
Abstract
Recent studies have identified enzymes that use NAD as a substrate, thus contributing to its net consumption. To maintain the intracellular pool, NAD is re-synthesized by a salvage pathway using nicotinamide, the by-product generated by the enzymatic cleavage of NAD. Enzymes involved in NAD re-synthesis include nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide mononucleotide adenylyltransferase. Our studies show, that NAMPT was substantially up-regulated by LPS in primary human monocytes, suggesting that it may be especially required during the process of monocyte activation. To evaluate the contribution of the NAD rescue pathway to LPS-induced biological responses in human monocytes, we used APO866, a well-characterized inhibitor of NAMPT. Concomitant with the inhibition of NAMPT, LPS-induced TNF-α protein synthesis declined, while TNF-α mRNA levels were minimally affected. Moreover, APO866 strongly decreased the production of reactive oxygen species (ROS), increased surface expression of the NAD-consuming enzyme CD38, and modified the production of selective eicosanoids. We further demonstrate that protein ADP-ribosylation was strongly reduced, indicating a possible link between this post-translational protein modification and human monocyte inflammatory responses. Despite a substantial reduction in intracellular NAD levels, activated monocytes were resistant to apoptosis, while resting monocytes were not. Taken together, our data suggest that activated monocytes strongly depend on the NAD salvage pathway to mount an appropriate inflammatory response. Their survival is not affected by NAD-depletion, probably as a result of LPS-mediated anti-apoptotic signals.
Introduction
NAD is generally considered as a coenzyme exclusively involved in redox reactions. However, recent studies have identified enzymes that use NAD as a substrate and thus contribute to the net consumption of this nucleotide. 1 – 3 Hence, NAD re-synthesis is required to maintain a stable cellular concentration. In mammals, NAD is formed by the de novo pathway from tryptophan via quinolinic acid and by the salvage pathway mainly via nicotinamide (Nam), a reaction product of NAD-consuming enzymes. 4 In addition, a new pathway which leads to NAD from nicotinamide riboside has recently been described. 5
Nicotinamide phosphoribosyltransferase (NAMPT) is the first enzyme in the salvage pathway, catalyzing the production of nicotinamide mononucleotide (NMN) from Nam and 5-phosphoribosyl-1-pyrophosphate (PRPP). NMN, together with ATP, is then used by nicotinamide mononucleotide adenylyltransferase (NMNAT) to yield NAD (Figure 1). In contrast to NAMPT, little is known about NMNAT localization and regulation in immune cells.
Schematic presentation of the NAD salvage pathway.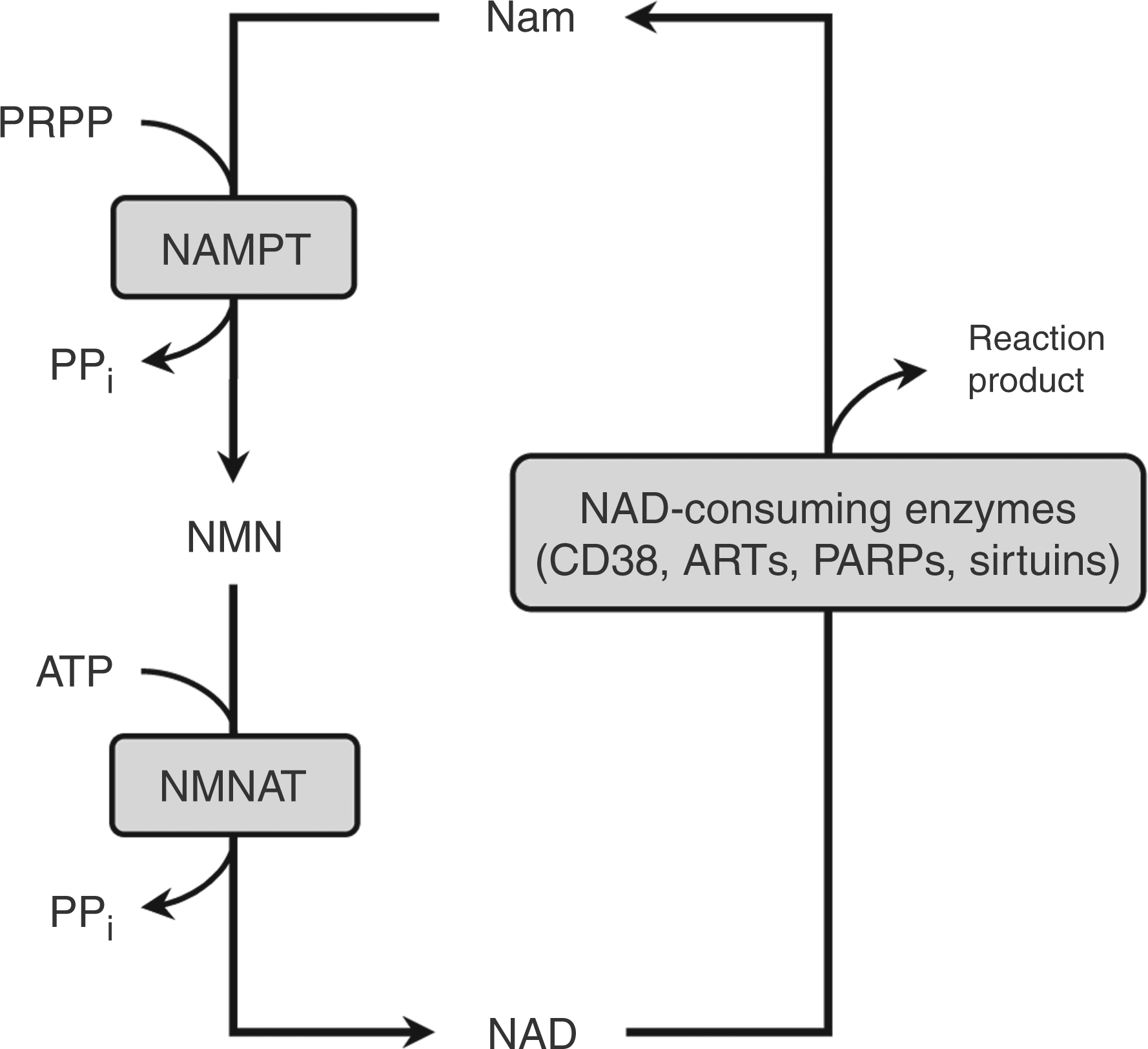
NAMPT has gained much attention because of its potential immunological properties. 6 The expression of NAMPT, also known as pre-B-cell colony-enhancing factor (PBEF) 7 , or visfatin 8 , is up-regulated during activation of immune cells such as monocytes, macrophages, dendritic cells, neutrophils, T and B cells. 9 – 11
In addition to its intracellular localization, NAMPT has also been detected in serum and culture supernatants.7,12– 14 As recombinant NAMPT has been shown to exert anti-apoptotic effects on macrophages 15 and activated neutrophils 14 , and to induce cytokine production in monocytes 16 , its extracellular occurrence seems to be of biological significance.
Over the last decade much evidence has emerged that NAMPT has potential implications in the pathogenesis of inflammatory diseases. Its expression was found to be increased in colonic biopsy specimens of patients with Crohn's disease and ulcerative colitis. 16 Furthermore, elevated NAMPT levels have been detected in synovial tissues and in serum and synovial fluid from rheumatoid arthritis patients. 17 Considering that monocytes are one of the major players in inflammatory diseases, we set out to study the role of NAMPT in regulating monocyte functions. To this end, we took advantage of APO866, an inhibitor of NAMPT able to reduce the intracellular NAD pool. This drug, which has been shown to induce apoptotic cell death in many cancer cell lines without any DNA damaging effects 18 – 20 is now under scrutiny in phase II trials for treatment of solid and hematologic malignancies (see also www.clinicaltrials.gov).21,22 How this treatment affects cells of innate immunity, such as monocytes, is unknown.
In the present study, we show that human monocytes up-regulated NAMPT in response to LPS and that inhibition of NAMPT resulted in drastic depletion of intracellular NAD levels. Blockage of NAMPT effectively reduced TNF-α and ROS production; it led to changes in the eicosanoid pattern and to an up-regulation of CD38, but had no consequences for the survival of activated monocytes.
Materials and methods
Reagents and antibodies
Unless otherwise indicated, materials used in this study were from the following manufacturers: Invitrogen GmbH (Karlsruhe, Germany): oligonucleotide synthesis, 100 bp DNA ladder; PAA (Pasching, Austria): RPMI 1640 (with
Cell separation and cell culture
Human peripheral blood mononuclear cells from healthy donors were obtained by centrifugation over a Ficoll-Isopaque (Amersham Biosciences, Freiburg, Germany) density gradient. After repeated washing in PBS containing 0.3 mM EDTA, the monocytes were isolated by counter-flow elutriation using the JE-6B elutriation system (Beckman Instruments, Palo Alto, CA, USA), as described previously. 23 The purity of the cell preparation was >90%, as assessed by morphological screening and immunofluorescence staining with a mAbagainst CD14 (BL-M/G14, DiaMak, Leipzig, Germany). Monocytes (2 × 106/ml) were suspended in RPMI 1640 medium supplemented with 10% (v/v) FBS, 100 U/ml penicillin and 100 µg/ml streptomycin, and incubated at 37°C/5% CO2, unless otherwise specified. Propylene glycol was used as a vehicle control for APO866 in concentrations ranging from 2.4 × 10−6% (1 nM APO866) to 0.24% (100 µM APO866).
RNA isolation and reverse transcription
Total RNA was isolated from monocytes (4 × 106) using the TRI reagent® (Sigma-Aldrich), according to the manufacturer's instruction. DNase I treatment and reverse transcription were performed as previously described. 24
Real-time PCR
The reaction mixture contained 10 µl of the SYBR Green PCR Mastermix (Applied Biosystems, Foster City, USA), 125 nM forward and reverse primers and 1 µl of cDNA template in a final volume of 20 µl. Expression of mRNA was analyzed with the annealing temperature of 60°C and the following primers in 5’–3’ orientation:
GAPDH fwd, CAGTCCATGCCATCACTGCCACCCAG; GAPDH rev, CAGTGTAGCCCAGGATGCCCTTGAG 25 ; GNB2L1 fwd, GAGTGTGGCCTTCTCCTCTG; GNB2L1 rev, GCTTGCAGTTAGCCAGGTTC; NAMPT fwd, GCAGAAGCCGAGTTCAACAT; NAMPT rev, TCTTCTTTTCACGGCATTCA; TNF-α fwd, TCAGCCTCTTCTCCTTCCTG; TNF-α rev, GGCTACAGGCTTGTCACTCG; CD38 fwd, ACAAACCCTGCTGCCGGCTCTC; CD38 rev, GCATCGCGCCAGGACGGTCT 26 .
The reactions were performed in the 7300 real-time PCR cycler system (Applied Biosystems) under the following conditions: initial denaturation at 95°C for 10 min, followed by 40 cycles of 15 s denaturation at 95°C, 30 s of primer annealing at 60°C and 30 s of extension/synthesis at 72°C. Product quantification was optimal at 72°C. Negative controls were performed with total RNA and water as template. Calculations (ΔΔCt−method) were carried out as previously described. 27
Western blot analysis
Western blot analysis was carried out as previously described with some modifications. 28 Briefly, cells (2–4 × 107/ml) were suspended in RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Nonidet P40, 0.5% deoxycholate, 0.1% SDS; pH 7.5) or for detection of TNF-α in permeabilizing buffer (10 mM Tris, 1 mM EDTA, 4 mM MgCl2; pH 7.8) and sonicated. Both buffers contained a complete protease inhibitor cocktail (Roche).
Samples were run on a 4%–20% tris-glycine gradient gel (Serva, Heidelberg, Germany) to detect poly-ADP-ribosylated proteins, and on a 12%–15% SDS-polyacrylamide gel (Protean II, Biorad, Munich, Germany) to detect NAMPT and TNF-α proteins and subsequently transferred to polyvinylidenfluorid
NAMPT activity assay
The NAMPT activity was measured as described by Elliot et al. and Rongvaux et al., with some modifications. 11 , 29 The assay is based on the differential solubility of Nam and NMN in acetone. Monocytes (4 × 107/ml) were suspended in ice-cold permeabilizing buffer, left on ice for 15 min and sonicated as described above. The supernatant (50 µl), cleared by centrifugation (10000 g; 5 min; 4°C), was added to a reaction mixturecontaining 50 mM Tris/HCl (pH 8.8), 2 mM ATP, 5 mM MgCl2, 0.5 mM 5-phosphoribosyl-1-pyrophosphate (PRPP) and 10–20 µM [carbonyl−14C] Nam (Hartenstein Analytic, Braunschweig, Germany) in a total volume of 1 ml. After incubation for 1 h at 37°C the reaction was stopped by adding 50 µl of the reaction mixture to 2 ml acetone. The acetone reaction mixture was collected on Whatman GF/A glass microfibre filters. After washing two times with acetone, filters were dried and counted by liquid scintillation spectrometry. While Nam was rinsed through the filter, NMN precipitated in acetone was trapped on the filter. NAMPT activity was normalized to the total protein content of cell lysates. Unless otherwise indicated, materials used in this method were from Sigma-Aldrich.
Determination of NAD concentration
The NAD concentrations in monocytes were measured by an enzymatic cycling assay as previously described by Zerez et al. and Zocchi et al. 30 , 31 Monocytes (4 × 107/ml) were suspended in ice cold cycling buffer (pH 10) containing 100 mM Na2CO3, 20 mM NaHCO3 and 10 mM Nam, left on ice for 15 min and sonicated as described above. After centrifugation (13,000 g; 1 min; 4°C), 50 µl of the supernatant were added to a reaction mixture [17 mM Tris (pH 8), 600 mM ethanol, 0.5 mM iodonitrotetrazolium chloride, 0.1 mM phenazine methosulfate, 0.1% BSA and 90.2 U alcohol dehydrogenase] in a total volume of 1 ml. Absorbance was measured at 520 nm. NAD content was quantified according to standard curves generated with NAD+ and normalized to the total protein content of cell lysates. All chemicals used in this method were obtained from Sigma-Aldrich.
Assay of cell viability
Apoptosis and necrosis of monocytes was assessed using the TACSTM Annexin V-FITC Apoptosis Detection Kit (R&D Systems), in which apoptotic cells are labelled with annexin V conjugated to FITC and necrotic cells are labelled with propidium iodide. The test was performed according to the manufacturer's instruction on a FACScan flow cytometer (Becton Dickinson, San Jose, CA,USA).
Antibodies and FACS analysis
After washing with PBS, monocytes were incubated with the anti-CD38 antibody (D2, DiaMak) or IgG1 isotype (Sigma-Aldrich) for 30 min at 4°C. After washing in PBS containing 0.1% sodium azide, cells were incubated for 30 min at 4°C with FITC-labelled goat-anti-mouse Ab (SIFIN, Berlin, Germany). After washing and fixation in 1% formaldehyde, cells were analyzed on a FACScan flow cytometer (Becton Dickinson) and values were isotype-corrected.
Detection of TNF-α in culture supernatants
Human monocytes (2 × 106/ml) were suspended in culture medium and incubated with various substances for 30 min before LPS (100 ng/ml) was added. After 4 h, the culture supernatants were tested for TNF-α content by ELISA as previously described. 32
Determination of the extracellular ROS production
Freshly-isolated human monocytes (2 × 106/ml) were suspended in RPMI1640 (without phenol red) containing 2.5% FBS and 5 mM HEPES, dispensed in a white, 96-well plate and loaded with 140 µM Luminol (Sigma-Aldrich) in a final volume of 250 µl. After 40 min ofequilibration, the cells were treated with either medium, LPS (100 ng/ml) or specific test substances and the chemiluminescence induced by the ROS-dependent oxidation of Luminol was continuously recorded for 30 min with data points being collected every minute. After the second stimulus, recordings were continued for a further 120 min. Duplicate determinations were performed for each set of measurements. The experiments were carried out at 37°C in a Luminoskan Ascent (Thermo Electron Corporation, Langenselbold, Germany) controlled by Ascent 2.6 software.
Determination of eicosanoid production by LC-MS/MS analysis
Human monocytes (2 × 106/ml) were suspended in culture medium and stimulated with various test substances for 30 min before adding medium or LPS (100 ng/ml). After incubation for 16 h at 37°C, culture supernatants were collected. Solid phase extraction with Strata X columns (Phenomenex, Aschaffenburg, Germany) was used for sample clean-up and concentration of the eicosanoids according to the manufacturer's instructions with minor modifications. 33 The supernatant, containing the isotope-labelled internal standards prostaglandin F2α (PGF2α)-d4, prostaglandin E2 (PGE2)-d4, thromboxane B2 (TxB2)-d4, leukotriene B4 (LTB4)-d4, 5-S-hydroxyeicosatetraenoic acid)5-S-HETE)-d8 and arachidonic acid (AA)-d8, was injected into the LC-MS/MS system and separated on a Vydac®-RP-HPLC column (Grace Vydac, Hesperia, CA, USA). For eicosanoid analysis, a triple quadrupole mass spectrometer with trap function (AB SCIEX QTrap® 5500, SCIEX Toronto, ON, Canada) was used after negative electrospray ionization. Multiple reaction monitoring transitions were used for detection of analytes and internal standards. A standard mixture containing 6-keto-prostaglandin F1α (6-keto-PGF1α), 8-iso-prostaglandin F2α (8-iso-PGF2α), TXB2, PGF2α, PGE2, prostaglandin D2 (PGD2), 5-S,6-S-lipoxin A4 (LXA4), LTB4, 5,6-dihydroxyeicosatrienoic acid (5,6-DHET), 12-S-hydroxyeicosatetraenoic acid (12-S-HETE), 11-S-hydroxyeicosatetraenoic acid (11-S-HETE), 5-S-HETE, 5-S-hydroperoxyeicosatetraenoic acid (5-HpETE), 5-oxo-eicosatetraenoic acid (5-oxo-ETE), 5,6-epoxyeicosatrienoic acid (5,6-EET) and AA was used for retention time correction. Analytes were quantified using the corresponding labelled internal standards. The standard mixture and the internal standards were purchased from Cayman chemical (Michigan, MO, USA). Data analysis was carried out using Analyst® 1.5 software (AB SCIEX, Darmstadt, Germany).
Statistical analyses
Statistical significance was calculated with the tests indicated and classified as follows:
* P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001
Results
Expression of NAMPT in response to LPS
LPS treatment led to a time-dependent increase in NAMPT expression at the mRNA and protein level in human monocytes (Figure 2). mRNA levels peaked at 10 h (Figure 2A), whilst protein expression was highest 16 h after LPS stimulation (Figure 2B).
Increase of NAMPT expression in response to LPS. Monocytes (2 × 106/ml) were incubated in the presence or absence of LPS (100 ng/ml) for the times indicated. (A) NAMPT mRNA was quantified by performing a quantitative real-time PCR. Relative mRNA levels (ΔΔCt-method) were standardized to the expression of the GAPDH housekeeping gene. Bars represent means ± SEM (n = 3); student's t-test calculated to the control (freshly isolated monocytes = 1, first bar). (B) Cells were lysed and proteins (75 µg) separated by SDS-PAGE were subjected to immunoblot analysis. One representative experiment out of three is shown.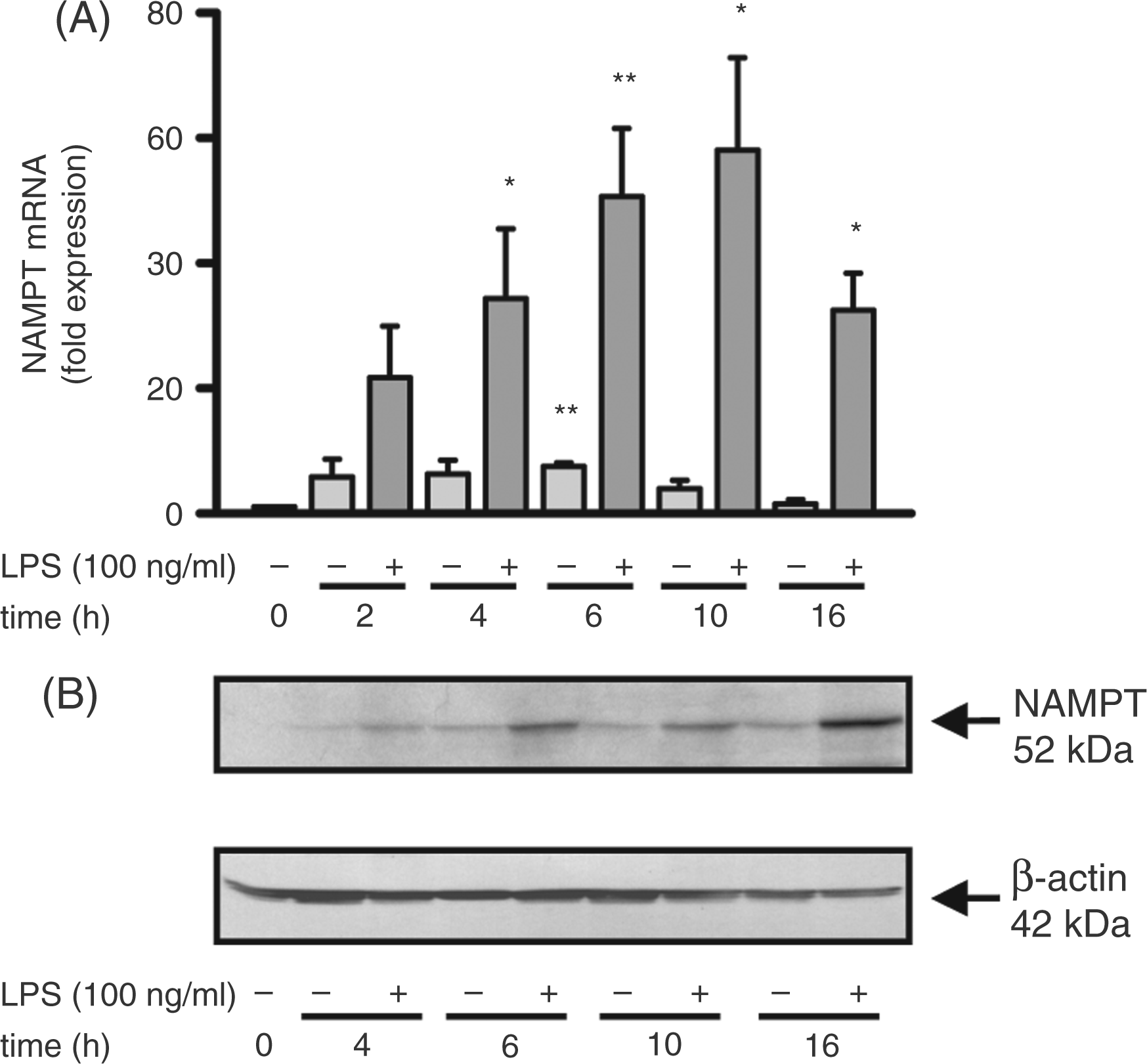
The remarkable up-regulation of NAMPT expression made us assume that the enzyme plays a significant role in LPS-regulated responses. To verify such a putative role, we blocked the enzyme activity of NAMPT by the use of the NAMPT inhibitor APO866.
Effect of APO866 on cell viability
According to previous reports in which APO866 was shown to induce apoptosis in hematologic cancer cells, 34 we first tested the effect of this inhibitor on the viability of primary human monocytes. In three independent experiments the cells were incubated with or without 100 µM APO866 for 4 h and 16 h in the presence and absence of LPS. Apoptosis was assessed by double staining with annexin V/propidium iodide. When incubated for 4 h, the percentage of apoptosis was hardly affected by the presence of 100 µM APO866 in both resting and LPS-activated monocytes (range 10.2%–16.9%). However, when prolonging the incubation time to 16 h, resting monocytes showed a slight increase in apoptosis (24.5% ± 1.8%), which was significantly elevated in the presence of 100 µM APO866 (51.0% ± 2.5% ***). In contrast, the viability of LPS-activated monocytes remained unaffected by the presence of APO866 (apoptosis rate ranged from 14.1%–16.5%). LPS has been suggested to rescue monocytes from apoptosis either directly through the activity of the CD14-TLR-complex or indirectly via the inductionof pro-inflammatory cytokines in an autocrine manner. 35 – 37 Interestingly, in activated monocytes, these anti-apoptotic mechanisms seem to overcome the cytotoxic effects of APO866 that include mitochondrialdysfunction, ATP depletion and/or excessive autophagy.18,20,34,38
APO866 inhibits NAMPT activity and reduces intracellular NAD concentrations
We next investigated the influence of APO866 on NAMPT activity and intracellular NAD levels. The enzyme activity was determined by measuring the formation of radioactive NMN in a reaction mixture containing [14C] Nam as substrate. Monocytes incubated for 16 h in the presence of LPS (100 ng/ml) displayed an enzyme activity of 4.5 ± 0.8 nmol/h/mg protein (n = 3), which was completely abolished in the presence of 100 µM APO866. To test whether the reduced NAMPT activity was mirrored by a decline in the intracellular NAD concentration, we evaluated intracellular NAD levels after 4 h and 16 h of incubation in the presence and absence of APO866 and LPS. As seen in Figure 3A, the NAD concentration of unstimulated cells treated with 100 µM APO866 decreased by 57.5% ± 23.9% after 4 h and 84.0% ± 3.4% after 16 h. In LPS-stimulated cells, the decline amounted to 66.4% ± 9.6% after 4 h and 85.5% ± 2.2% after 16 h. This robust decrease demonstrates a high turnover of NAD in unstimulated and LPS-activated monocytes. When adding 10 mM NMN, the product of the NAMPT-catalyzed reaction, to monocytes treated for 4 h with LPS and APO866 the decline in NAD concentrations was nearly prevented (Figure 3B). To test whether NAD depletion had any effect on NAMPT protein expression, we carried out Western blot analyses with lysates of cells treated for 16 h with LPS and APO866. As seen in Figure 3C, APO866 had no influence on NAMPT expression.
Effect of APO866 on intracellular NAD levels. (A) Monocytes (2 × 106/ml) were treated with 100 µM APO866 for 30 min and incubated in the presence or absence of LPS (100 ng/ml) for the times indicated. Cells were lysed and intracellular NAD levels were measured by an enzymatic cycling assay. NAD contents were quantified according to standard curves generated with NAD+ and normalized to the total protein content of cell lysates. Bars represent means ± SEM (n ≥ 3); paired t-test calculated to the PG control. (B) Monocytes (2 × 106/ml) were treated with 100 µM APO866 in the presence or absence of NMN (10 mM) for 30 min before LPS was added for 4 h. NAD levels were measured as described in (A). Bars represent means ± SEM (n = 3); student’s t-test calculated to the PG control (=100%, black bar). (C) Monocytes (2 × 106/ml) were pre-incubated with 100 µM APO866 for 30 min before LPS (100 ng/ml) was added for 16 h. Cells were lysed and proteins (50 µg) separated by SDS-PAGE were subjected to immunoblot analysis using the goat anti-NAMPT and mouse anti-human β-actin Abs. One representative experiment out of three is shown.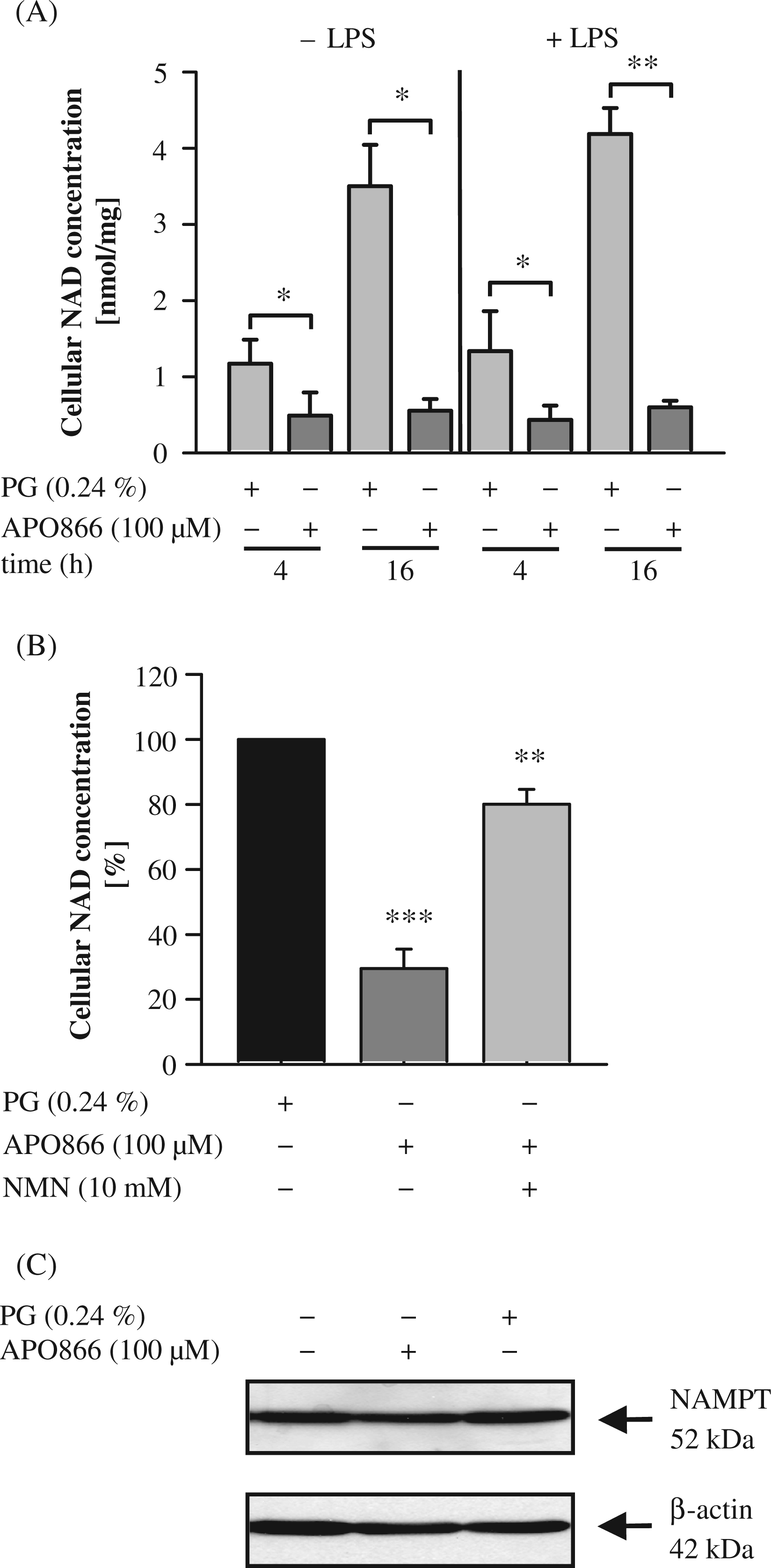
APO866 reduces TNF-α protein production, without affecting mRNA expression
Having verified the inhibitory effect of APO866 on NAMPT activity in monocytes, we next studied its influence on TNF-α production. To this end, monocytes were cultured for 30 min with increasing doses of APO866 and then stimulated with LPS (100 ng/ml) for 4 h. As seen in Figure 4A, APO866 inhibited TNF-α protein production in a concentration-dependent manner. However it had no effect on TNF-α mRNA levels measured after 1–3 h of incubation (Figure 4B). To test whether the reduced TNF-α protein production was caused by a depletion of NAD, monocytes were incubated with APO866 and NMN prior to the addition of LPS. As seen in Figure 4C, although NMN almost replenished the cellular NAD pool (Figure 3B), it failed to prevent the inhibitory effect of APO866. In the absence of APO866, NMN slightly promoted LPS-induced TNF-α secretion. These data indicate that either the replenishment of NAD (80%) was insufficient or that inhibition of TNF-α biosynthesis may have been caused by an increase in intracellular nicotinamide concentrations. Nicotinamide, a potent inhibitor TNF-α,
39
is likely to accumulate when NAMPT is inhibited, irrespective of the intracellular NAD concentrations.
Effects of APO866 on TNF-α and pro-TNF-α expression in LPS-activated monocytes. Monocytes (2 × 106/ml) were incubated with APO866 (A–E) and, if indicated, in the presence of NMN (C) for 30 min before LPS (100 ng/ml) was added. After 4 h of incubation, TNF-α concentrations in the culture supernatants were determined by ELISA (A, C) and after 1 h, 2 h and 3 h TNF-α mRNA expression was analyzed by quantitative real-time PCR. Relative mRNA levels (ΔΔCt-method) were standardized to the expression of the GNB2L1 housekeeping gene (B). Cells were lysed 4 h after incubation and proteins (100 µg) separated by SDS-PAGE were subjected to immunoblot analysis using a polyclonal goat anti-human TNF-α antibody and a mouse anti-human β-actin antibody. One representative experiment out of eight is shown (D, E). For statistical analysis, band intensities were quantified using Adobe® Photoshop® (E). Bars represent means ± SEM (n ≥ 3); paired t-test calculated to the PG control (= 100 % or 1, black bar).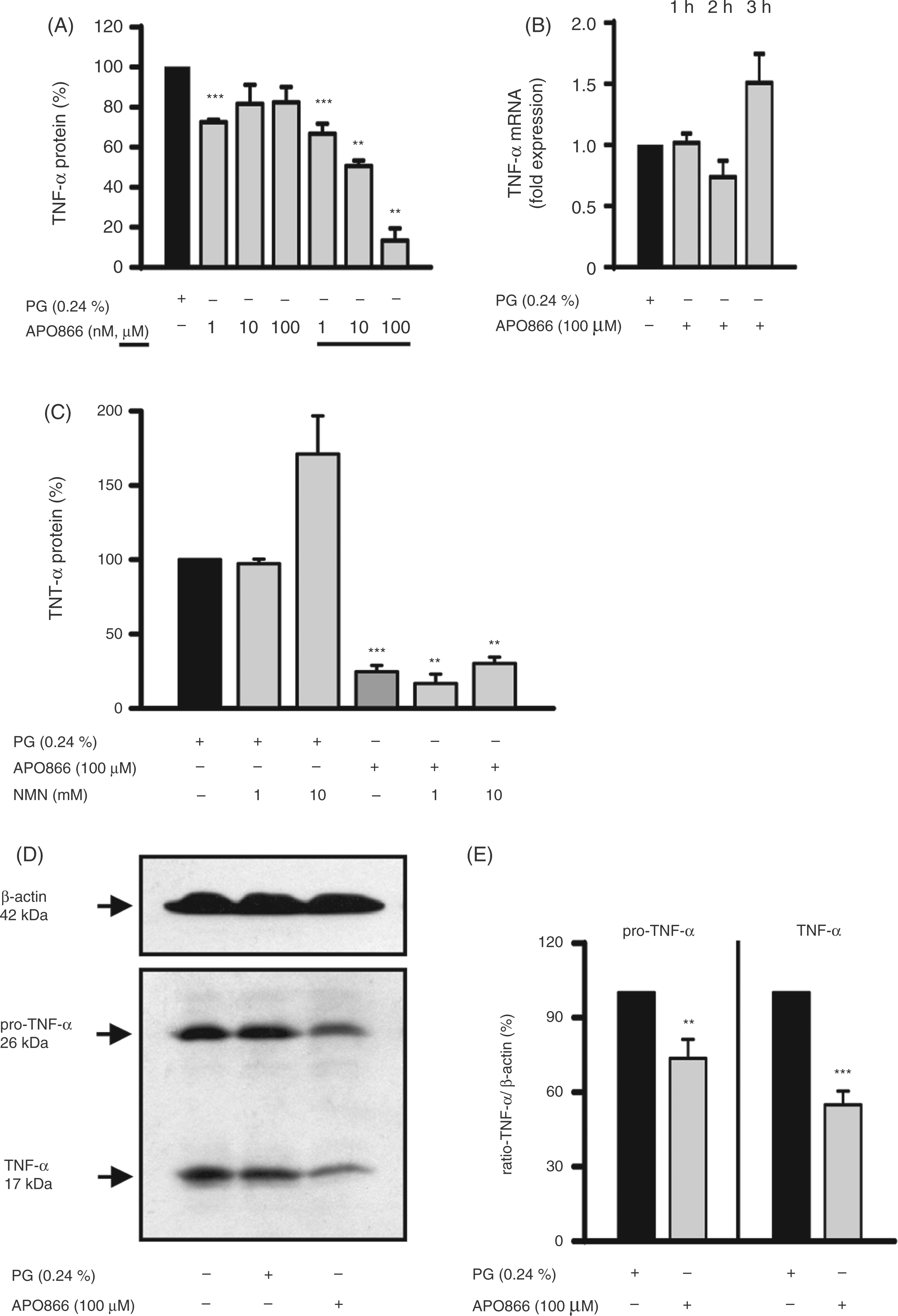
As APO866 reduced TNF-α protein secretion but did not affect mRNA levels, we tested whether the effect of APO866 was caused by diminished production of the precursor form of TNF-α (pro-TNF-α), a 26 kDa transmembrane type II polypeptide from which the soluble 17 kDa polypeptide is derived by proteolytic cleavage. 40 Western blot analyses revealed that the amount of intracellular pro-TNF-α protein was significantly reduced inAPO866-treated monocytes (Figure 4D, E). Taken together, these studies suggest that APO866 affects TNF-α protein production by acting at a post-transcriptional step, possibly by interfering with TNF-α mRNA translation. The unexpected presence of the 17 kDa TNF-α in the cell lysates most likely reflects the binding of soluble TNF-α to TNF-α receptors on the cell surface, so that the amount of detectable 17 kDa TNF-α in lysates mirrored the level of TNF-α secretion.
APO866 decreases protein ADP-ribosylation
The APO866-triggered depletion in intracellular NAD concentrations prompted us to hypothesize that NAD-consuming enzymes, such as mono-ADP-ribosyltransferases (mARTs) and poly-ADP-polymerases (PARPs) would be affected. While mARTs transfer one ADP-ribose moiety from NAD to specific target proteins, PARPs catalyze the transfer of multiple moieties and branching of the ADP-ribose chain. This post-translational protein modification, called ADP-ribosylation, results in an alteration of the target protein’s functional properties.
41
By Western blot analyses we showed that there was extensive ADP-ribosylation of cellular proteins in LPS-activated monocytes. When cells were exposed to APO866, ADP-ribosylation levels decreased (Figure 5), indicating that substantial amounts of NAD are required for optimal ADP-ribosylation of target proteins.
Effect of APO866 on the protein ADP-ribosylation in LPS-activated monocytes. Monocytes (2 × 106/ml) were incubated in the absence and presence of APO866 (100 µM) for 30 min before LPS (100 ng/ml) was added. After 16 h, cells were lysed and proteins (50 µg) separated by SDS-PAGE were subjected to immunoblot analysis using the mouse anti-poly(ADP-ribose) antibody and mouse anti-human β-actin Ab. One representative experiment out of three is shown.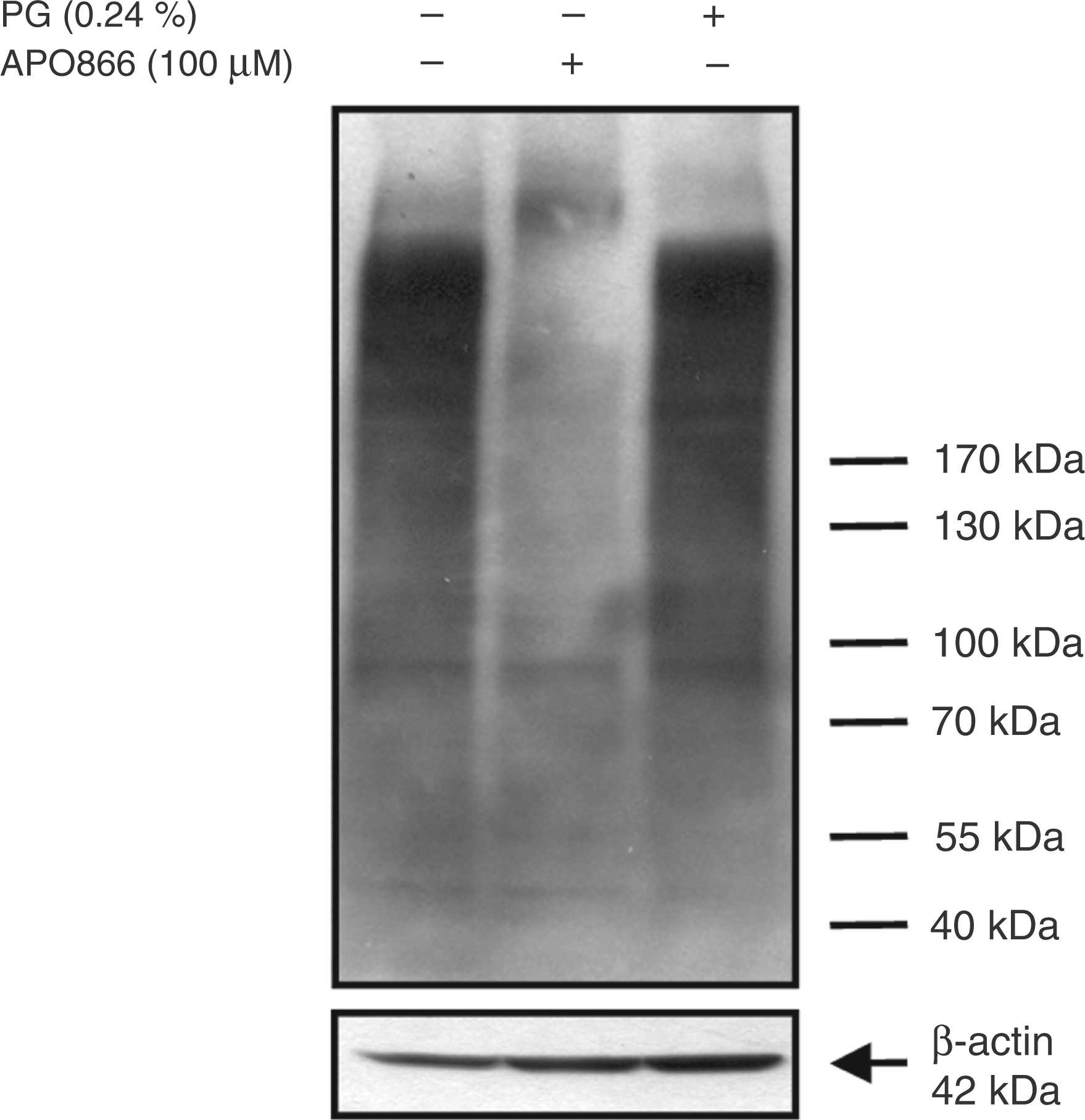
APO866 up-regulates CD38 expression
When studying the effect of APO866 on the expression of CD38, another NAD-consuming enzyme, we found an up-regulation of CD38 at the protein and mRNA level in activated, but not in resting, monocytes (Figures 6A, B). In the presence of NMN and NAD, CD38 expression returned to normal levels (Figure 6C), indicating that NAD depletion was involved in the up-regulation of CD38 expression. A similar effect was observed after the addition of Nam, which has been reported to compete with APO866 on binding to the NAMPT catalytic pocket.42,43
Effect of APO866 on CD38 protein and mRNA expression. (A, B) Monocytes (2 × 106/ml) were treated with APO866 (100 µM) for 30 min and incubated in the absence and presence of LPS (100 ng/ml). (A) After 6 h, CD38 mRNA expression was analyzed by quantitative real-time PCR. Relative mRNA levels (ΔΔCt−method) were standardized to the expression of the GAPDH housekeeping gene. Bars represent means ± SEM (n ≥ 4); student's t-test calculated to the PG control (= 1, black bar). (B) After 16 h of incubation, CD38 protein expression was analyzed by flow cytometry. Bars represent means ± SEM (n ≥ 4); student’s t-test calculated to the PG control [= 1 (A) or 100 % (B), black bar]. (C) Monocytes (2 × 106/ml) were incubated with NAD+, NMN or Nam for 30 min treated with APO866 (100 µM) for another 30 min and incubated for 16 h in the absence and presence of LPS (100 ng/ml). CD38 protein expression was analyzed by flow cytometry. Bars represent medians ± SEM (n ≥ 4); student's t-test calculated to the PG control (black bar).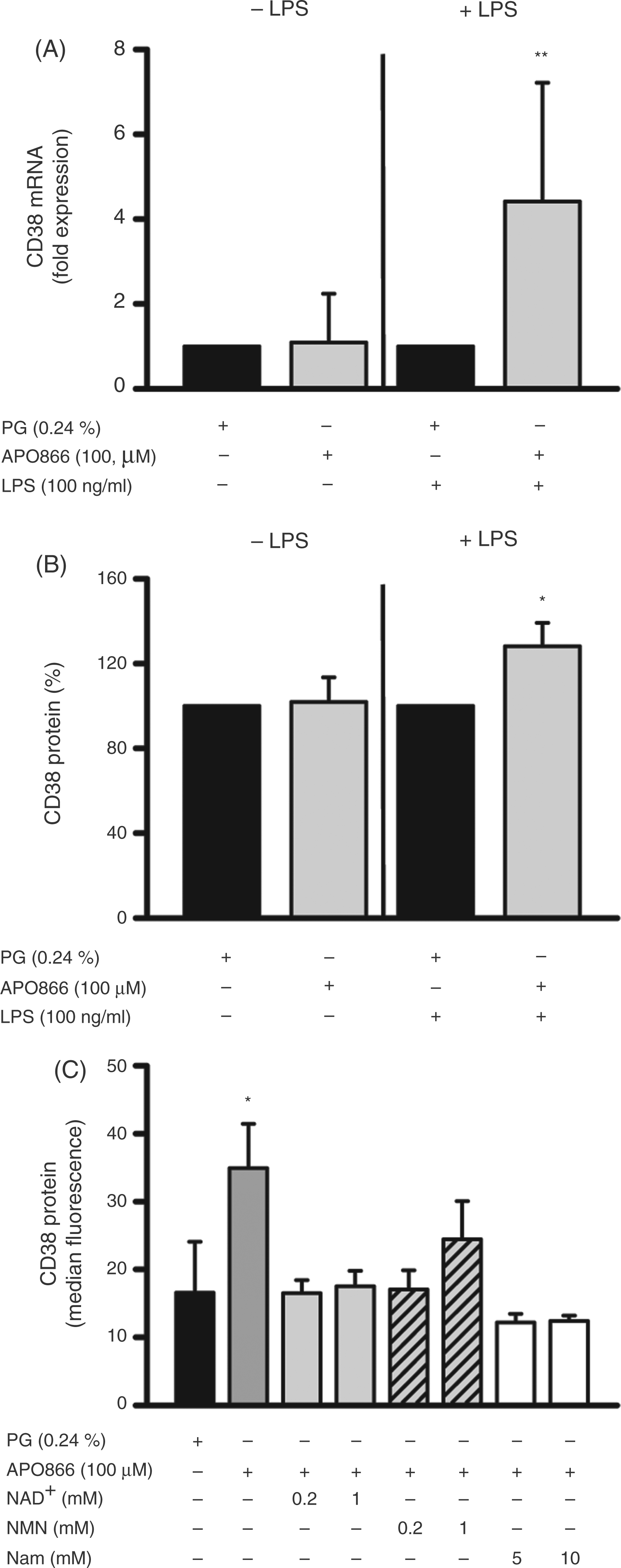
APO866 modulates eicosanoid production
Eicosanoid concentrations in monocytes
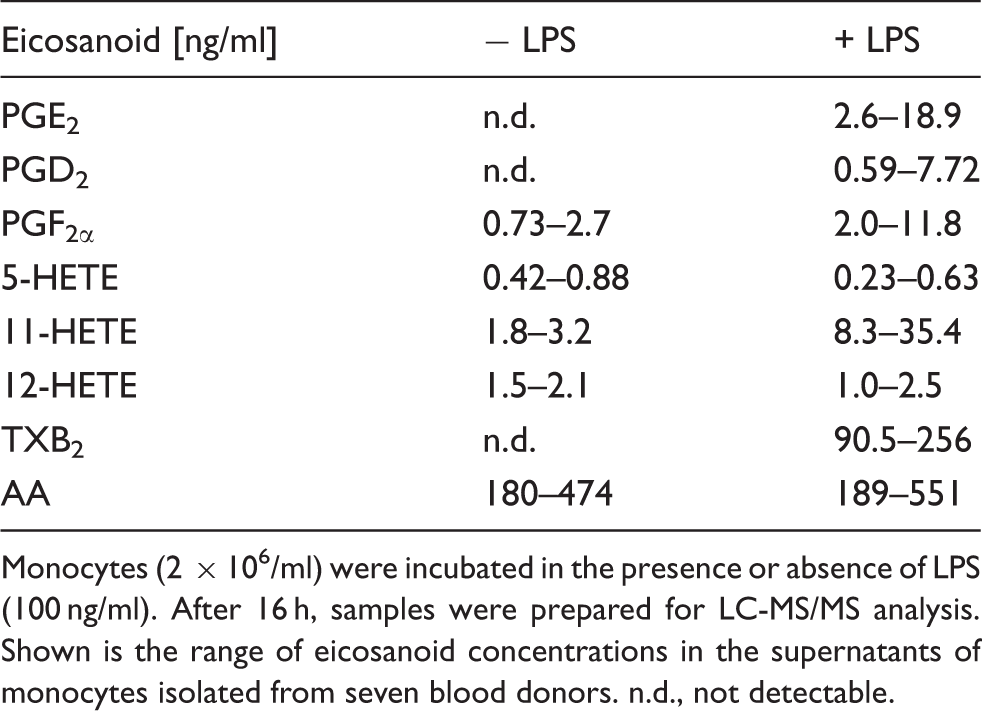
Monocytes (2 × 106/ml) were incubated in the presence or absence of LPS (100 ng/ml). After 16 h, samples were prepared for LC-MS/MS analysis. Shown is the range of eicosanoid concentrations in the supernatants of monocytes isolated from seven blood donors. n.d., not detectable.
Effect of APO866 on eicosanoid production
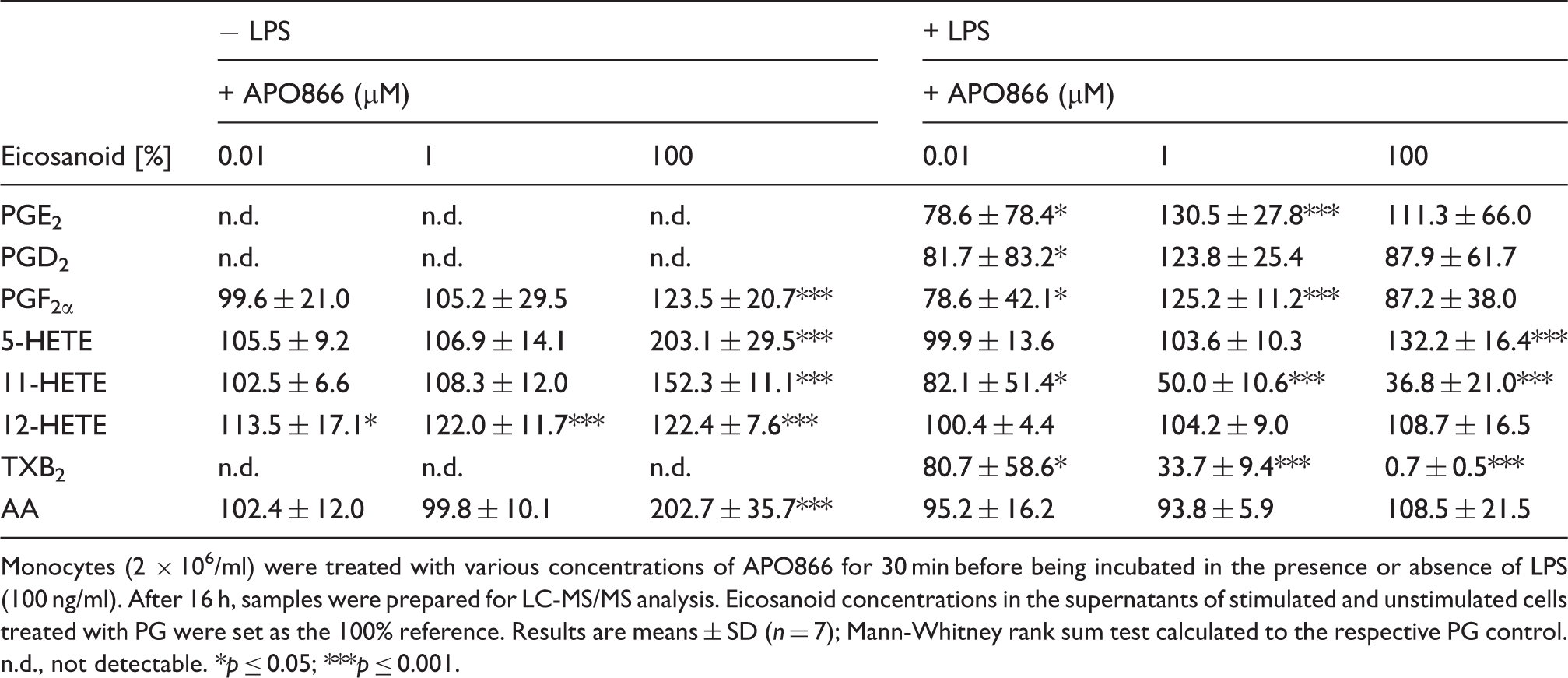
Monocytes (2 × 106/ml) were treated with various concentrations of APO866 for 30 min before being incubated in the presence or absence of LPS (100 ng/ml). After 16 h, samples were prepared for LC-MS/MS analysis. Eicosanoid concentrations in the supernatants of stimulated and unstimulated cells treated with PG were set as the 100% reference. Results are means ± SD (n = 7); Mann-Whitney rank sum test calculated to the respective PG control. n.d., not detectable. *p ≤ 0.05; ***p ≤ 0.001.
APO866 inhibits LPS-induced ROS production
The enzyme involved in the production of ROS is NAD(P)H oxidase, a membrane-located multi-subunit oxidoreductase complex that requires NAD(P)H for its enzyme activity. As seen in Figure 7A, APO866, in a concentration-dependent manner, inhibited the LPS-induced ROS production almost completely at a concentration of 100 µM (Figure 7A, B). Adding NAD and LPS to the cells did not abolish the inhibitory effect of APO866 on ROS production (Figure 7C). When switching the order of stimulation, the LPS-induced burst was immediately reduced by the subsequent addition of APO866, 30 min after LPS activation (Figure 7D). Again, APO866 showed a concentration-dependent inhibitory effect starting at a concentration of 25 µM. The possibility that APO866 acts purely by scavenging ROS or quenching ROS detection was excluded because changes in luminescence induced by H2O2 in the absence of cells were not affected by APO866 (data not shown).
Effect of APO866 on the LPS-induced ROS production of monocytes. (A) Monocytes (2 × 106/ml) were incubated with APO866 in the given concentrations for 30 min before LPS (100 ng/ml) was added. (B) Representative original registrate of one of the experiments summarized in (A). (C) Monocytes (2 × 106/ml) were incubated with APO866 (100 µM) for 30 min before LPS (100 ng/ml) and NAD+ (0.1 mM, 1 mM) were simultaneously added. (D) Monocytes (2 × 106/ml) were stimulated with LPS (100 ng/ml) for 30 min before APO866 in the given concentrations was added. (A–D) The production of ROS was monitored for a further 120 min using a luminometric assay. Results are presented as the integrals of the time-course curves, mean ± SEM (n ≥ 3); paired t-test calculated to the solvent control (= 100%, black bar).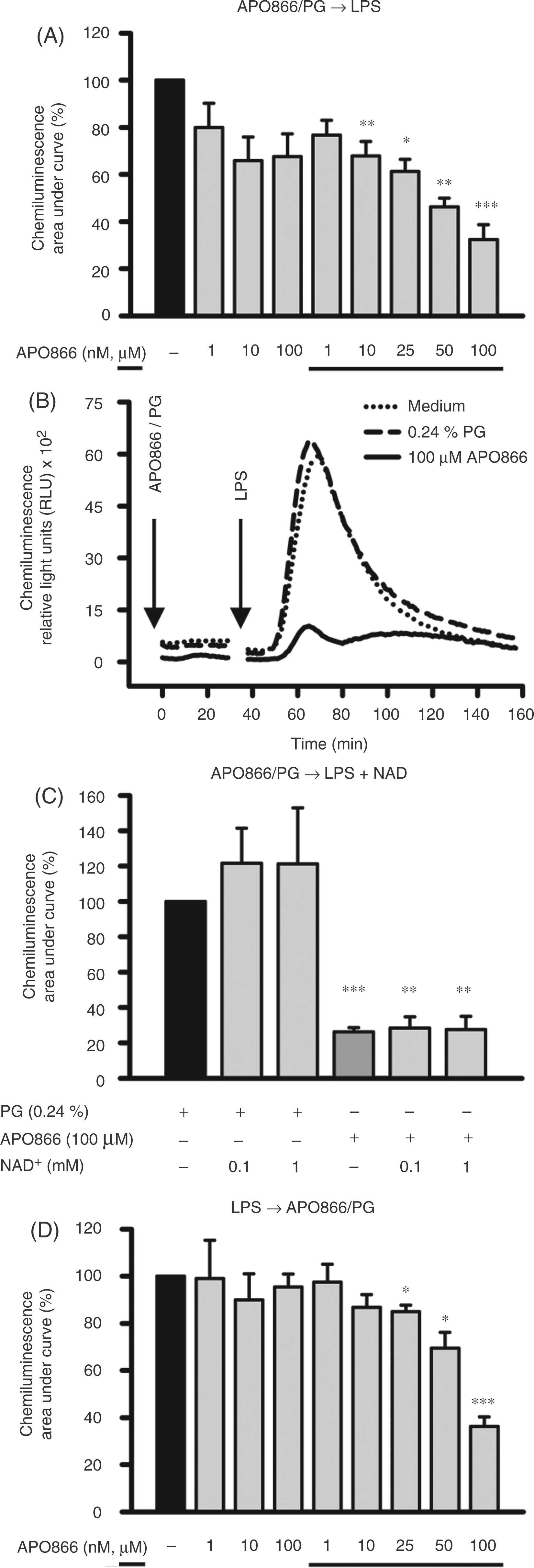
Discussion
LPS, the major component of the outer membrane of Gram-negative bacteria, is a highly potent activator of monocytes. It not only induces the expression of multiple genes encoding inflammatory mediators to combat infection, but, as shown here, it also affects the expression of NAMPT, an enzyme directly involved in NAD metabolism.
Whereas LPS strongly up-regulated NAMPT expression, it had only minor effects on mRNA and protein levels of NMNAT, the enzyme that catalyzes the final step in the NAD salvage pathway (data not shown). Thus, consistent with its housekeeping role, NMNAT contributes to a stable cellular concentration and rapid availability of NAD required for many NAD-dependent processes.
As described previously and as shown here, the inhibition of NAMPT activity led to a decrease in intracellular NAD concentrations and resulted in diminished TNF-α production.10,11 This reduction was not caused by apoptosis, as no loss in viability was detected in LPS-activated monocytes. It is surprising that, despite massive NAD depletion, activated monocytes are almost resistant to cell death, whereas unstimulated cells are not. Considering that metabolic processes are increased in activated monocytes, one would expect these cells to be more susceptible to NAD depletion but, obviously, the anti-apoptotic effects displayed by LPS overcome the demands for sufficient NAD supply. This is not a common feature of all immune cells. As reported by Bruzzone et al., activated, but not resting, T cells are crucially dependent on NAMPT activity for their function and survival. 44
Compared with our studies, the concentrations of APO866 used by Busso et al. to achieve maximum inhibition of TNF-α expression were much lower ranging in nanomolar concentrations. 9 The reason for this discrepancy may lie in different experimental conditions. Whereas we only incubated the cells for 4.5 h, Busso et al. exposed the cells to APO866 for 20 h. As the kinetics of LPS-induced TNF-α secretion show a maximum at 4 h, we favoured this time-point for our investigations. To achieve sufficient NAD depletion under these conditions, the use of higher APO866 concentrations was necessary.
Although the molecular mechanisms linking NAMPT inhibition by APO866 and blockage of TNF-α production are largely unknown, there is some evidence that NAD-consuming enzymes with multiple roles in cellular regulation, such as PARPs, mARTs and sirtuins, are involved in this process. 45
Bruzzone et al., who showed that APO866 inhibits TNF-α production in T cells, relate the defective production to an impaired activity of SIRT6, a member of the sirtuin family. 44 In line with these findings, Van Gool et al. suggest a role of SIRT6 in the post-transcriptional control of TNF-α expression. 46 We recently reported that LPS induces an increase in ADP-ribosyltransferase activity in human monocytes and that inhibitors of ADP-ribosylation interfere with phosphorylation events and cytokine production.24,39
Moreover, our studies showing that exhaustion of intracellular NAD correlates with a marked reduction in PARP/ART activity and a reduced capacity to produce TNF-α also point to a link between ADP-ribosylation and TNF-α synthesis. While some studies favoured the view that PARP1, through inhibition of nuclear factor-κB transcriptional activity, regulates TNF-α production; 47 others dismissed a role of PARP1 in this process. 46
In addition to its function as an enzyme, extracellular NAMPT has been reported to act as a pro-inflammatory mediator triggering the release of cytokines. As NAMPT secreted by monocytes could regulate cytokine production in an autocrine fashion, 16 we reasoned that, should APO866 inhibit NAMPT synthesis, its capacity to act as a mediator may be impaired. However, by showing thatAPO866 has no impact on NAMPT synthesis, this mode of action does not account for the decrease in TNF-α production.
Depletion of intracellular NAD not only affected TNF-α synthesis but also the production of eicosanoids, another group of mediators that influence inflammation and immune responses. In resting monocytes, APO866 increased the production of all detectable eicosanoids, the highest values being reached by 5-HETE and AA. As AA is released by cells undergoing apoptosis,48,49 its increase may very well be attributed to the catabolic state of resting monocytes. The elevation of the remaining eicosanoids could be a mere consequence of the increase of AA, which gives rise to prostanoids (prostaglandins, leukotriens) and HETEs. Alternatively, 12-HETE and 5-HETE, which have been associated with anti-apoptotic activities,50,51 could rise in an attempt to counteract apoptosis. The up-regulation of 5-HETE may also be caused by the limited availability of NAD/NADP, which serve as cofactors of enzymes involved in 5-HETE metabolism. 52 – 56 APO866-induced changes in the eicosanoid pattern of activated monocytes include a modest increase in 5-HETE, PGE2 and PGF2α and a pronounced decline in 11-HETE and TXB2 concentrations. As TXA2 has a relatively short half-life and is rapidly converted to the more stable TXB2, 57 TXA2 production is measured by monitoring TXB2. In addition to PGE2 and 11-HETE, TXA2 is a quantitatively important product of stimulated human monocytes. 58 – 61 While TXA2 has been identified as an eicosanoid that facilitates TNF-α synthesis, PGE2 seems to suppress the production of this cytokine.62,63 Given that the treatment of LPS-activated monocytes with APO866 results in decreased levels of TXA2 and elevated levels of PGE2 an anti-inflammatory effect of APO866 may be anticipated. The accumulation of prostaglandins may be explained by diminished activity of the NAD-dependent 15-hydroxyprostaglandin dehydrogenase, the key enzyme responsible for the biological inactivation of prostaglandins. 64 Whatever the precise mechanisms that underlie the observed effects of APO866 on eicosanoid production are, our data indicate that the response of resting monocytes differs from that of activated monocytes, and that selected pathways are affected in the latter cells.
Similar to eicosanoid production, expression of CD38 was measured after 16 h of incubation with APO866, a time at which NAD levels were drastically reduced. As supplementation with NAD and NMN reversed the effect, depletion of NAD is clearly associated with the up-regulation of CD38. The CD38 molecule is widely known as a multifunctional ecto-enzyme that metabolizes NAD and NADP generating cyclic ADP-ribose (cADPR), ADPR and nicotinic acid adenine dinucleotide phosphate (NAADP). These reaction products are essential for the regulation of intracellular Ca2+, the most ancient and universal cell-signalling system. 65 Should the concentration of these products fall below a critical value, the cell may try to compensate for this loss by up-regulating the expression of CD38 with the option of cleaving additional NAD and internalizing the reaction products cADPR and NAADP via CD38.66,67 As a side-effect, up-regulation of CD38 activity would also result in an increase in Nam, which stabilizes cellular NAD concentrations by inhibiting NAD-consuming enzymes. We could, indeed, show that, in the presence of Nam, CD38 expression returned to basal levels.
Not only TNF-α and eicosanoid production, but also LPS-induced ROS production, was subject to inhibition by APO866. The production of ROS is a short-term effect starting soon after encounter of the stimulus. The immediate effect of APO866 applied before and after initiation of the oxidative burst, as well as the failure of NAD to restore ROS production, suggests that APO866 described as a specific inhibitor of NAMPT 20 may directly affect NAD(P)H oxidase activity independent of its ability to reduce intracellular NAD levels. However, should Nam accumulate as a consequence of NAMPT blockage, it has the potential to suppress the generation of ROS as described previously. 32
In conclusion, our data clearly indicate that the NAD rescue pathway is of great significance for human monocytes to mount an appropriate immune response. Considering that APO866 is now under scrutiny in phase II trials for the treatment of solid and hematological malignancies, it would be of interest to monitor the response of monocytes to this treatment, especially under conditions of primary and secondary infection.
Footnotes
Acknowledgements
This study was supported by the Deutsche Forschungs-gemeinschaft (HA 2484 / 3-1).
APO866 was a kind gift from Topo Target Germany AG (Frankfurt am Main, Germany) and the rabbit anti-NAMPT antibody was kindly provided by Amgen (Thousand Oaks, CA, USA).
We are indebted to Dr Matt Sweet (Institute for Molecular Bioscience, The University of Queensland, Australia) for critically reading the manuscript.