
Abstract
The vascular endothelium is integrally involved in the host response to infection and in organ failure during acute inflammatory disorders such as sepsis. Gram-negative and Gram-positive bacterial lipoproteins circulate in sepsis and can directly activate the endothelium by binding to endothelial cell (EC) TLR2. In this report, we perform the most comprehensive analysis to date of the immune-related genes regulated after activation of endothelial TLR2 by bacterial di- and triacylated lipopeptides. We found that TLR2 activation specifically induces the expression of the genes IL-6, IL-8, CSF2, CSF3, ICAM1 and SELE by human umbilical vein ECs and human lung microvascular ECs. These proteins participate in neutrophil recruitment, adherence and activation at sites of inflammation. Significantly, our studies demonstrate that TLR2-mediated EC responses are specifically geared towards recruitment, activation, and survival of neutrophils and not mononuclear leukocytes, that ECs do not require priming by other inflammatory stimuli to respond to bacterial lipopeptides and, unlike mononuclear leukocytes, TLR2 agonists do not induce ECs to secrete TNF-α. This study suggests that endothelial TLR2 may be an important regulator of neutrophil trafficking to sites of infection in general, and that direct activation of lung endothelial TLR2 may contribute to acute lung injury during sepsis.
Introduction
Although endothelial cells (ECs) are not classically viewed as central regulators of immune responses, they do express receptors and contain signaling pathways that are involved in inflammation. As the endothelium is in continuous contact with circulating blood, it is uniquely situated to contribute to the host’s early response to a disseminated infection or to inflammatory disorders, such as sepsis. Additionally, systemic activation of the endothelium at sites remote from the source of the infection likely contributes to organ failure during sepsis. 1 – 3 The endothelium actively participates in processes that facilitate the localization of leukocytes and other cells to sites of infection and injury, which ultimately lead to the clearance of infectious agents and the healing of damaged tissues. 4 – 6
Vascular ECs express pattern recognition receptors (PRRs) such as TLRs and also express receptors for host inflammatory mediators, such as TNF-α. Once activated, these receptors stimulate ECs to secrete cytokines and chemokines and up-regulate adhesion molecules that promote the targeting, activation and adherence of leukocytes at sites of endothelial inflammation.6,7 TLRs are expressed at the cell surface or in intracellular compartments in a variety of cell types and are activated through the binding of conserved pathogen-associated molecular patterns (PAMPs), such as dsRNA, ssRNA, flagellin, LPS, CpG-DNA and lipoproteins, or endogenous damage-associated molecular patterns (DAMPs), such as mitochondrial DNA or HMGB1, that are released by microbes and injured host cells, respectively.5,8– 10 TLR2 recognizes a diverse set of microbial lipopeptides and lipoproteins that are present on Gram-positive and Gram-negative bacteria. 11 TLR2 forms heterodimers with either TLR1 to bind to triacylated lipoproteins (e.g. peptidoglycan-associated lipoprotein (PAL) and the synthetic peptide agonist Pam3Cys) or with TLR6 to bind diacylated lipoproteins (e.g. macrophage-activating lipoprotein 2kDa (MALP-2) and the synthetic agonist fibroblast-stimulating lipopeptide (FSL-1)). 12 – 16 The majority of research thus far has focused on the biological function of TLR2 expressed in classic immune cells. However, several groups, including our own, have demonstrated that low basal expression of TLR2 is present on the EC surface and many agonists have been found to induce up-regulation of TLR2 in ECs, including histamine, HMGB1, Pam3Cys, MALP-2, TNF-α, IFN-γ, LPS, and IL-1β. 17 – 23
The vascular endothelium actively participates in the recruitment of leukocytes to sites of inflammation. During microbial infection and sterile injury, neutrophils are the first leukocytes to accumulate at these sites.24,25 They eliminate pathogens or necrotic cellular debris through phagocytosis or through the generation of neutrophil extracellular traps (NETs), which directly bind, kill, and prevent the spread of invading microorganisms. 26 Another fundamental function of neutrophils is to promote wound repair; however, the relatively indiscriminate nature of their activity can also mediate collateral tissue damage. 27 Additionally, neutrophils can release chemokines and induce ECs to express proteins that recruit other leukocyte cell types, such as monocytes/macrophages and dendritic cells (DCs).24,28
Upon release from ECs, chemokines such as IL-8 and CCL2 form intravascular gradients along the endothelium near the site of inflammation that directly recruit leukocytes to the site of infection or injury.29,30 In addition to acting as chemoattractants, these molecules also activate signaling pathways in leukocytes that lead to higher affinity conformational changes in β2- and α4-containing integrins. 31 – 33 At the site of EC activation, P- and E-selectin mediate leukocyte capture and rolling through relatively weak associations with molecules such as P-selectin glycoprotein 1 (PSGL1) and L-selectin present on the leukocyte cell surface. These associations also increase integrin affinity for its cognate adhesion molecule receptor, such as VCAM-1 or ICAM-1, to facilitate a more firm attachment of leukocytes and their eventual arrest on the vascular endothelium. 34 – 36 After their arrest, leukocytes migrate out of the vasculature to sites of infection and inflammation using para- or transcellular routes. 37
We have previously reported that treatment of ECs with the naturally-occurring TLR2 agonist PAL increases the expression levels of IL-6, IL-8, and E-selectin, suggesting that the endothelium plays a direct role in the inflammatory process, and can recruit leukocytes as a result of bacterial infection. 20 In the current report, we extend these observations by utilizing a human immune array to obtain a more comprehensive list of immune-related genes whose regulation is dependent on TLR2 activation in ECs. Out of 92 genes analyzed, we identified six genes that were robustly induced by Pam3Cys treatment of HUVEC: IL-6, IL-8, CSF2, CSF3, ICAM1, and SELE. Using both Pam3Cys and FSL-1, we show that the gene induction correlates with a TLR2-dependent increase in expression of the mature proteins at the EC cell surface or in the extracellular supernatant. We also provide evidence that these same six proteins are up-regulated after TLR2 activation in human lung microvascular ECs (HMVEC-L). Interestingly, these proteins are intimately involved in neutrophil proliferation, differentiation, survival, trafficking and activation, suggesting that the primary role of TLR2-dependent activation of the vascular endothelium is to recruit and activate neutrophils during infection.
Materials and methods
Cell lines
Human ECs were incubated at 37°C under humidified 5% CO2. HUVEC (passage 2–6) and human lung microvascular endothelial cells (HMVEC-L; passage 4–6) were purchased from Lonza (Walkersville, MD, USA). HMVEC-L were prepared from both male and female cadaver donors. HUVEC were grown in EGM-2 and HMVEC-L were grown in EGM-2MV (Lonza). EGM was supplemented with 2% FCS. The monocytic cell line THP-1 was grown and maintained in RPMI 1640 supplemented with 10% FBS,
siRNA transfection of HUVEC
TLR2 siRNA (D-001810-10-05), control siRNA (L-005120-01-0005), siRNA 5× Buffer (B-002000-UB-100), and DharmaFECT 4 Transfection Reagent (T-2004-04) were all purchased from Thermo Scientific (Rockford, IL, USA), and used in accordance with the manufacturers suggested protocol for HUVEC transfections. mRNA and protein cell lysates (see below) were prepared either 48 or 72 h post-siRNA transfection, respectively. Cell culture supernatants were collected 68 h post-siRNA transfection (48 h + 20 h agonist treatment). We confirmed TLR2 knockdown by siRNA using Western blots at 72 h post siRNA transfection so that it would coincide with the time point at which the supernatants were collected in the siRNA studies in Figure 3.
Immunoblots
HUVEC were lysed with PLC-Lysis Buffer [50 mM Hepes, pH 7.5; 150 mM sodium chloride; 10% glycerol; 1% triton-X 100; 1.5 mM MgCl2; 1 mM EGTA; 100 mM sodium fluoride; 0.5 mM sodium vanadate; plus protease inhibitor cocktail (P8340, Sigma)] and protein concentrations of the lysates were estimated using the RCDC Protein Assay kit (500-0120; Bio-Rad, Hercules, CA, USA). Total proteins were separated by SDS-PAGE and then transferred to PVDF membranes (28148-750; Pall Corp, Ann Arbor, MI, USA). The membrane was blocked in 3% BSA for 45 min at room temperature (22°C, RT) and then cut in half. The top half was incubated with anti-human TLR2 (0.5 µg/ml; AF2616; R&D Systems, Minneapolis, MN, USA) and the bottom half with anti-actin rabbit polyclonal Ab (1:10000; Sigma-Aldrich, St Louis, MO, USA) for 18 h at 4°C. Binding of the primary Abs was determined by adding suitable peroxidase-conjugated secondary Abs (Jackson ImmunoResearch, West Grove, PA, USA). Immunoblots were developed using SuperSignal West Dura Extended Duration Substrate (34076; Thermo Scientific), and the signal was detected using a Gel Logic 2200 Imaging System (Kodak, Rochester, NY, USA) run on Carestream Imaging Software (Carestream Health, Rochester, NY, USA).
Immunofluorescence
Surface expression of E-selectin and ICAM-1 by HUVEC and HMVEC-L was assessed by performing immunofluorescence after 16 h of stimulation with FSL-1. The cells were grown on collagen-I-coated coverslips. After removing the supernatants, the cells were gently washed with PBS containing Ca2+ and Mg2+, and were further washed after subsequent steps. Cells were fixed with 4% paraformaldehyde for 10 min at RT and permeabilized with 0.5%Triton X-100 in PBS for 5 min at RT. After permeabilization, cells were blocked with 2% BSA in PBS [with 1 µg/coverslip of human IgG (1-001-A; R&D Systems)] for 30 min at RT. The cells were then incubated with either 10 µg/ml goat anti-human E-selectin (BBA18; R&D Systems), mouse anti-human ICAM-1 (BBA3; R&D Systems), normal goat IgG (NI02; Calbiochem, Darmstadt, Germany), or mouse IgG1 (MAB002; R&D Systems) diluted in 2% BSA in PBS for 1 h at RT. The cells were then incubated with either 5 µg/ml bovine anti-goat-FITC or goat anti-mouse-FITC (AP180F, AQ303F; Millipore; Billerica, MA, USA) diluted in 2% BSA in PBS for 30 min at RT. Coverslips were stained with 0.1 µg/ml of 4',6-diamidino-2-phenylindole [(DAPI) 95059-474; VWR] for 10 s prior to mounting on microscope slides with Vectashield (NC9029228; Fisher).
Cell-based ELISAs
Surface expression of E-selectin and ICAM-1 by HUVEC and HMVEC-L was assessed using a cell-based ELISA at intervals up to 24 h of stimulation with Pam3Cys or FSL-1. The cells were grown on collagen-I-coated 48-well plates. After removing the supernatants and gently washing the cells with PBS containing Ca2+ and Mg2+, cells were incubated with either 2.5 µg/ml goat anti-human E-selectin (BBA18; R&D Systems), mouse anti-human ICAM-1 (BBA3; R&D Systems), normal goat IgG (NI02; Calbiochem), or mouse IgG1 (MAB002; R&D Systems) diluted in 1% FBS in Iscove's Modified Dulbecco's Media (IMDM) for 1 h at 37°C and were washed again with PBS containing Ca2+ and Mg2+. The cells were then incubated with either a 1:2000 dilution bovine anti-goat-HRP or goat anti-mouse-HRP (805-035-180, 115-035-062; Jackson Immunoresearch) diluted in 1% FBS in IMDM for 1 h at 37°C. The cells were then washed again with PBS containing Ca2+ and Mg2+, developed using a tetramethyl benzidine (TMB) reagent (95059-154; KPL, Gaithersburg, MD, USA) and the absorbance (OD) was read at a wavelength of 450 nm and values corrected at 570 nm. Absorbance values were normalized based on the crystal violet staining of adhered cells in each well where appropriate.
Sandwich ELISAs
Confluent monolayers of HUVEC or HMVEC-L were grown and treated with Pam3Cysfor or FSL-1 as described in the previous section. Supernatant concentrations of IL-6, CSF-2, and CSF-3 were detected using Human DuoSet kits from R&D Systems (DY206; DY215; DY214). IL-8 levels were detected using a human IL-8 ELISA kit (555244; BD Biosciences, San Jose, CA) and TNF-α levels were detected using a human TNF-α Quantikine ELISA Kit (DTA00C; R&D Systems). All assays were performed according the manufacturers supplied protocol.
Flow cytometry
Confluent monolayers of HUVEC were treated for 20 h with medium or FSL-1 (10 µg/ml) before detaching them at 37°C using Accutase Cell Detachment Solution (AT104; Innovative Cell Technologies, San Diego, CA, USA). The HUVEC were then passed through a 40 µM filter, counted and aliquoted at 1 × 106cells per sample. The cells were then washed using flow cytometry (FC) staining buffer (FCSB) (FC001; R&D Systems) and incubated with 10 µg of human IgG (1-001-A; R&D Systems) in 0.2 ml of FCSB for 15 min at 4°C. After washing twice with FCSB, the cells were incubated for 45 min at 4°C with primary Abs, which included PE-conjugated mouse anti-human E/P-selectin and mouse IgG1 control (FAB6169P and IC002P; R&D Systems, respectively; 1:10 dilution) or FITC-conjugated mouse anti-human ICAM-1 and mouse IgG1 control (mab-BBA20; R&D Systems and 556649; BD Pharmingen, respectively; 2 µg/sample). All samples were washed two more times with FCSB and then were analysis on a BD LSRII Flow Cytometer (BD Biosciences).
Quantitative real-time PCR (qPCR)
TaqMan Human Immune Arrays (4418718), specific gene expression assays (HPRT1 (Hs01003267_m1), SELE (Hs00950401_m1), ICAM1 (Hs00164932_m1), IL6 (Hs00174131_m1), IL8 (Hs00174103_m1), CSF2 (Hs00929873_m1), CSF3 (Hs00357087_g1), TNF (Hs00174128_m1) and PF4 (Hs00427220_g1), and the manufacturers suggested assay reagents were purchased from Applied Biosystems (Foster City, CA, USA). HUVEC monolayers were lysed and mRNA isolated using Trizol according to the manufacturers supplied protocol (Invitrogen). mRNA concentrations were determined with a ND-1000 (NanoDrop®/Thermo Fisher Scientific) and mRNA was reverse transcribed to cDNA using a High Capacity RNA-to-cDNA Kit using 2 µg of mRNA per reaction (Invitrogen). An input of 2.5 ng of cDNA in 10 µl total reaction volume per well containing TaqMan® Fast Advanced Master Mix (Applied Biosystems) was used in all qPCR experiments and qPCR was performed using the ABI Prism 7000 Sequence Detection System (Applied Biosystems). Run method: PCR activation at 95°C for 20 s was followed by 40 cycles of 1 s at 95°C and 20 s at 60°C. The average Ct value of two technical replicates was used in all calculations with the PamCys time course samples. The average Ct value of the internal controls HPRT1 and GusB was used to calculate ΔCt values for the array samples as this combination of reference genes displayed the lowest standard deviation amongst groups. HPRT1 alone was used to calculate ΔCt values for the Pam3Cys time course samples. If the average Ct value of a group was > 35.00, the gene was determined to be low expressing. For the human immune gene arrays, the initial data analysis was performed using the 2 -ΔΔCt method and the data was corrected for statistical analysis using log transformation, mean centering, and autoscaling, as described by Willems et al.39,40 However, if all samples in a group recorded ‘undetected’, or if a group contained only one sample with a detectable Ct value after qPCR, the data for that gene was not used in statistical analysis. For the Pam3Cys time course samples, statistical analyses were calculated from the uncorrected ΔCt values. The methods of calculation utilized assume an amplification efficiency of 100% between successive cycles. The qPCR array data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus 41 and are accessible through GEO Series accession number GSE30117 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE30117).
Statistics
Data are expressed as mean ± SD. The data were analyzed using t-tests (unequal variance) when comparing 2 conditions or one-way ANOVA with Dunnett’s multiple comparison post-test when comparing more than two conditions. GraphPad Prism or Excel® 2008 were used for statistical analyses. P-values ⩽0.05 were considered significant.
Online supplementary material
Supplementary Table 1 contains the corrected human immune gene array qPCR Relative Quantification (RQ) data and the statistical analyses used to ascertain the TLR2-dependent genes. Supplementary Table 2 contains the RQ qPCR data for TNF and PF4 expression after Pam3Cys treatment of HUVEC. Supplementary Table 3 contains the ΔCt qPCR data for the individual gene expression assays shown in Table 2 and Supplementary Table 2. Supplementary Figure 1 shows the efficacy of TLR2 knockdown in HUVEC transiently transfected with the TLR2 siRNA and Supplementary Figure 2 depicts the ELISA results to determine the amount of TNF-α present in HUVEC and THP-1 supernatants after Pam3Cys treatment.
Results
TLR2 activation induces the expression of IL-6, IL-8, CSF2, CSF3, ICAM1, and SELE
Human immune gene array relative quantification (RQ) data.
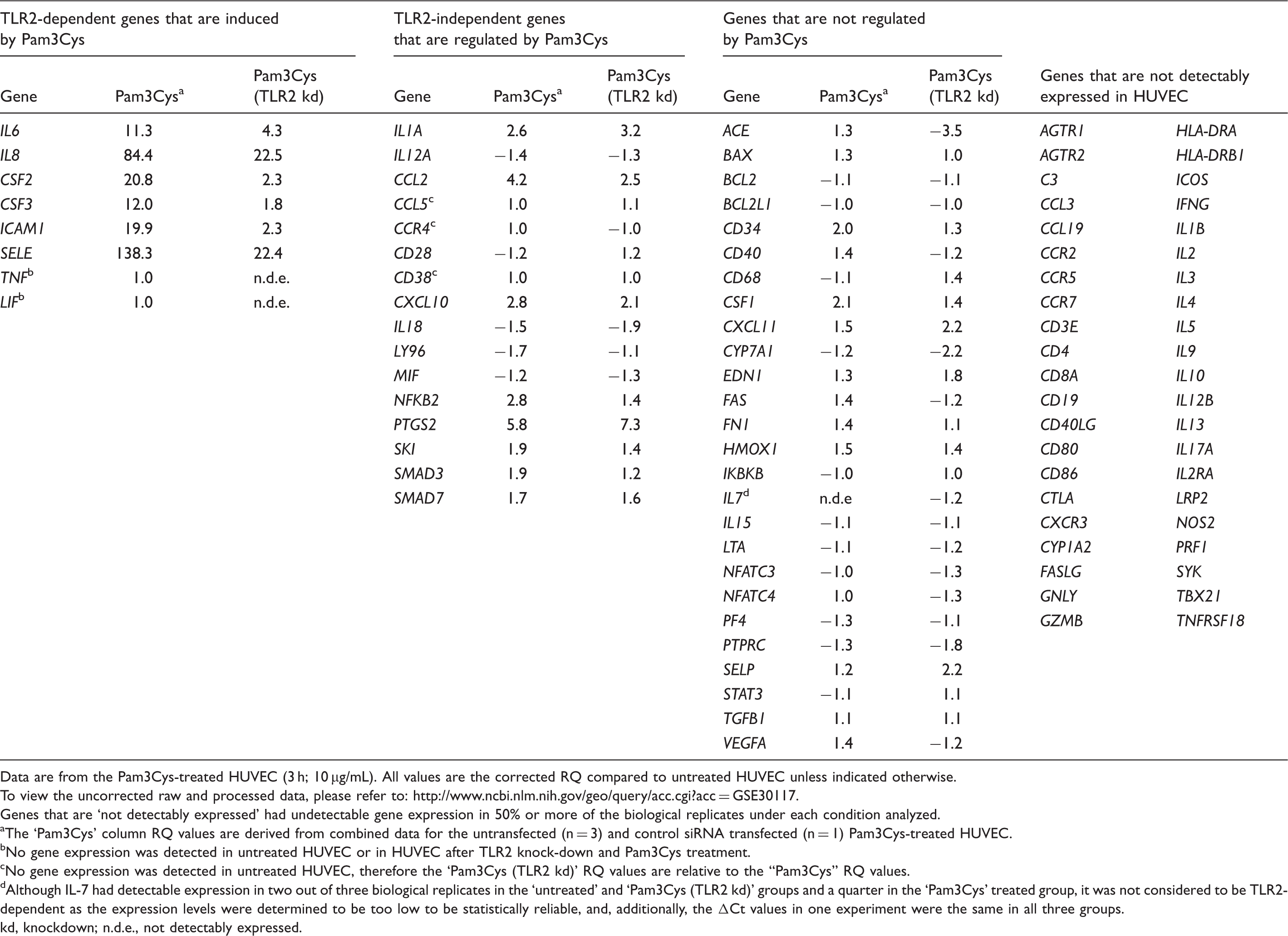
Data are from the Pam3Cys-treated HUVEC (3 h; 10 µg/mL). All values are the corrected RQ compared to untreated HUVEC unless indicated otherwise.
To view the uncorrected raw and processed data, please refer to: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE30117.
Genes that are ‘not detectably expressed’ had undetectable gene expression in 50% or more of the biological replicates under each condition analyzed.
The ‘Pam3Cys’ column RQ values are derived from combined data for the untransfected (n = 3) and control siRNA transfected (n = 1) Pam3Cys-treated HUVEC.
No gene expression was detected in untreated HUVEC or in HUVEC after TLR2 knock-down and Pam3Cys treatment.
No gene expression was detected in untreated HUVEC, therefore the ‘Pam3Cys (TLR2 kd)’ RQ values are relative to the “Pam3Cys” RQ values.
Although IL-7 had detectable expression in two out of three biological replicates in the ‘untreated’ and ‘Pam3Cys (TLR2 kd)’ groups and a quarter in the ‘Pam3Cys’ treated group, it was not considered to be TLR2-dependent as the expression levels were determined to be too low to be statistically reliable, and, additionally, the ΔCt values in one experiment were the same in all three groups.
kd, knockdown; n.d.e., not detectably expressed.
We chose HUVEC as a model EC line due to its availability, relatively low cost, ease of growth in culture and manipulation (i.e. transfection efficiency), and its extensive publication profile. For the immune array analysis, HUVEC were left untreated or were treated with the synthetic tri-acylated TLR1/2 agonist Pam3Cys in the presence or absence of either a control siRNA or TLR2 siRNA. We chose to stimulate HUVEC for 3 h with Pam3Cys, based on our previously published experiments and pilot mRNA expression experiments (data not shown). 20 Each condition, with the exception of the Pam3Cys-treated control siRNA transfected cells, was independently performed and analyzed three separate times. TLR2 knockdown was utilized to control for gene regulation induced by Pam3Cys independent of TLR2 activity (Supplementary Figure 1).
By using the 2 -ΔΔCt method of qPCR analysis coupled with corrective methods to standardize data from independent biological replicates, we observed that only six genes on the array displayed a statistically significant TLR2-dependent induction after Pam3Cys treatment: IL-6, IL-8, CSF2, CSF3, ICAM1 and SELE (Table 1 and Supplementary Table 1).39,40 A TLR2-dependent gene was defined as one that showed a significant induction after Pam3Cys treatment (P-value of ⩽ 0.05), coupled with a less significant induction in the Pam3Cys-treated TLR2 knockdown group. Also, the reduction in Pam3Cys-mediated gene induction in the presence of the TLR2 siRNA had to be significant (P-value of ⩽ 0.05) (Supplementary Table 1). No genes showed a significant TLR2-dependent reduction after Pam3Cys treatment. Additionally, we identified several genes that were regulated independently of TLR2; however, we did not focus on these genes for the present studies, as they were not robustly induced or reduced, and the intended focus of this work was on TLR2-mediated responses.
IL6, IL8, CSF2, CSF3, ICAM1 and SELE gene expression profiles after Pam3Cys treatment of HUVEC (relative quantification (RQ) values).

P < 0.05; **P < 0.01. ns, not significant.
TNF-α is not up-regulated by Pam3Cys treatment of HUVEC
In the immune gene assays, the messengers of two genes that were not detected in untreated HUVEC, leukemia inhibitory factor (LIF) and TNF-α, displayed a small, but detectable, induction in all samples (n = 4) that were treated by Pam3Cys in the immune arrays, which appeared to depend on the presence of TLR2 (Table 1). We decided to test the expression profile of one of these genes, TNF-α, along with a gene that did not show any significant up- or down-regulation in the immune array as a negative control, Platelet Factor 4 (PF4). The results show that neither TNF-α nor PF4 were significantly induced by treatment with Pam3Cys through a 9 h time course (Supplementary Tables 2 and 3). Consistent with the mRNA data, we found that TNF-α protein was not detectable by ELISA in the supernatants of HUVEC treated with Pam3Cys (Supplementary Figure 2). The monocytic cell line THP-1 was used as a positive control as it is known to produce TNF-α in response to treatment with a TLR2 agonist. 42 Based on these results, and the fact that LIF recorded an even lower expression than TNF-α after Pam3Cys-treatment in the immune arrays, we felt confident in not pursuing these genes further.
IL-6, IL-8, GM-CSF, G-CSF, ICAM-1, and E-selectin are up-regulated at the protein level
We next analyzed the genes of interest at the protein level to confirm the relevance of the immune array data, as quantitative mRNA expression data may not accurately reflect protein expression levels.
43
In these experiments, we treated cells with the diacylated TLR2/6 agonist, FSL-1, in addition to Pam3Cys. We found that both Pam3Cys and FSL-1 significantly up-regulate the expression and secretion of IL-6, IL-8, GM-CSF (CSF-2) and G-CSF (CSF-3) proteins (Figures 1A–D). The higher expression levels observed at later time points most likely reflects the accumulation of protein in the supernatants over time.
TLR2 agonists promote endothelial cells to up-regulate the surface adhesion molecules ICAM-1 and E-selectin, as well as secrete IL-6, IL-8, GM-CSF, and G-CSF. HUVEC monolayers were incubated with Pam3Cys or FSL-1 (10 µg/ml) for intervals through 24 h (n = 4). The equivalent media control values were subtracted out for each time point. One-way ANOVA with Dunnett’s multiple comparison post-test [untreated (0 h) vs. specific time point]: *P < 0.05; **P < 0.01. (A–D) Levels of IL-6, IL-8, GM-CSF, and G-CSF were quantified using sandwich ELISAs on HUVEC culture supernatants. (E, F) Surface expression of adhesion molecules ICAM-1 and E-selectin was assessed using a cell-based ELISA. ns, not significant.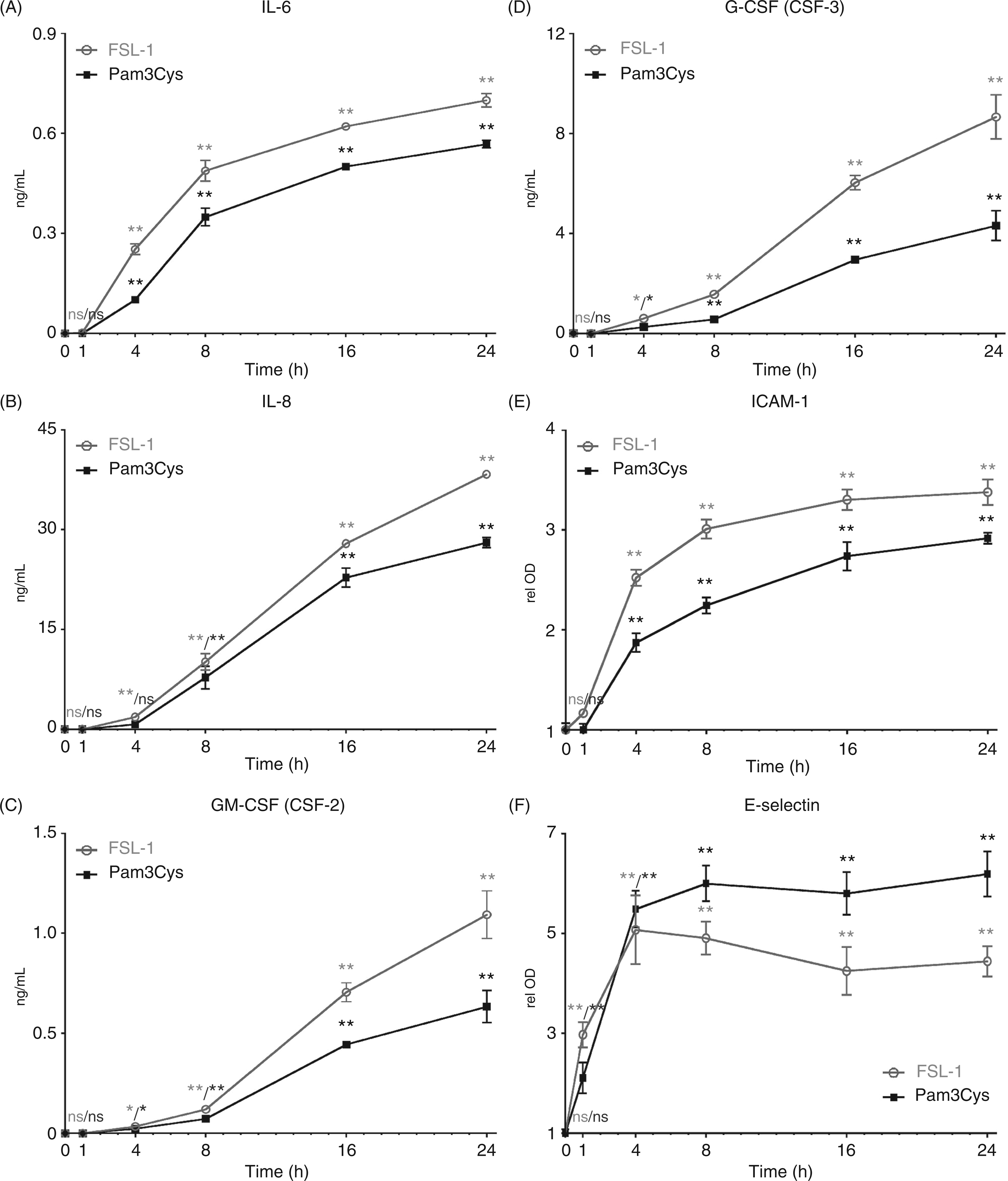
As ICAM-1 and E-selectin are expressed at the surface of the cell, we analyzed their relative expression using a cell-based ELISA method. We observed that both Pam3Cys and FSL-1 treatment of HUVEC caused a rapid increase in surface expression of ICAM-1 and E-selectin (Figures 1E, F). To verify the results of the cell ELISA, we also analyzed the surface expression of ICAM-1 and E-selectin by FC using the TLR2/6 agonist FSL-1. The results demonstrate very low E-selectin expression levels on untreated HUVEC, which increases in cells treated for 16 h with FSL-1 (Figure 2A). In contrast, ICAM-1 is expressed on the cell surface of some cells prior to agonist treatment, and is dramatically up-regulated by FSL-1 treatment (Figure 2B). These data are consistent with our previously published data showing up-regulation of E-selectin by HUVEC treated with the TLR1/2 agonists Pam3Cys and PAL.
20
In addition, the FC expression profiles for both ICAM-1 and E-selectin suggest that there is a wide range of expression levels in the cell population. The heterogeneity of E-selectin and ICAM-1 expression was further confirmed using immunofluorescence microscopy (IF) on HUVEC, and using human lung microvascular endothelial cells (HMVEC-L), which were derived from a single donor. Indeed, the results show that the individual cells express varying amounts of E-selectin and ICAM-1, with some showing high levels and others showing little or no detectable up-regulation of the adhesion molecules (Figure 2C). Using vascular endothelial (VE)-cadherin IF staining, we confirmed that all of the cells, including those expressing low and high levels of the adhesion molecules, were indeed ECs (data not shown). We speculate that the differential expression might correlate with cells being at different stages of the cell cycle.
HUVEC and HMVEC-L treated with FSL-1 express heterogeneous levels of the surface adhesion molecules ICAM-1 and E-selectin. (A, B) Flow cytometry was performed to assess surface expression of E/P-selectin and ICAM-1 by HUVEC after 16 h of treatment with FSL-1 (10 µg/ml). (C) E-selectin and ICAM-1 expression as detected by immunofluorescence staining of HUVEC and HMVEC-L monolayers after 16 h of treatment with FSL-1 (10 µg/ml).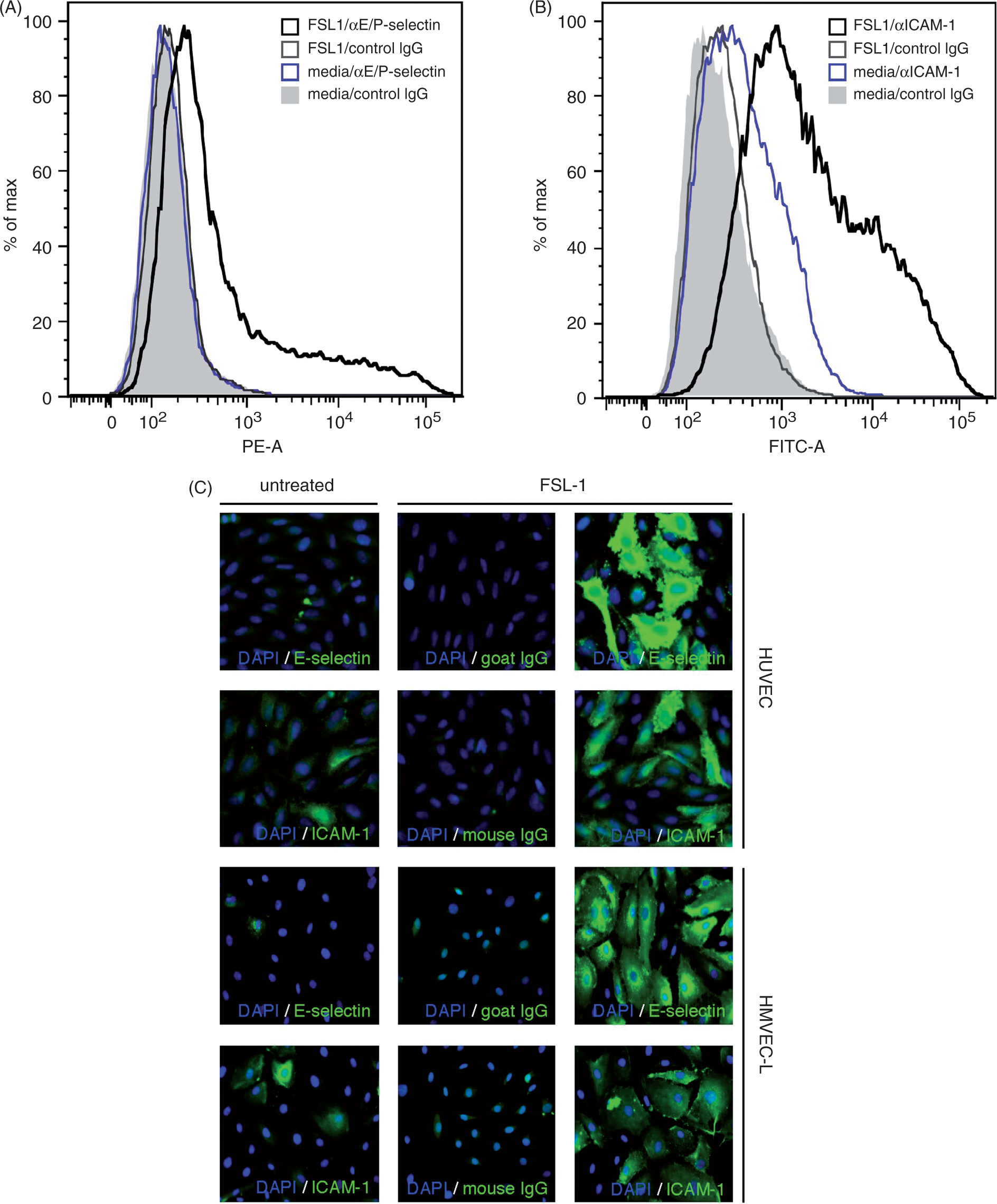
IL-6, IL-8, GM-CSF, G-CSF, ICAM-1, and E-selectin up-regulation is dependent upon TLR2 expression
siRNA knockdown of TLR2 expression in HUVEC was used to verify the dependence on TLR2 of IL-6, IL-8, GM-CSF, G-CSF, ICAM-1, and E-selectin protein up-regulation by Pam3Cys and FSL-1. Consistent with the qPCR immune array data shown in Table 1, TLR2 knockdown in HUVEC significantly attenuated the up-regulation of the aforementioned proteins after treatment with Pam3Cys and FSL-1 (Figure 3). This data further suggests that TLR2 activation on ECs may directly modulate the inflammatory response to microbial infection. Although TLR2 was clearly knocked down at 72 h after transfection based on Western blots, we suspect that there many not have been complete knockdown of TLR2 based on the fact that there was a low, albeit significant, protein response for most samples treated with the TLR2 siRNA (Supplementary Figures 1 and 3). This may have been due, in part, to the fact that we began the stimulation 48 h after introducing the TLR2 siRNA, before the knockdown was complete. Nonetheless, we believe that at 48 h there was a sufficient down-regulation to verify any TLR2-dependent effects.
Pam3Cys and FSL-1-induced surface adhesion molecule and cytokine expression is dependent on the presence of TLR2. HUVEC monolayers were pre-incubated with TLR2 or control siRNA for 48 h then treated with Pam3Cys or FSL-1 (10 µg/ml) for 20 h (n = 3). Student’s t-test of TLR2 siRNA versus control siRNA for each condition: ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; one-way ANOVA with Dunnett’s multiple comparison post-test of control siRNA-treated cells (media vs. TLR2 agonists): #P < 0.05; ##P < 0.01; one-way ANOVA with Dunnett’s multiple comparison post-test of TLR2 siRNA-treated cells (media vs. TLR2 agonists): δP < 0.05; δδP < 0.01. (A–D) Levels of IL-6, IL-8, GM-CSF, and G-CSF were quantified using sandwich ELISAs on HUVEC culture supernatants. (E, F) Surface expression of adhesion molecules ICAM-1 and E-selectin was assessed using a cell-based ELISA. ns, not significant.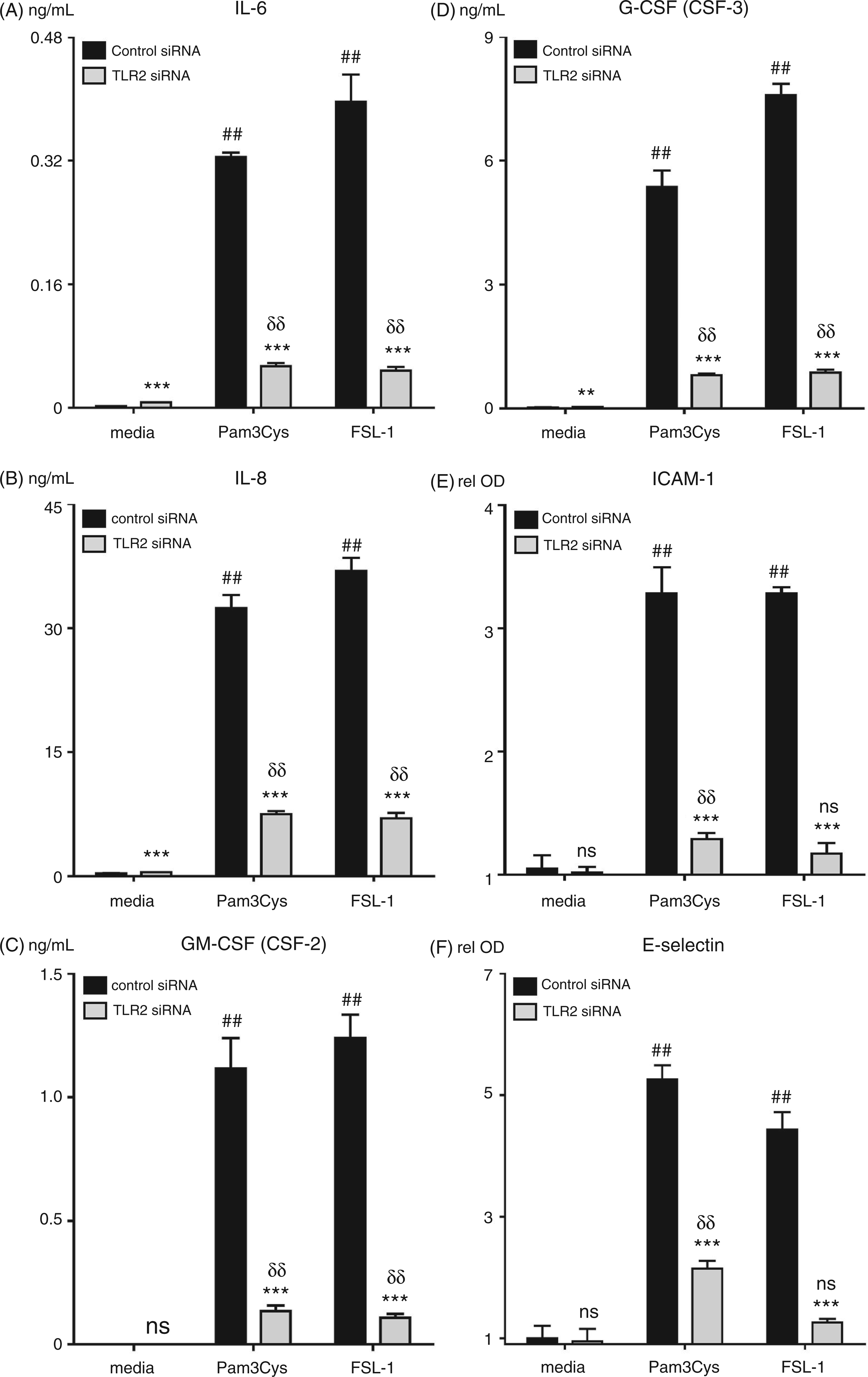
IL-6, IL-8, GM-CSF, G-CSF, ICAM-1, and E-selectin are up-regulated in lung microvascular cells after Pam3Cys and FSL-1 treatment
We have previously reported that treatment of HMVEC-L with the TLR2 agonist PAL increases the expression levels of IL-6, IL-8, and E-selectin, suggesting that the lung endothelium itself may play a role in inflammation of this organ.
20
We extended these findings by analyzing HMVEC-L expression of IL-6, IL-8, GM-CSF, G-CSF, ICAM-1, and E-selectin in response to Pam3Cys, which activates via TLR 2/1 dimers, and FSL-1, which activates via TLR 2/6 dimers. We observed that both TLR2 agonists caused a significant up-regulation in the expression of these factors; of note, significant variability in response between individual HMVEC-L donors that did not appear to depend on passage number was observed (Figure 4 and data not shown). These results also indicate that TLR2 activation up-regulates the same immune-related genes in HMVEC-L and HUVEC, suggesting that the immune response elicited by TLR2 agonists may be conserved in endothelial cells from different vascular beds.
Pam3Cys and FSL-1 induce surface adhesion molecule and cytokine expression in HMVEC-L cells. HMVEC-L monolayers were treated with Pam3Cys or FSL-1 (10 µg/ml) for 20 h (n = 4). One-way ANOVA with Dunnett’s multiple comparison post-test (media vs. TLR2 agonists): **P < 0.01. (A–D) Levels of IL-6, IL-8, GM-CSF, and G-CSF were quantified using sandwich ELISAs on HMVEC-L culture supernatants. (E, F) Surface expression of adhesion molecules ICAM-1 and E-selectin was assessed using a cell-based ELISA.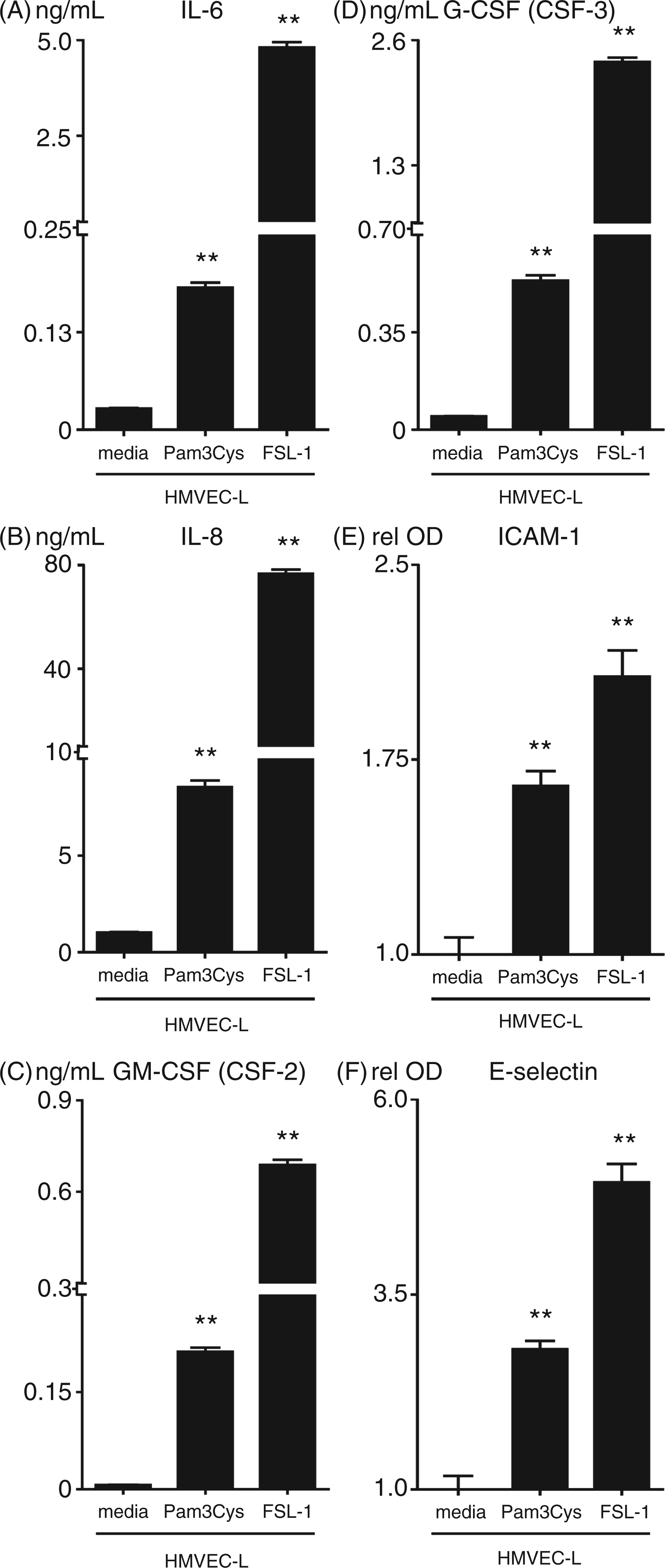
Discussion
Our data indicate that diacylated and triacylated bacterial lipopeptides directly activate TLR2 on human ECs to up-regulate inflammatory proteins involved in facilitating the neutrophil response to infection. There are several aspects of the present studiy that are novel and distinguish it from previous work. Whereas we previously established that TLR2 activation induces inflammatory endothelial responses in a general way, in the present study, we delved more deeply into the nature and breadth of the EC inflammatory response. We have shown that TLR2 activation up-regulates additional cytokines and adhesion molecules (ICAM-1, CSF2, CSF3), but, more importantly, that the direct activation of endothelial TLR2 induces responses in both HUVEC and lung microvascular EC that are specifically geared towards the recruitment, activation, and survival of neutrophils, rather than mononuclear leukocytes. Additionally, in contrast to the effects of TLR2 agonists on mononuclear leukocytes, we found that TLR2 agonists do not induce ECs to secrete TNF-α. Although these studies were focused on gene expression rather than on functional effects, we believe that the up-regulation of neutrophil-specific EC responses is functionally relevant based on our prior work showing augmented binding of neutrophils to monolayers of HUVEC
20
and HMVEC-L (data not shown) that have been pretreated with TLR2 agonists. This work extends the current understanding of the effects of TLR2 activation by bacterial lipoproteins on the human lung endothelium, and suggests that activation of lung EC TLR2 may be important in the initial neutrophil influx and activation that occur within the lung during bacterial sepsis. In Figure 5 we present a model which, although speculative, attempts to summarize our results with TLR2 in the context of other published data on neutrophil-specific endothelial responses to infection.
Theoretical model summarizing the neutrophil-specific endothelial response to TLR2. Bacterial-derived diacylated or triacylated lipopeptides activate TLR2-dependent signaling pathways in vascular ECs that result in the up-regulation of IL-8, E-selectin, ICAM-1, GM-CSF, G-CSF, and IL-6 proteins. IL-8 acts as a chemoattractant for neutrophils, while E-selectin mediates the capture and rolling of neutrophils along the EC surface via relatively weak associations with sialylated carbohydrates present on P-selectin glycoprotein 1 (PSGL1) or L-selectin on the neutrophil surface. E-selectin and IL-8 binding to neutrophil receptors also activates signaling pathways in neutrophils that induce a high-affinity conformational state of the integrins LFA-1 or MAC-1. These integrins then form strong bonds with ICAM-1 on the EC surface, resulting in the arrest of neutrophils at the site of inflammation. Upon arrest, neutrophils can migrate out of the vasculature directly towards the sites of inflammation. Additionally, secreted GM-CSF and G-CSF promote the survival, and activate the microbial activities, of neutrophils. Finally, activated neutrophils release soluble IL-6 receptor α (sIL-6Rα), which, after binding IL-6, associates with gp130 on the EC surface (i.e. IL-6 trans-signaling). This activates additional signaling pathways in ECs that lead to the up-regulation of mononuclear leukocyte chemoattractants, and adhesion molecules, such as CCL2 and VCAM-1, respectively.
67
Recruited monocytes engulf apoptotic neutrophils, thus signaling the end of the acute inflammatory phase. This cartoon was adapted from a figure in Kaplanski et al.
65
EC, endothelial cell; ECM, extracellular matrix; gp130, common glycoprotein 130 subunit; sIL-6Rα, soluble IL-6 receptor α; VCAM-1, vascular cell adhesion molecule-1.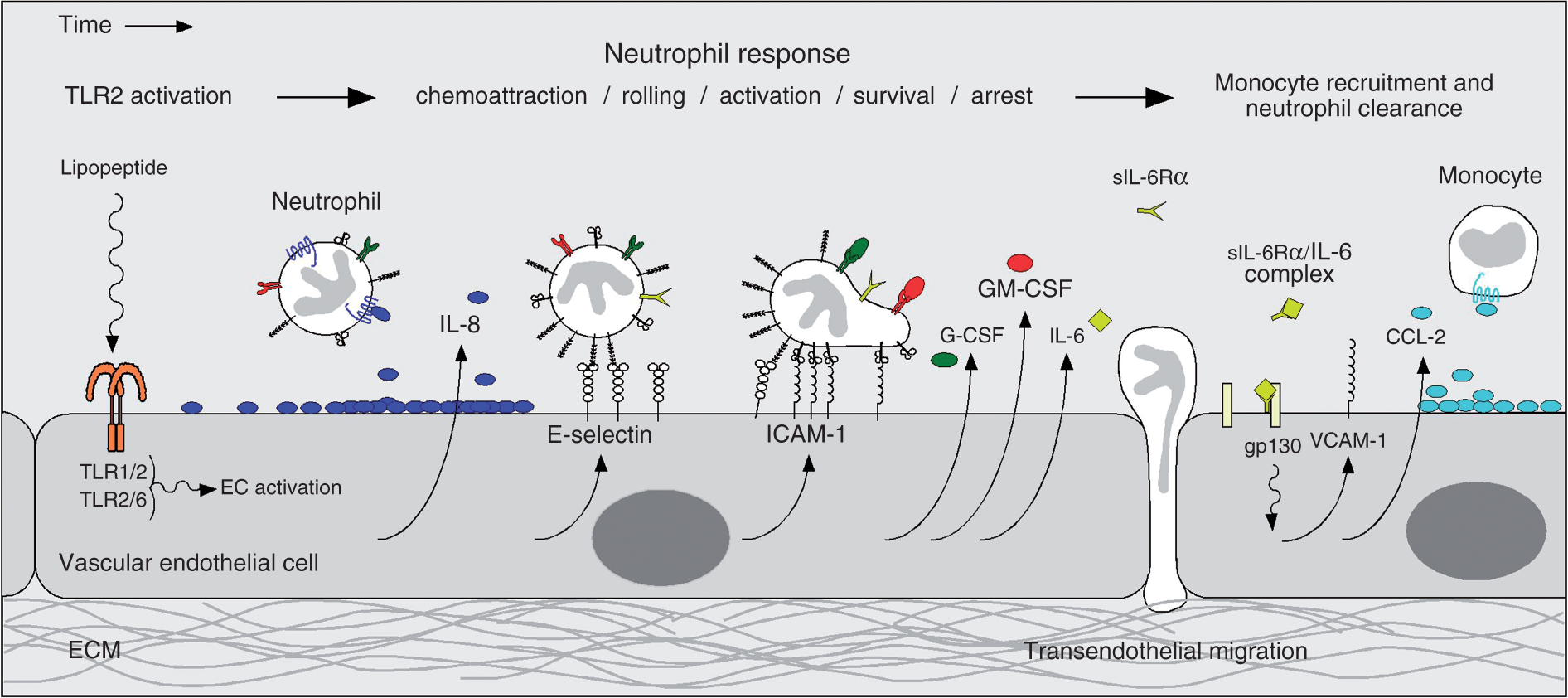
Six out of 92 genes tested displayed TLR2-dependent induction after agonist treatment in HUVEC. The expression of these genes was confirmed at the protein level and shown to be dependent upon the expression of the TLR2 protein. Interestingly, the six proteins up-regulated—IL-6, IL-8, GM-CSF (CSF-2), G-CSF (CSF-3), ICAM-1, and E-selectin—are intimately involved in the processes of neutrophil proliferation, differentiation, survival, activation, trafficking, and adhesion. These same proteins are also up-regulated in human lung microvascular ECs treated with diacylated and triacylated TLR2 agonists. We have previously reported that TLR2 agonists cause lung inflammation, altered lung vasoconstrictive responses to alveolar hypoxia, arterial hypoxia, and increased neutrophil adherence to EC monolayer; other investigators have reported that endothelial TLR4 expression is necessary to sequester neutrophils in the lungs after systemic LPS treatment.20,44– 46 Taken together, these findings support the hypothesis that lung microvascular ECs are the sentinel cells responsible for the early recognition and response to systemic microbial infection within the pulmonary vasculature. An important potential clinical implication of these studies is that during systemic infection, direct activation of lung endothelial TLR2 by bacterial lipoproteins may participate in neutrophil recruitment to, and activation within, the lung, and thereby contribute to acute lung injury during systemic infections.
Contrary to previous reports suggesting that ECs have to be primed by other inflammatory stimuli, such as INF-γ, LPS, TNF-α or IL-1β in order to respond to TLR2 agonist, we clearly demonstrate that ECs can directly respond to TLR2 lipopeptide agonists without the need for prior priming events.21,47 We suspect that this discrepancy is primarily attributable to the use of lipoteichoic acid (LTA) as the TLR2 agonist in these other studies. Although LTA can associate with TLR2, it is not a potent activator of TLR2. Moreover, recent reports suggest that LTA may not even act as a TLR2 agonist, which is supported by data showing that LTA does not induce the heterodimerization of either the TLR1/2 or TLR2/6 extracellular domains.11,14,48 It is plausible that the up-regulation of TLR2 observed after INF-γ, LPS, TNF-α, or IL-1β pre-treatment of ECs is prerequisite for LTA to activate these cells.
Our data support the conclusion that TLR2 activation in the vascular endothelium specifically targets responses that promote mobilization, trafficking, and activation of neutrophils, rather than mononuclear leukocytes. Activation of TLR2 potently induced IL-8, ICAM-1, and G-CSF, each of which is involved in different aspects of neutrophil function. The chemokine IL-8 is a strong chemoattractant for neutrophils; 49 ICAM-1 is necessary for neutrophil arrest on the endothelium;34,50 and G-CSF is the primary cytokine responsible for maturation, survival, and activation of neutrophils. 51 Conversely, there was no significant induction of CSF1, which is important in activation and function of mononuclear leukocytes, and only relatively minor induction of the gene for the monocyte chemoattractant protein CCL2, which we found was not even dependent on the presence of TLR2.52,53 Nonetheless, we cannot entirely rule out the possibility that TLR2 activated ECs can attract and retain specific populations of mononuclear leukocytes at the site of inflammation as our array did not cover all of the genes involved in their recruitment. Additionally, we did not detect a significant induction of genes involved in the recruitment and activation of eosinophils, including IL4, IL5, IL13 or CCL5. 54
The colony stimulating factors M-CSF (CSF-1), GM-CSF, G-CSF, and IL-3 (multi-CSF) are essential for the proper development of the myeloid blood cell lineages. 55 IL-3 predominantly affects the development of the earliest hematopoietic stem cells and GM-CSF the development of common myeloid progenitor cells and cells of the granulocyte-macrophage lineage. As mentioned above, M-CSF primarily influences cells of the monocyte lineage, while G-CSF primarily influences those of the granulocyte lineage.51,56,57 As G-CSF and GM-CSF also activate survival pathways and the antimicrobial properties of neutrophils, their secretion by ECs in response to TLR2 agonists may be necessary to regulate these functions at the site of infection.58,59 Of note, IL-1 also stimulates ECs to specifically to express G-CSF and GM-CSF, and not M-CSF or IL-3, suggesting that this property of ECs is conserved after activation by other inflammatory stimuli. 60 Additionally, both G-CSF and GM-CSF have been reported to induce human ECs to proliferate and migrate. 61 It is plausible that another function of these two cytokines is to promote wound healing after infection-induced tissue injury through their effects on viable ECs at the site of infection. This is further supported by a recent report showing that the activation of TLR2 by MALP-2 induced ECs to secrete GM-CSF, which was shown to promote angiogenesis. 19
IL-6 has been described to have both pro-inflammatory and anti-inflammatory effects, and to be involved in such divergent roles as leukocyte recruitment, activation, cytokine repression, and apoptosis. IL-6 is also involved in the switch from innate to acquired immune responses. 62 In this regard, IL-6 has been shown to have an important role in the resolution of the acute phase of inflammation. IL-6 and its soluble receptor (sIL-6R) suppress neutrophil recruitment and promote neutrophil apoptosis, and facilitate mononuclear leukocyte attraction to sites of inflammation, where they subsequently participate in clearance of the apoptotic neutrophils. 63 – 65 Interestingly, the disruption of this process, as occurs with IL-6 dysregulation, can cause a chronic inflammatory condition characterized by the accumulation of large numbers of neutrophils and mononuclear leukocytes at sites of infection. 66 It therefore appears that the balance of cytokines secreted by ECs in response to TLR2 activation can influence the number and type of leukocytes present at the site of infection: G-CSF and GM-CSF secretion promote the survival and activity of neutrophils, while IL-6 may promote neutrophil clearance and the subsequent resolution of the acute inflammation phase.
During inflammatory conditions, such as seen in severe sepsis, the widespread activation of the endothelium by both circulating microbial components and inflammatory mediators such as TNF-α leads to the migration of activated leukocytes out of the vascular space and into surrounding tissues at sites that can be located remotely from the inciting source of inflammation. It is believed that these activated leukocytes contribute to organ injury through the production of injurious mediators, such as reactive oxygen species (ROS). Lipoprotein TLR2 agonists are abundantly expressed by bacteria and are shed into the blood during bacterial sepsis. As such, we have previously found that administration of TLR2 agonists to mice induces responses that are characteristic of those that occur in sepsis, including systemic and lung inflammation, disturbances in coagulation pathways, impaired lung vascular responses to low oxygen conditions, and reduced arterial oxygenation levels. 45 Furthermore, we have reported that systemic treatment of mice with TLR2 agonists up-regulates E-selectin and myeloperoxidase levels and the production of ROS in the lungs, and that several TLR2 agonists promote the adherence of neutrophils to EC monolayers.20,44,45 Together, the data in this report, along with our previously published data, suggest that, during sepsis, circulating bacterial lipoproteins may directly activate lung endothelial TLR2, leading to up-regulation of specific neutrophil responses, and support the hypothesis that endothelial TLR2 activation may contribute to sepsis-induced acute lung injury.
Footnotes
Funding
This work was supported by a grant from the NIH [NIAID R01AI058106/Hellman] and the UCSF Department of Anesthesia and Perioperative Care.
Acknowledgements
We would to thank Dr Kirsten Copren from the UCSF Genome Analysis Core Facility for help with qPCR data analysis.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.