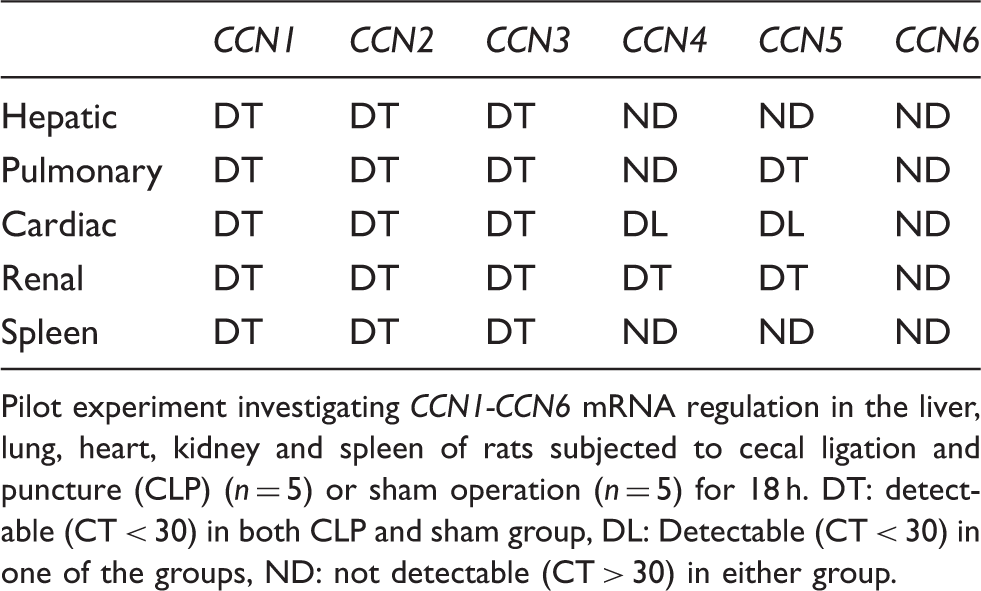
Pilot experiment investigating CCN1-CCN6 mRNA regulation in the liver, lung, heart, kidney and spleen of rats subjected to cecal ligation and puncture (CLP) (n = 5) or sham operation (n = 5) for 18 h. DT: detectable (CT < 30) in both CLP and sham group, DL: Detectable (CT < 30) in one of the groups, ND: not detectable (CT > 30) in either group.
Analysis of hepatic CCN1-CCN3 mRNA expression (Figure 2A–C) revealed an average 7.4-fold CCN1, a 2.1-fold CCN2 and a 2.4-fold CCN3 mRNA induction in CLP animals at 18 h.
Hepatic CCN1, CCN2, and CCN3 mRNA expressions are induced in early sepsis. Gene expressions were analyzed in whole liver homogenates by means of RT-PCR and are expressed as fold induction of sham mean. Individual animals represented by dots amid, average as horizontal lines (A) CCN1, (B) CCN2, and (C) CCN3. LI-CLP: low Inflammation- cecal ligation and puncture (CLP) group, HI-CLP: high Inflammation-CLP group. Correlation of CCN1 and TNF-α mRNA expressions in the CLP animals (D). Animals in the LI group (•) scatter in the lower left corner, while HI animals (+) spread towards the upper right. Statistics were done by Kruskal-Wallis test, followed by Mann-Whitney U post-test with Bonferroni correction. Correlations (D) were calculated by Spearman rho. Results were considered significant at alpha level (or adjusted alpha level) *P < 0.05, **P < 0.01, ***P < 0.001.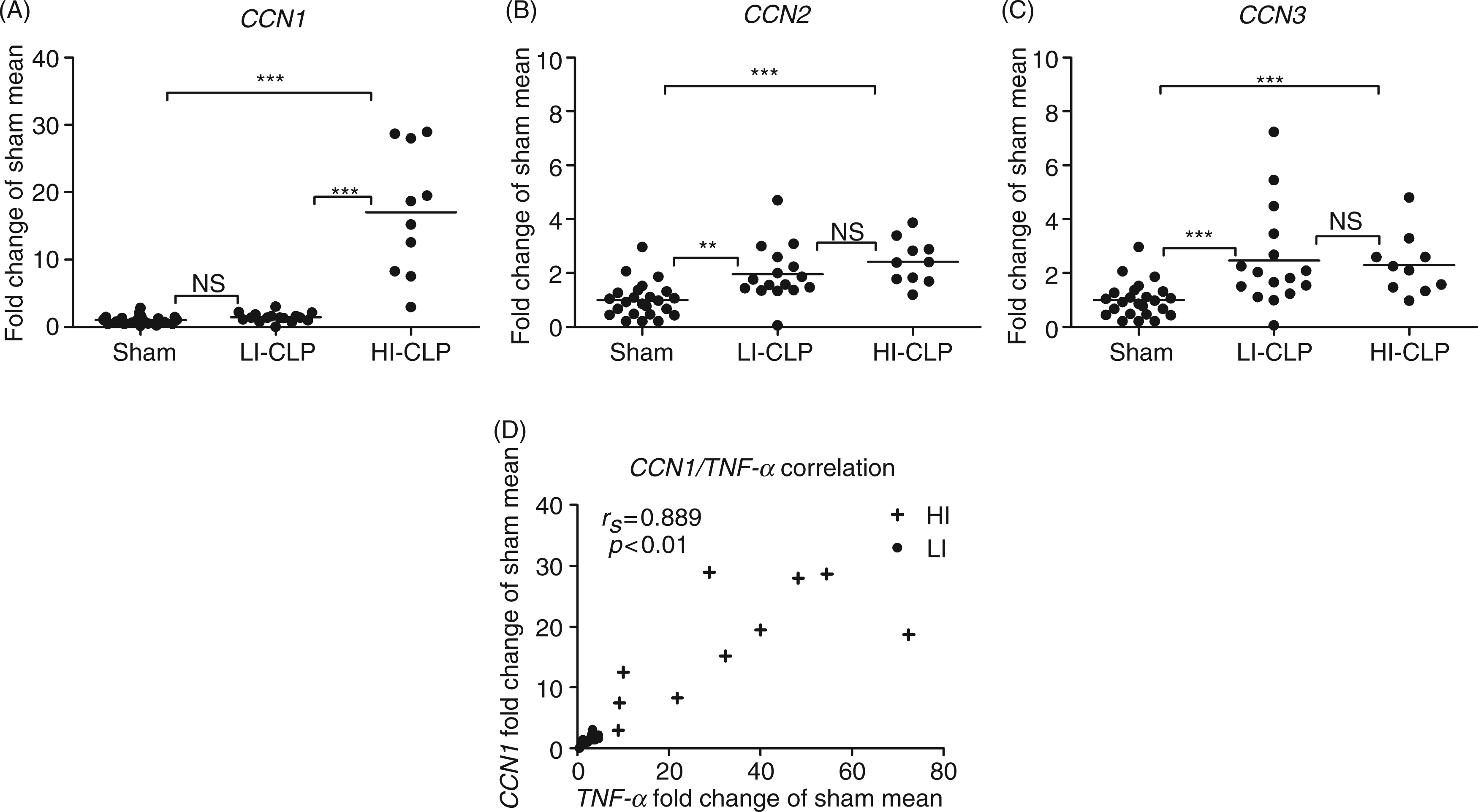
Interestingly, the LI/HI groups defined by inflammatory activation were also uncovered for CCN1 mRNA expression, which was not reflected in CCN2 or CCN3 mRNA patterns. The CCN1 mRNA expression in the HI group exhibited a 17-fold induction while the mRNA expression in the LI group remained at the level of sham. Statistical analysis confirmed strong correlations between CCN1 mRNA expression and the gene expression of TNF-α (0.889, P < 0.01) (Figure 2D), IL-1β (0.811, P < 0.01), IL-6 (0.813, P < 0.01), CXCL1 (0.711, P < 0.01), and IL-10 (0.868, P < 0.01). Contradicting this, CCN2 mRNA presented with weak correlations to TNF-α (0.397, P < 0.05) and IL-10 (0.400, P < 0.05) only, and CCN3 mRNA with IL-6 (0.424, P < 0.05).
Western-blot analysis confirmed CCN2 protein regulation, but failed to detect changes in CCN1 and CCN3 protein levels (data not shown).
Pulmonary CCN expressions (Figure 3A–C) revealed an average 3.3-fold CCN1 mRNA induction, a twofold CCN2 mRNA reduction and a 2.2-fold CCN3 mRNA increase. The HI/LI distribution was also maintained for CCN1 mRNA expression in this tissue. The HI group exhibited a 4.8-fold mRNA induction and, in contrast to the hepatic situation, the LI group exhibited a 2.3-fold mRNA induction. As for the liver, neither CCN2 nor CCN3 mRNA expression reflected the HI/LI pattern. Meanwhile, evident correlations between CCN1 mRNA and gene expression of tested inflammatory markers were only observed for IL-1β (0.537, P < 0.01) and IL-6 (0.594, P < 0.01) in this tissue. In contrast to hepatic CCN2 mRNA induction, the pulmonary CCN2 gene was repressed, manifesting tissue-specific CCN2 mRNA responses (Figures 2B and 3B).
Pulmonary CCN1, CCN2 and CCN3 expressions. CCN1 (A), CCN2 (B) and CCN3 (C) gene expressions were analyzed by means of RT-PCR and are expressed as fold change of sham mean. Individual animals represented by dots amid average as horizontal lines. LI-CLP and HI-CLP indicates LI and HI cecal ligation and puncture (CLP) animals. Regulation on the protein level was confirmed by Western-blot (D) in randomly selected animals, with gene expression at the level of the average mean. CCN1-specific bands corresponding to the glycosylated full-length CCN1 (50 ku) and a un-glycosylated form at 40 ku was observed. Regulation was detected at the 40 ku band as shown. Statistical analyses were done by Kruskal-Wallis test, followed by Mann-Whitney U post-test with Bonferroni correction. Results were considered significant at the alpha level (or adjusted alpha level): *P < 0.05, **P < 0.01, ***P < 0.001.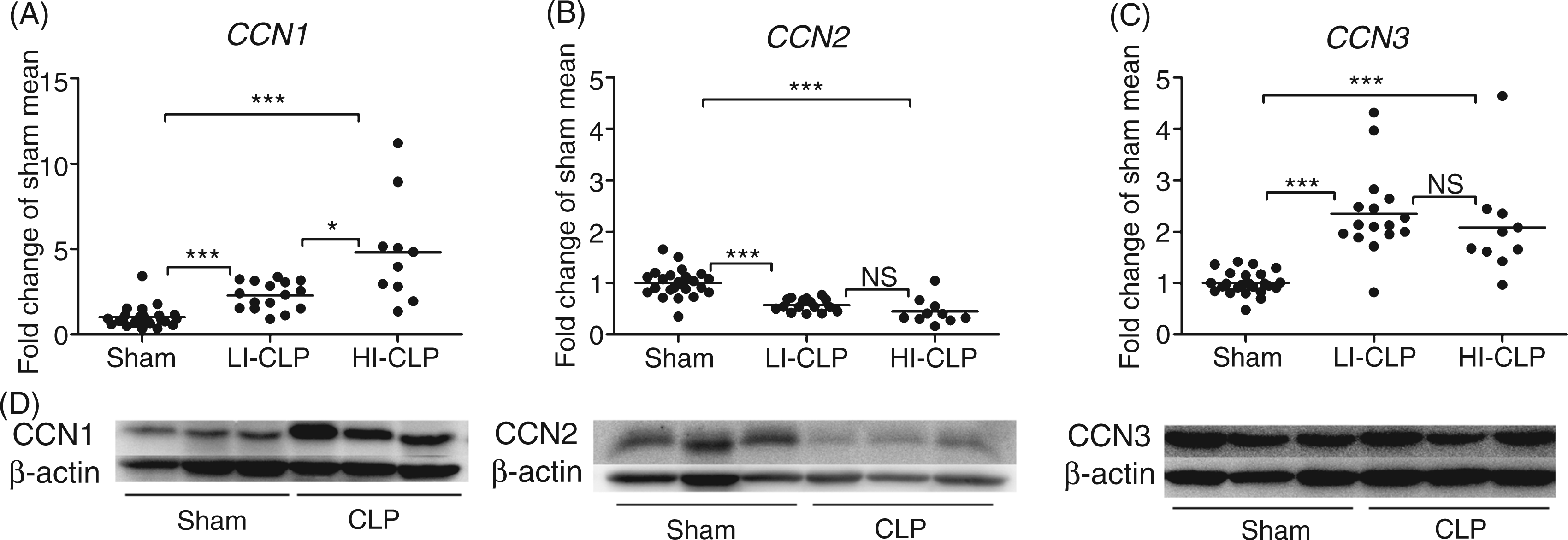
Pulmonary CCN1 and CCN2 protein regulation was confirmed by means of Western blot, while CCN3 protein was unchanged (Figure 3D). To identify the potentially involved and affected cell types, CCN2 protein expression was evaluated by immunohistochemistry (Figure 1B–D). Staining was seen around the minor bronchia (arrow 2), in the ECM (arrow 3) and the endothelial lining of larger vessels (arrow 1) in the central lung parts. Furthermore, protein expression was observed in vascular and airway smooth muscle cells (not shown). In contrast, no staining was observed in the distal alveolar part of the lungs (Figure 1B).
Cardiac CCN1 and CCN3 expression were unchanged, while CCN2 gene expression was repressed by 30% (P < 0.05, data not shown).
Recombinant TNF-α regulates hepatocyte CCN1 but not CCN2 or CCN3 mRNA levels
With the aim to further address the molecular mechanisms behind the observed hepatic CCN1–CCN3 mRNA regulation, and to provide evidence for CCN regulation in non-immune cells, as well as investigating its relevance in a human cell-type, in vitro studies were conducted in HepG2 cells (Figure 4). The cells were stimulated with increasing dosages of recombinant TNF-α for 24 h and analyzed for CCN1-CCN3 mRNA expression.
Hepatocyte CCN1 expression responds to TNF-α exposure. HepG2 cells were stimulated 24 h with increasing dosages of TNF-α (1, 10 and 100 ng/ml) and CCN1 (A), and CCN2 (B) gene expression profiles were analyzed by means of RT-PCR. A pulse-chase experiment was conducted to confirm the specificity of CCN1 induction by TNF-α. The cells were stimulated for 1 h with TNF-α and CCN1 expression tested after 1, 6 and 24 h. Data are obtained from three independent experiments conducted in triplicate. Values standardized to control mean and expressed as mean ± SEM. Statistical tests were performed by one-way ANOVA with Bonferroni post-test. Results were considered significant at the level of: *P < 0.05, **P < 0.01, ***P < 0.001.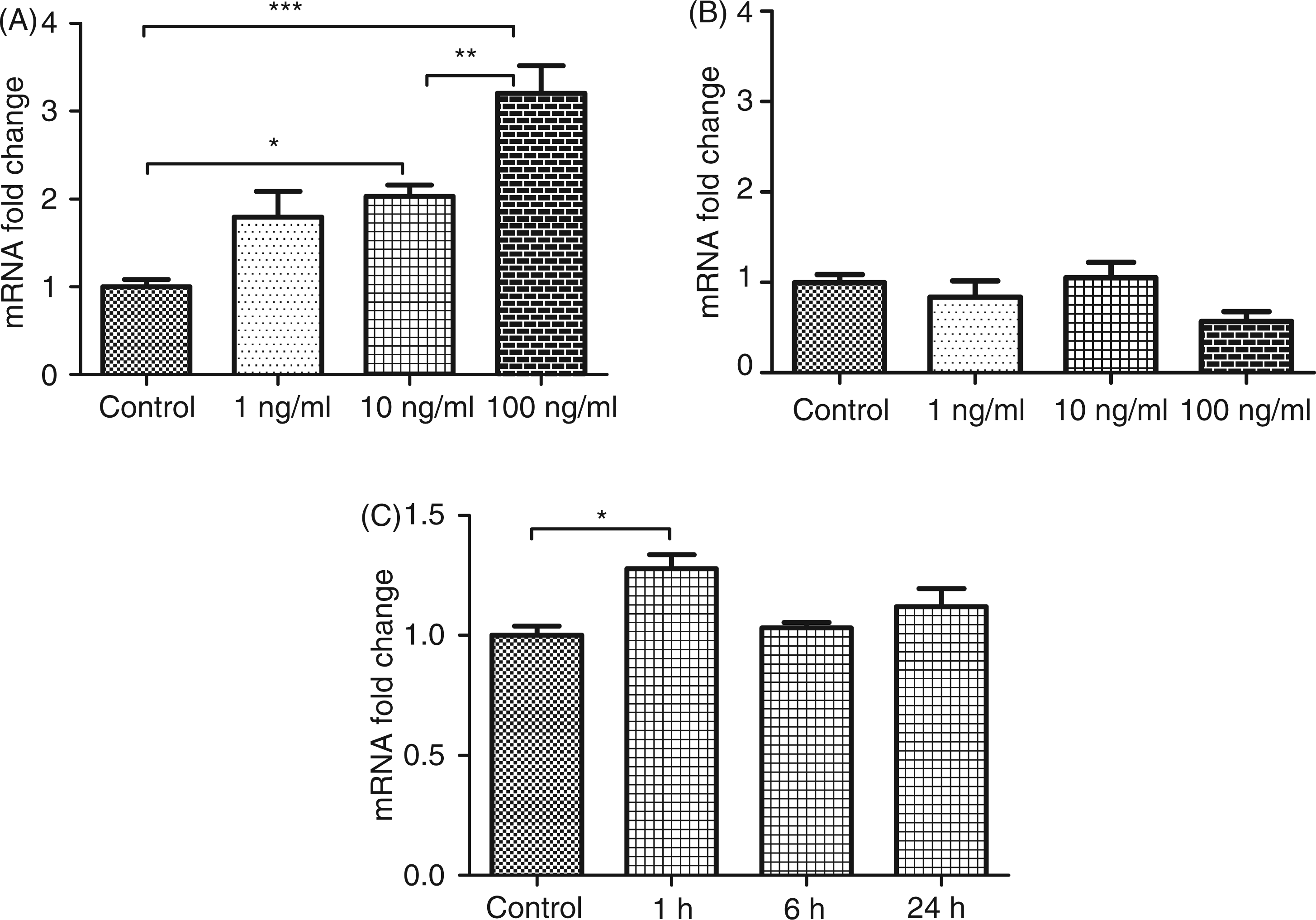
CCN1 mRNA expression (Figure 4A) showed a clear, dose-dependent response while CCN2 mRNA expression (Figure 4B) was unaffected. CCN3 persisted below the detection limit of the assay system used (not shown). To investigate the potential unspecific effects of TNF-α stimulation on cell metabolism a pulse-chase experiment was conducted (Figure 4C). The cells were stimulated with 10 ng of TNF-α for 1 h and CCN1 mRNA expression was analyzed consecutively for 24 h. At 1 h, CCN1 was significantly induced while returning to baseline after 6 h.
Discussion
The present study provides novel evidence for simultaneous regulation of the matri-cellular CCNs in different organs as a response to sepsis-induced impairment. It provides clear evidence for a connection between CCN1 regulation and the expression of both pro-inflammatory and anti-inflammatory cytokines, as well as chemokines. The data indicate that this connection is mediated through cytokines and chemokines provoking CCN1 responses. The experiments also demonstrate CCN2 to be regulated in a tissue-specific manner that is adverse in some organs. Furthermore, it also implies that CCN3 is regulated. The underlying mechanisms of CCN2 and CCN3 regulation seem, however, more complicated than for CCN1. Lastly, our pilot study identified all members of the CCN-family, except for CCN6, as potentially affected in the early stages of sepsis-induced organ impairment.
The gene expression profiles of cytokines and chemokines were analyzed to delineate the environment in which we studied CCN expression. In accordance with the notion of a compartmentalized inflammatory response in sepsis,32,33 tissue-specific profiles were observed in the three organs. However, isolated evaluation of the pro-inflammatory genes revealed hepatic and cardiac profiles to be similar, though different in magnitude, while the pulmonary profile was disparate. Similar observations have been made also by others34,35 and could indicate pulmonary inflammation to be different from the inflammation in other organs. Moreover, the septic rats divided into LI and HI responders, which is likely to reflect different stages of disease progression or inter-individual differences in susceptibility to the infection.
Interestingly, these inflammatory-based LI and HI groups were also seen in hepatic and pulmonary CCN1 mRNA expression. Simultaneously, CCN1 mRNA regulation was not detected in the heart which exhibited limited inflammation. Furthermore, the mRNA levels correlated strongly to tested inflammatory markers in the liver. These observations provide strong evidence for a biological dependency between CCN1 and multiple cyto- and chemokines in vivo. In the lungs, however, the correlations were evident only for IL-6 and IL-1β. However, this may well be explained by a relatively small difference between CCN1 mRNA induction in the pulmonary LI and HI groups. Conversely, the hepatic LI group elicited CCN1 expression at the level of sham thereby presenting a more substantial difference between the LI and HI groups. That CCN1 mRNA was induced in the lung LI group but not in the liver LI group, was likely caused by the distinct nature of the inflammatory response or tissue differences of CCN1 susceptibility.
The causal connection between CCN1 and cytokine/chemokine expressions, as well as the cellular source of the CCN1 responses, remains to be identified. Human peripheral blood monocytes demonstrate increased CCN1 mRNA expression after challenge with endotoxin in vitro. 36 This, in conjunction with CCN1 obeying the LI/HI patterns and displaying close associations to inflammatory markers, could indicate the observed CCN1 response to originate from inflammatory cell types. However, several lines of evidence also support expression of the autocrine and paracrine acting CCNs from other cells-types, both stromal and parenchymal ones.8,9 Our studies clearly demonstrate a CCN1 mRNA response in hepatocytes upon TNF-α exposure. This implies that the observed CCN1 regulation in vivo might partially emerge from parenchyma cells, and that the parenchymal CCN1 regulation can be a consequence of environmental cytokine/chemokine exposure. Hereby, the data implicate a role for CCN1 in the parenchymal response to sepsis.
The observed CCN2 regulation appears to be more complicated. The mRNA levels correlated with few inflammatory markers, the LI/HI division was absent and TNF-α did not elicit a CCN2 mRNA response in the hepatocytes. This merely excludes cytokines and chemokines as main regulators of CCN2 in the utilized model. Despite the elusiveness of regulatory factors, an interesting tissue-specific response was observed for CCN2. Such tissue-specific CCN2 responses have also been observed previously.37,38 In these studies, TNF-α inhibited CCN2 expression in skin fibroblasts and conversely, induced CCN2 in mesangial cells in vitro. As such, the observed tissue-specific differences in CCN2 expression are likely to occur through similar mechanisms and this observation is encouraging further investigations.
The regulation of CCN1 and CCN2 on the protein level revealed a complex pattern. Our experiments uncovered CCN2 but not CCN1 protein regulation in the liver, while both proteins were regulated in the lungs. With a few exceptions, CCN proteins are regulated at the transcriptional level. 39 Moreover, in vitro studies have demonstrated that CCN2 responds more instantly to TNF-α stimulation than CCN1 does. 40 This indicates that the lack of CCN1 protein regulation in the liver is likely a result of a premature time point of analysis. The regulation of CCN1 in one organ and not in the other at 18 h could implicate tissue-specific differences in CCN1 sensitivity to environmental perturbations. Alternatively, it could be caused by tissue-specific differences in inflammatory profiles.
As the functional properties of the CCN proteins are diverse, current knowledge implicates potentially beneficial, as well as adverse, effects of these proteins in sepsis and MOF. CCN1 protects pulmonary epithelial cells from hyperoxia and nutrient deprivation which, in addition to its pro-angiogenic properties, implies a protective role in sepsis.11,41 Furthermore, a recent study demonstrated cardio-protective properties of CCN1 over-expression in a mouse model of infectious myocarditis, promoting an organ-preserving role of CCN1. 42 CCN2 is also a pro-angiogenic factor, processing cardio-protective capabilities. 43 As such, the observed pulmonary and cardiac CCN2 suppression could be part of the vascular damage observed in sepsis.3,5,24,44 Meanwhile, neonatal mice, over-expressing CCN2 in type-II epithelial cells, have recently been shown to suffer massive macrophage and neutrophil infiltration of the alveolar space. 45 Hence, the down-regulation of CCN2 may well represent a protective response.
In contrast to these potentially beneficial effects, the CCN proteins exert functions that may be detrimental in sepsis. The support of platelet adhesion to endothelial cells hints to involvement in intravascular clot formation and micro-vascular dysfunction. 17 Moreover, macrophages adhere to distinct CCNs on endothelial cells and some of the proteins induce macrophage pro-inflammatory signaling and chemotaxis.18–20,46 Given the critical role of tissue infiltration with dysfunctional leukocytes, this promotes possible harmful effects of CCN proteins.23,26,27
The current study is the first multi-organ description of CCN proteins in sepsis-induced organ-impairment. It provides evidence for CCN1 regulation in early stages of sepsis and implies the eruption of cytokines/chemokines as a potential mechanism. Evidence is also presented for tissue-specific CCN2 regulation and for uniform CCN3 mRNA regulation but seemingly through different mechanisms than for CCN1. Furthermore, the study indicates a potential regulation of CCN4 and CCN5 in sepsis. However, the diversity of CCN functions necessitates functional studies to assess the pathophysiologic impact of the presented data.
Conclusion
The current study is the first multi-organ investigation of CCNs in early sepsis. It provides evidence that CCN1 and CCN2 are regulated in rodents with CLP-induced sepsis. Furthermore, a close association between cytokine and CCN1 gene expression was observed, and evidence was presented for TNF-α-induced CCN1 mRNA expression in vitro. The study is encouraging for functional investigations on the role of CCN proteins in sepsis and sepsis-induced multiple organ dysfunction.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Acknowledgements
The authors sincerely thank Dr Wynne Jones for his comments and corrections to this article and Professor Finn P. Reinholt for his expert pathology assistance.
Conflict of interests
The authors declare that there are no conflicts of interest.