
Abstract
Neutrophils are among the first cells to arrive at the site of injury. Chemokines secreted by neutrophils affect the migration of both neutrophils and other inflammatory cells, such as monocytes. It has been reported that LPS-induced release of IL-8 (CXCL-8) by neutrophils is amplified by neutrophil-derived TNF-α. We hypothesize that chemokine release by neutrophils is altered in chronic obstructive pulmonary disease (COPD) compared with healthy controls and that TNF-α may be involved in this alteration. Peripheral blood neutrophils isolated from smokers with COPD (n = 12), smokers without COPD (n = 12) and healthy, non-smokers (n = 12) were stimulated with LPS, TNF-α or organic dust. Anti-TNF-α Ab (infliximab) was used to study the effect of neutrophil-derived TNF-α. Release of CXCL-8, macrophage inflammatory protein-1 α (MIP-1α, CCL-3), monocyte chemotactic protein-1 (MCP-1, CCL-2) and TNF-α was measured. Neutrophils spontaneously released CXCL-8, CCL-2 and CCL-3. Inhibition of TNF-α reduced the spontaneous release of CXCL-8 and CCL-3. Stimulation with LPS and organic dust increased the release of CXCL-8 and CCL-3 (but not CCL-2) which was reduced by inhibition of TNF-α. In the COPD group, inhibition of TNF-α failed to inhibit the release of LPS-induced CXCL-8. The role of neutrophils as cytokine and chemokine producers was confirmed. Neutrophil-derived TNF-α contributed to the release of chemokines after stimulation with LPS and organic dust, as the response was inhibited by infliximab. In the COPD group, infliximab did not significantly inhibit the release of CXCL-8, suggesting that the role of TNF-α is altered in COPD.
Keywords
Introduction
Neutrophils are the most abundant leukocyte in human blood and play a central role in the first line of defence against invading pathogens. Traditionally, the main function of neutrophils has been considered to be the killing and phagocytosis of invading bacteria by the generation of reactive oxygen species (ROS) and release of enzymes. However, it has also become clear that neutrophils contribute to inflammatory responses, being a source of cytokines and chemokines, such as IL-1β, IL-8 (CXCL-8), TNF-α, macrophage inflammatory protein-1α (MIP-1α, CCL-3) and monocyte chemotactic protein (MCP-1, CCL-2). 1 – 3
Chronic obstructive pulmonary disease (COPD) is characterised by inflammation of the large and small airways and progressive tissue destruction, causing airflow obstruction. 4 In COPD, there are increased numbers of neutrophils in the circulation, as well as in central and peripheral airways, as demonstrated by analyses of blood, sputum and bronchoalveolar lavage (BAL) fluid. 5 – 7 Moreover, the number of neutrophils in biopsies and sputum correlates with disease severity.5,8 A major chemo-attractant for neutrophils is CXCL-8 which is produced by neutrophils but also by other cell types, such as macrophages and bronchial epithelial cells. Neutrophils release CXCL-8 upon stimulation by exogenous agents, such as LPS but also in an autocrine manner when stimulated by other pro-inflammatory cytokines, such as TNF-α.9,10 Furthermore, sputum levels of CXCL-8 and TNF-α are increased in patients with COPD compared with healthy controls and there is a correlation between CXCL-8 levels and the number of neutrophils in BAL fluid.6,8
COPD has been described as a process of several steps where the innate immune response with neutrophils constitutes the first step which is then followed by activation of the adaptive immune system with T-cells. 11 Chemokines released by neutrophils act upon other inflammatory cells, such as monocytes and lymphocytes. Thus, neutrophil-derived CCL-3 and CCL-2 are involved in macrophage recruitment into inflamed tissue. 12 It has also been suggested that CCL-2 and MIP-1β (CCL-4) are involved in lung and airway inflammation in humans as the levels of these chemokines are increased in BAL fluid from smokers with chronic bronchitis. 13
Exposure of healthy subjects to the environment within swine confinement buildings causes intense airway and systemic inflammation with a cellular predomination of neutrophils, a huge increase in bronchial responsiveness, and systemic effects, including fever. 14 , 15 Furthermore, dust from swine confinement buildings is a potent inducer of cytokine and chemokine release in airway epithelial cells and alveolar macrophages in vitro. 16
In the present study, we hypothesized that the release of chemokines that direct the migration of neutrophils and mononuclear cells is altered in COPD compared with healthy non-smokers. We also hypothesized that TNF-α plays a central role in the regulation of chemokine release and that this pro-inflammatory cytokine may be important in the possible alteration of chemokine release in COPD. Therefore, isolated peripheral blood neutrophils from smokers with and without COPD, and healthy non-smokers were collected and chemokine production and release was measured following stimulation with LPS, organic dust and TNF-α in the presence and absence of an anti-TNF-α Ab (infliximab).
Materials and methods
Subjects and study design
Characteristics of the subjects. Data are presented as mean and 95% confidence intervals
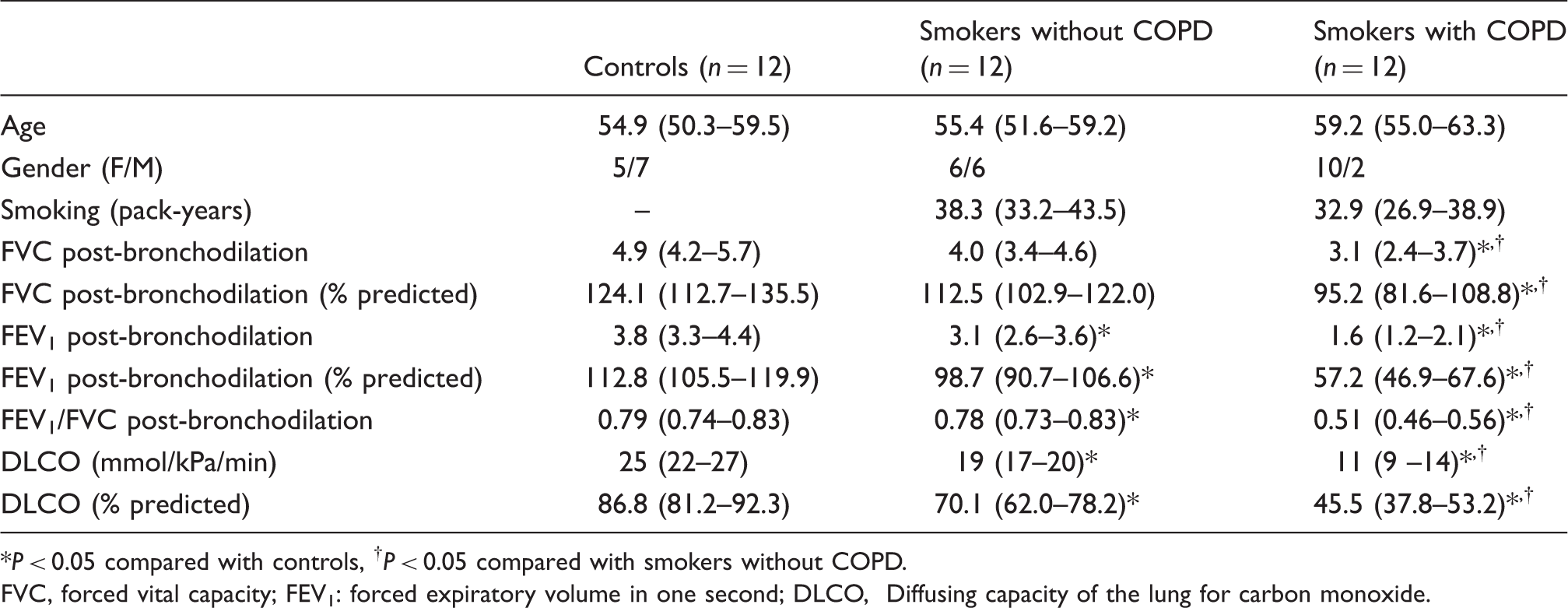
P < 0.05 compared with controls, †P < 0.05 compared with smokers without COPD.
FVC, forced vital capacity; FEV1: forced expiratory volume in one second; DLCO, Diffusing capacity of the lung for carbon monoxide.
Spirometry was performed according to current guidelines (the American Thoracic Society/European Respiratory Society) and diffusion capacity [transfer factor, diffusing capacity of the lung for carbon monoxide (DLCO)] was performed using carbon monoxide single breath technique.17,18
The study was approved by the local ethics committee at Karolinska Institutet (Stockholm, Sweden) and all subjects gave their informed consent.
Isolation of peripheral blood neutrophils
Blood was collected in heparin Venoject® tubes (Terumo Sweden AB, Västra Frölunda, Sweden) and neutrophils were isolated within 1 h of collection. The blood was mixed with Dulbecco's PBS (D-PBS) without Ca2+ and Mg2+ (GIBCO, Invitrogen, Carlsbad, CA, USA) containing 2% dextran (Pharmacosmos, Holbaek, Denmark) and allowed to sediment for 40 min. Next, the leukocyte-containing portion was carefully layered onto Lymphoprep™ (Medinor AB, Lidingö, Sweden). After centrifugation at 600 g for 25 min, the granulocyte-containing fraction was collected. This was followed by hypotonic lysis of the remaining erythrocytes. The cells were then washed twice with D-PBS before they were re-suspended in RPMI 1640 medium (Sigma, St Louis, MO, USA) supplemented with
Stimulation of peripheral blood neutrophils
Cells were seeded into 24-well plates (Nunc A/S, Roskilde, Denmark) at a density of 106 cells/well and incubated at 37°C in 5% CO2 for 4 or 16 h. One of the following stimuli were added to the wells with or without infliximab (Schering-Plough, 5 µg/ml, Stockholm, Sweden): LPS (Escherichia coli serotype O111:B4, 1 µg/ml; Sigma-Aldrich, Stockholm, Sweden), TNF-α (5 ng/ml; R&D Systems, Abingdon, UK), organic dust from pig barns (collected from pig barn shelves and window ledges about 1.2 m above the floor, 10 µg/ml). The supernatants were collected and centrifuged at 1000 g for 10 min to remove cell debris and stored at −70°C until analysis. Viability, as assessed by Trypan blue exclusion (Sigma), averaged >90% after 16 h of incubation. No significant differences in viability were noted between subject groups or treatments.
Preparation of mRNA and real-time PCR
Extracted mRNA was prepared from isolated peripheral blood neutrophils from five subjects in each group. After 4 h stimulation, as described above, the supernatant was removed and total mRNA was isolated from the cells using PureLink™ Micro-to Midi Total RNA Purification System (Invitrogen, Stockholm, Sweden). First strand cDNA was synthesised from 0.3 µg of total RNA using QuantiTect®Reverse Transcriptase (Qiagen, Sollentuna, Sweden). Real-time PCR was performed using ABI Power SYBR Green Master mix (Applied Biosystems, Stockholm, Sweden) with primers for CXCL-8, CCL-2 and CCL-3. Beta-actin was used as an internal control gene. Primers were designed using Primer Express 3.0 (Applied Biosystems) and purchased from CyberGene AB (Stockholm, Sweden).
Data were analysed using 7500 Software v.2.0.1 (Applied Biosystems) and transformed using the ΔCt-method. The results are expressed as 2−ΔCt.
Assays for cytokine and chemokine measurement
Cytokines (IL-1β, TNF-α) and chemokines (CXCL-8, CCL-2) were analysed using Bender MedSystems FlowCytomix™ Technology. Analyses were performed using FACSCalibur™ and concentrations of the different chemokines were determined using FlowCytomix Pro 2.2 Software (Bender MedSystems, Vienna, Austria). Results are presented as pg/ml. The range of the standard curve was 0–2000 pg/ml for IL-1β, 0–10,000 pg/ml for CXCL-8, 0–3000 pg/ml for CCL-2 and 0–20,000 pg/ml for TNF-α. TNF-α was only measured in samples not containing recombinant TNF-α and/or infliximab.
CCL-3 was analyzed by ELISA using a Human CCL3/MIP-1 alpha DuoSet kit (R&D Systems, Abingdon, UK) according to the manufacturer's instructions. The range of the standard curve was 7.8–500 pg/ml.
Statistics
Subject characteristics are presented as mean with 95% confidence intervals and comparisons were made using analysis of variance (ANOVA). Cytokine and chemokine concentrations and mRNA levels were not normally distributed, as assessed by Shapiro-Wilk test, and are therefore presented as median with 25th–75th percentiles. Comparisons between groups were performed using the Kruskal-Wallis test followed by the Mann-Whitney U-test when appropriate. Within-group comparisons were performed using the Wilcoxon signed rank test. A P-value below 0.05 was considered significant. All data was analysed using STATISTICA 9 software (StatSoft, Uppsala, Sweden).
Results
Summary of CXCL-8 and CCL-3 results. Results are presented as median and 25th–75th percentiles
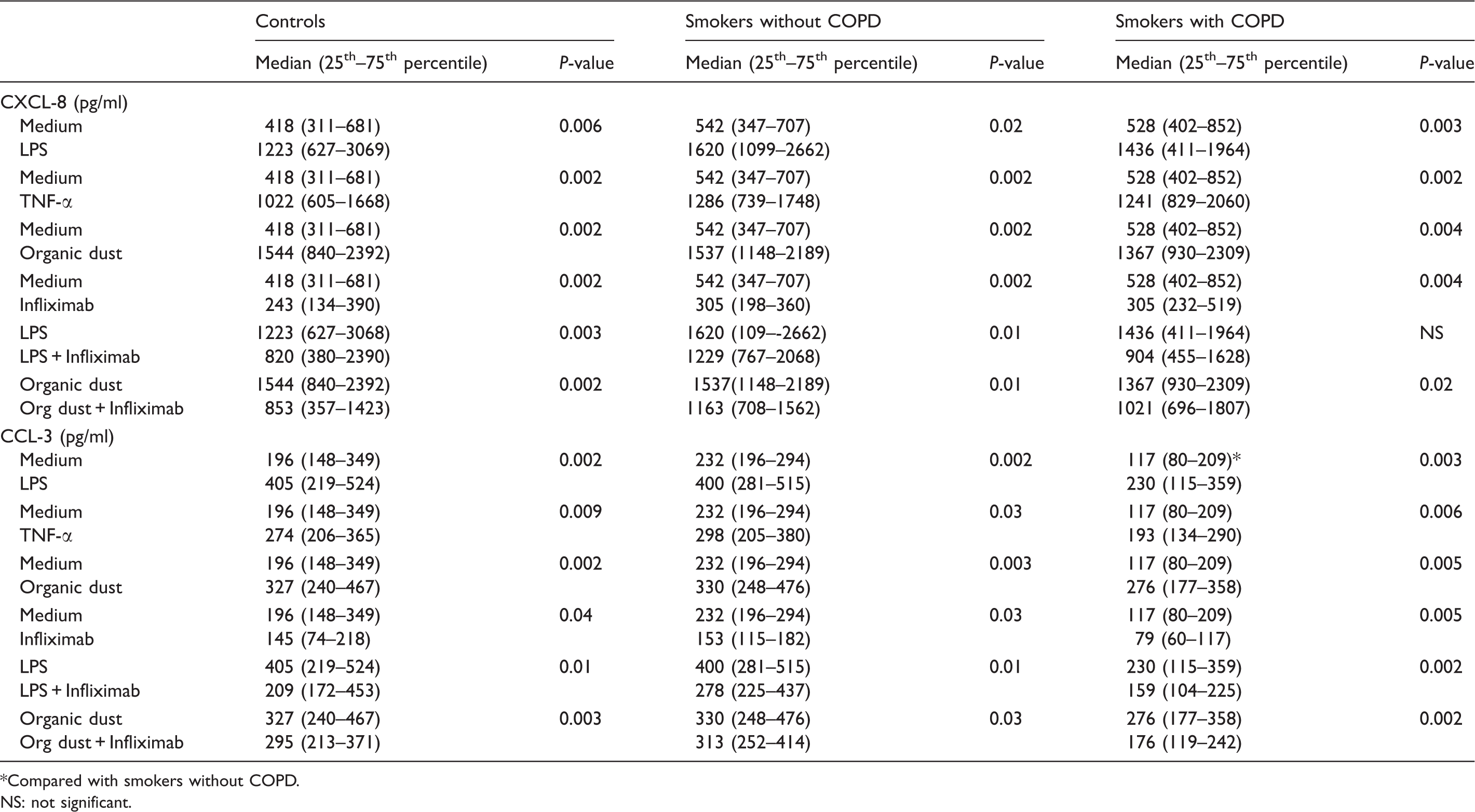
Compared with smokers without COPD. NS: not significant.
Non-stimulated neutrophils (medium controls)
The spontaneous release of CCL-3 was significantly lower in the COPD group compared with the smokers without COPD (P = 0.009, Figure 1). Furthermore, the spontaneous release of CXCL-8 and CCL-3 (Figure 1), but not of CCL-2 (data not shown), was inhibited by infliximab in all groups.
Release of (A) CXCL-8 and (B) CCL-3 from unstimulated cells and cells treated with TNF-α (5 ng/ml) or infliximab (5 µg/ml). *P ≤ 0.05, ** P ≤ 0.01 effect of TNF-α compared with unstimulated control at the same time point. # P ≤ 0.05, ## P ≤ 0.01 effect of infliximab compared with unstimulated control at the same time point.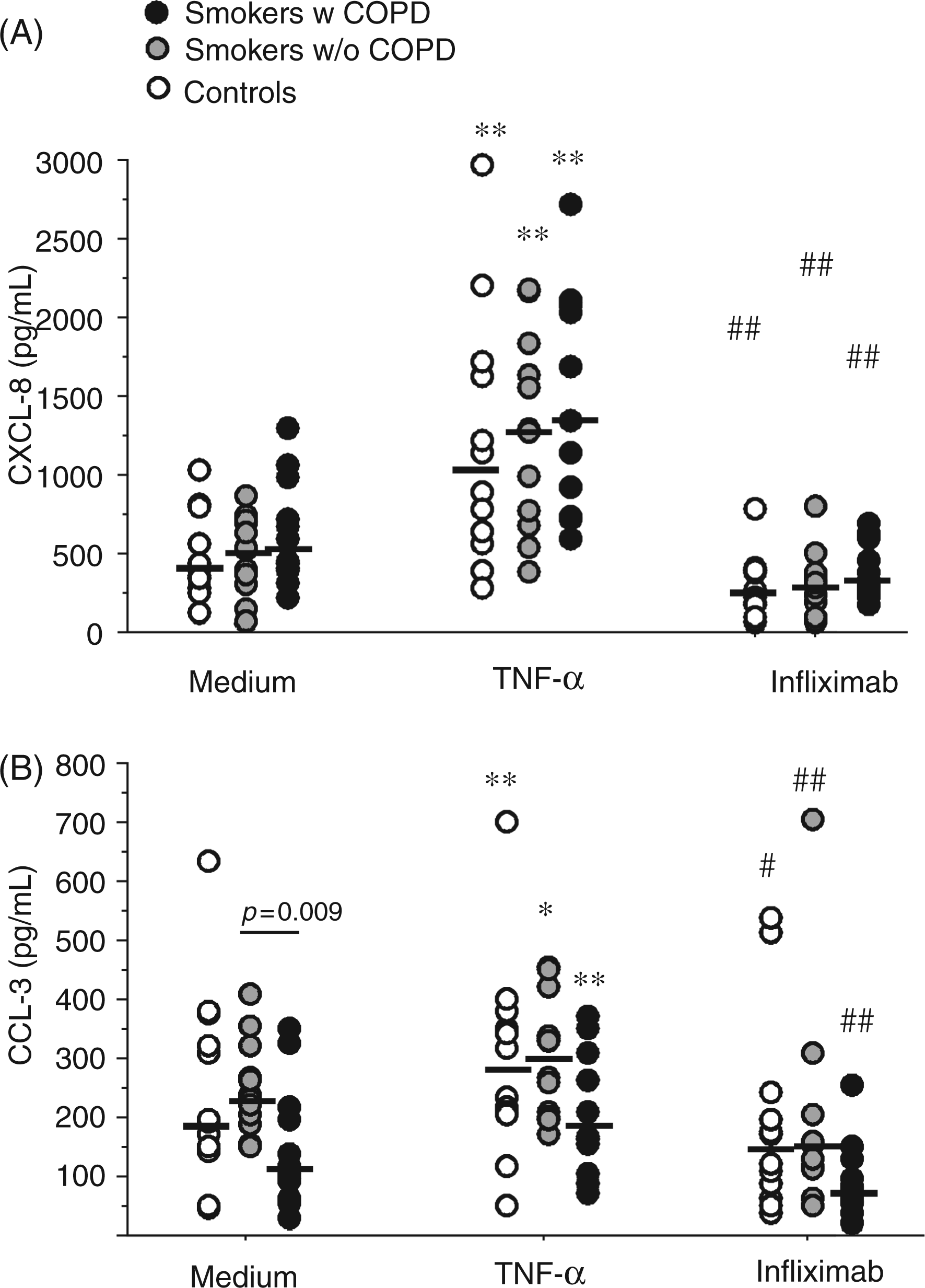
Neutrophils stimulated with TNF-α
The release of CXCL-8 and CCL-3 was stimulated by TNF-α in all three groups (Figure 1); however, TNF-α did not alter CCL-2 release in any group (data not shown).
Neutrophils stimulated with LPS
Stimulation with LPS increased CXCL-8 and CCL-3 in all groups (Figure 2), whereas CCL-2 release was unaltered by LPS (data not shown).
Release of (A) CXCL-8 and (B) CCL-3 from LPS-stimulated (1 µg/ml) cells and cells treated with LPS (1 µg/ml) and infliximab (5 µg/ml). * P ≤ 0.05, ** P ≤ 0.01 compared with unstimulated control at the same time point. # P ≤ 0.05, ## P ≤ 0.01 compared with LPS-stimulated cells.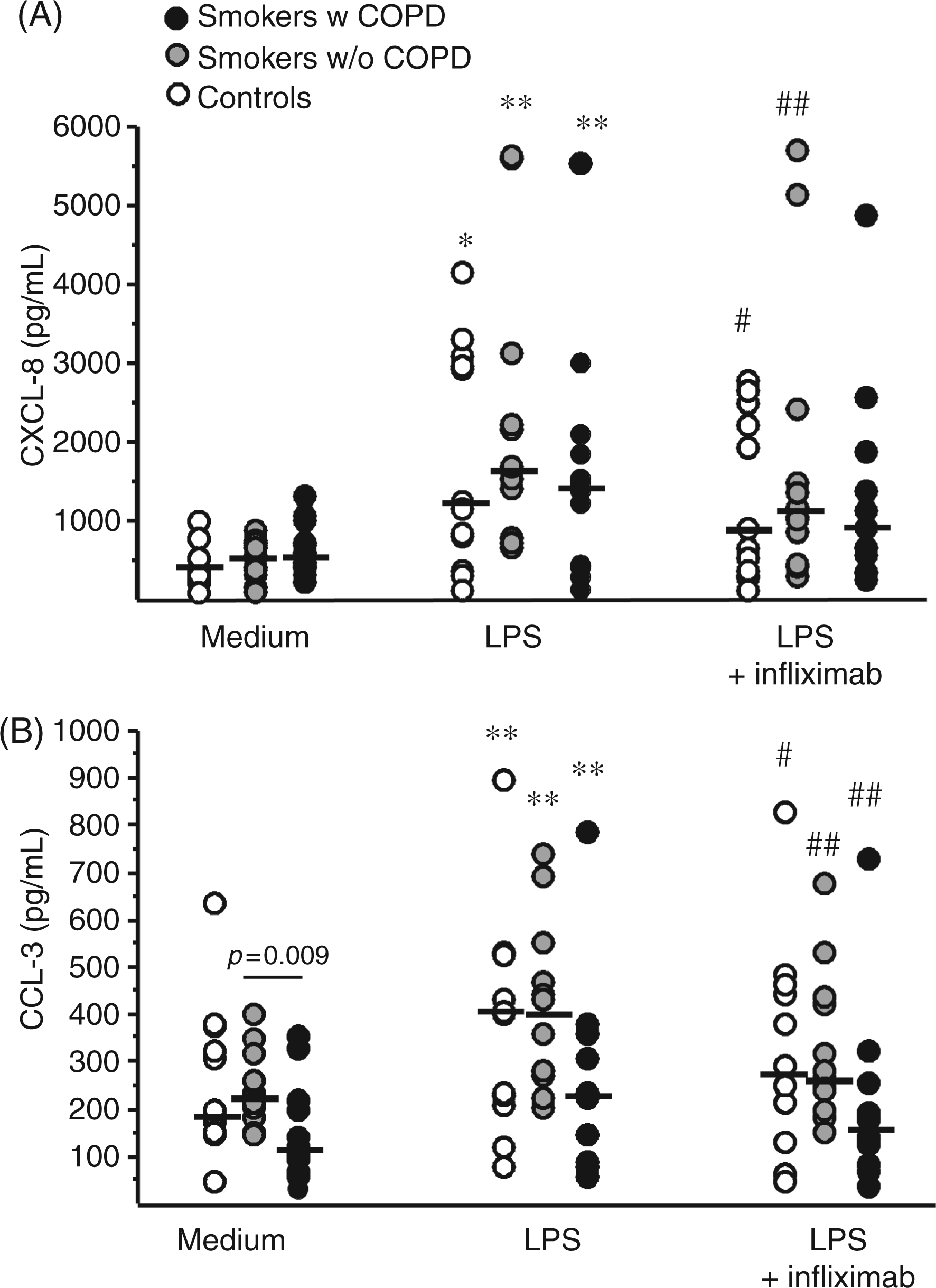
Infliximab inhibited the LPS-induced release of CCL-3 in all three groups (P < 0.05), and inhibited LPS-induced release of CXCL-8 in the control group (P = 0.003) and in smokers without COPD (P = 0.01), but not in smokers with COPD (P = 0.11).
Neutrophils stimulated with organic dust
Dust increased CXCL-8 and CCL-3 release in all groups (Figure 3) but did not alter CCL-2 release (data not shown). Infliximab inhibited dust induced release of CXCL-8 and CCL-3 in all groups (P < 0.05, Figure 3).
Release of (A) CXCL-8 and (B) CCL-3 from dust-stimulated (10 µg/ml) cells and cells treated with organic dust (10 µg/ml) and infliximab (5 µg/ml). * P ≤ 0.05, ** P ≤ 0.01 compared with unstimulated control at the same time point. # P ≤ 0.05, ## P ≤ 0.01 compared with swine dust-stimulated cells.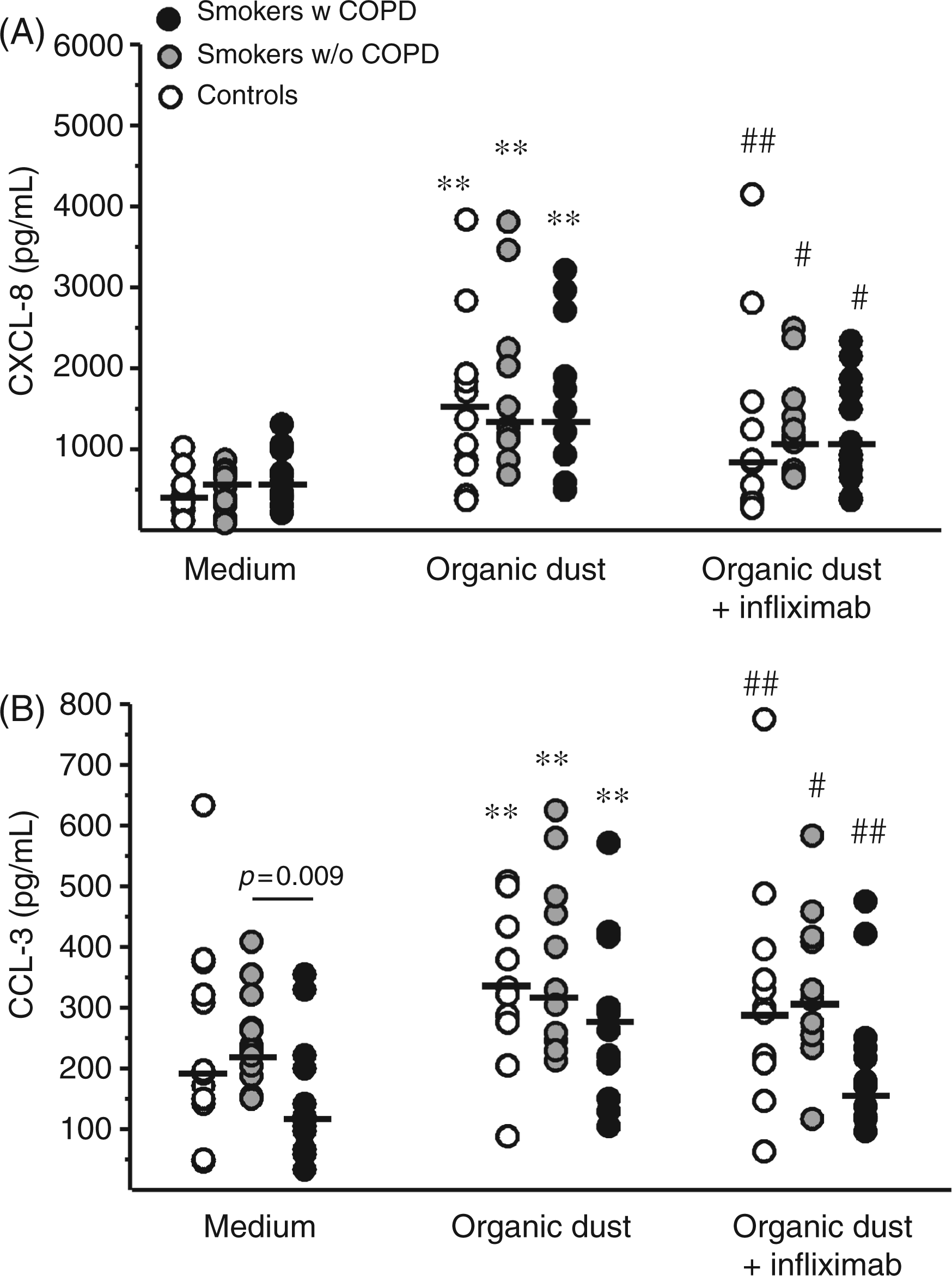
Combined data analyses
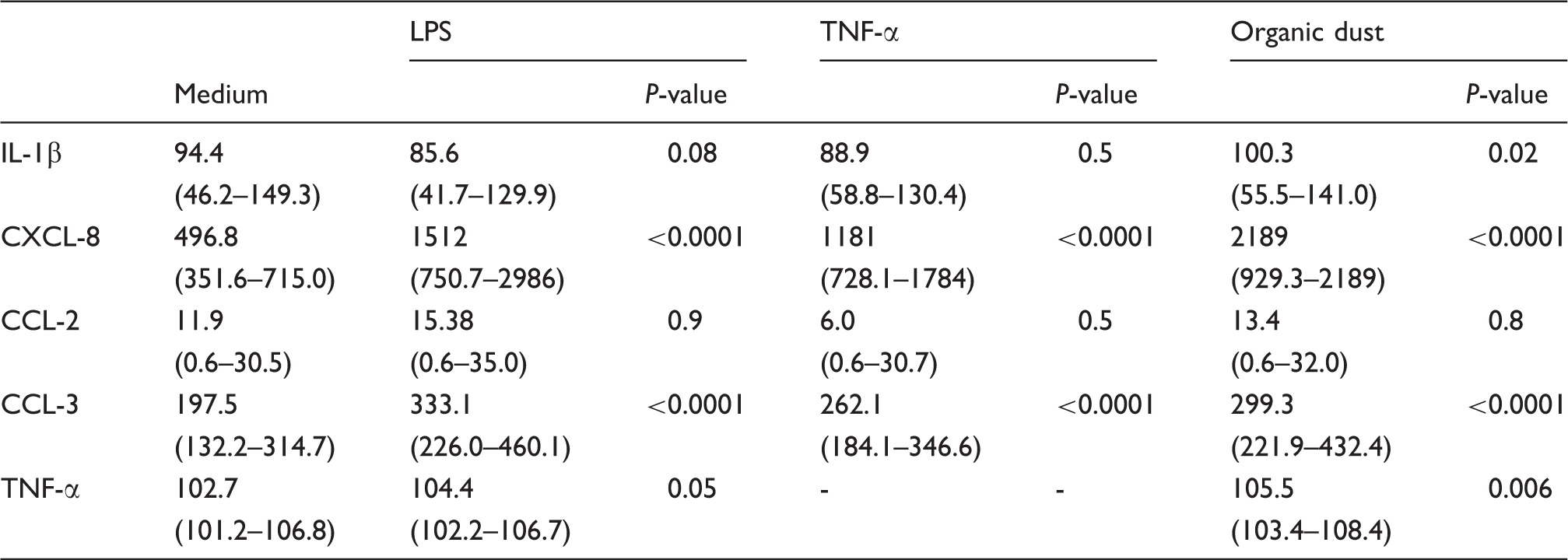
Comparisons are made between medium control and stimuli at 16 h [combined data from three groups (n = 36)]. Results are expressed as pg/ml. Data are presented as median (25th–75th percentiles). P-values indicate comparison with medium
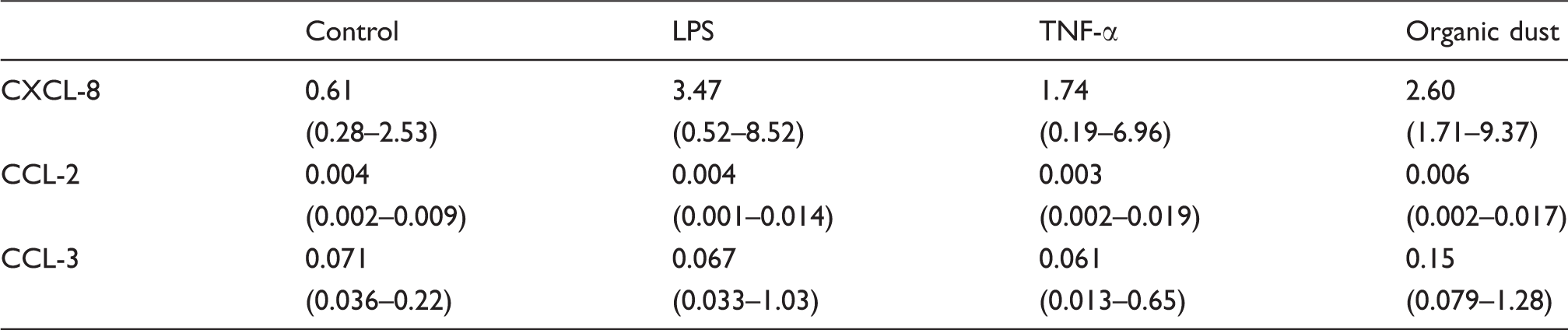
Chemokine mRNA expression at 4 h [combined data from the three groups (n = 14)]. Results are expressed as 2−ΔCt and presented as median and 25th–75th percentiles
Discussion
In the current study, we confirm that neutrophils release CXCL-8, CCL-2 and CCL-3 and that endogenous TNF-α is involved in LPS- and organic dust-induced release of CXCL-8 and CCL-3 as the release of these chemokines was partly inhibited by the anti-TNF-α Ab (infliximab). Interestingly, infliximab was less effective in reducing the release of CXCL-8 in the COPD group.
The LPS-induced release of CXCL-8 from neutrophils has been described as a bi-phasic response, where the first phase has a direct effect of LPS and the second phase is induced by endogenously-produced mediators (e.g. TNF-α). 9 We found that the addition of infliximab inhibited CXCL-8 release in controls and smokers without COPD, while inhibition of TNF-α did not influence CXCL-8 release in the COPD group. The reason for this lack of inhibition is not clear and there are several possible explanations. First, LPS-induced TNF-α release may be enhanced in COPD—an interpretation that is supported by a tendency towards higher TNF-α levels in the COPD group compared with the other groups after 4 h of LPS stimulation. Second, another possible explanation for the lack of inhibition by infliximab in the COPD group is that neutrophils from the COPD patients are more sensitive to stimulation and therefore display a stronger response to endogenous TNF-α than do neutrophils from the other two groups. This priming phenomenon, which has been demonstrated in neutrophils from COPD patients,19,20 is caused by a prior low level exposure of the cells to a priming agent (e.g. TNF-α) which greatly increases the cells' ability to respond to an activating agent (e.g. LPS). 21 If these are the mechanisms behind the lack of inhibition of CXCL-8 release by infliximab it is possible that a higher dose of infliximab may have caused inhibition. However, in pilot experiments on neutrophils from healthy volunteers, inhibition by infliximab reached a plateau at the selected dose (5 µg/ml) and increasing doses did not enhance inhibition further. Irrespective of this, the finding still indicates that the response pattern to LPS is shifted in the COPD group compared with the control groups. Third, one may speculate that the relative importance of TNF-α as a mediator of chemokine release is reduced in COPD, and that agents other than TNF-α become increasingly important in the disease pathophysiology.
The chemokines CCL-2 and CCL-3 have been previously shown to be produced and released by neutrophils and it has been suggested that CCL-2 plays a role in the pathogenesis of COPD as it is increased in BAL fluid from smokers with chronic bronchitis.2,22,23 However, in the present study, CCL-3 was far more responsive to stimulation than CCL-2 was. While mRNA and protein for CCL-2 were detected at baseline, none of the stimuli tested in this study induced an increase in CCL-2. Previous studies show conflicting data with regard to CCL-2 release by neutrophils and it seems as if co-stimulation with IFN-γ is required for CCL-2 release following stimulation with low doses of LPS. In addition, CCL-2 expression has also been described as a late event, peaking after up to 48 h of incubation.24,25 It is therefore possible that the increased levels of CCL-2 observed in BAL fluid from COPD subjects are produced by a cell type other than neutrophils. However, the findings of the current experiments would suggest that the release of CCL-2 by circulating neutrophils in smokers is influenced by endogenous stimuli and activation with exogenous stimuli is without effect.
The spontaneous release of CXCL-8, CCL-2 and CCL-3 detected in medium controls was inhibited by infliximab for CXCL-8 and CCL-3. Consequently, this low spontaneous activity, which may, in part, be caused by the isolation process, includes an effect mediated by endogenous TNF-α as it was reduced by infliximab.
Somewhat surprisingly, CCL-3 release in unstimulated neutrophils was significantly lower in the COPD group compared with the smokers without COPD. This may be indicative of a changed chemokine pattern in COPD. The migration of inflammatory cells to the site of injury is a carefully regulated process and even small changes in chemokine levels could lead to changes in the recruited subsets of inflammatory cells. It has previously been reported that CCL-3 affects the migration of different lymphocyte subsets in a concentration-dependent manner, with low concentrations mainly attracting CD8+ T-cells and higher concentrations primarily attracting CD4+ T-cells. 26
IL-1β is released in response to LPS stimulation and has an effect similar to that of TNF-α on the second phase of LPS induced CXCL-8 release. 9 However, we were unable to detect a significant increase in IL-1β after stimulation with LPS. It is possible that this can be explained by a difference in measuring techniques or stimulation protocols between our study and previous studies. However, when data from the three groups were combined, organic dust induced a substantial release of IL-1β indicating that IL-1β is more important to the cytokine response after dust stimulation than after stimulation with LPS. It is not known which dust components are responsible for inducing the production and release of inflammatory mediators, but it seems likely that microbial products are of importance. The LPS content in the organic dust used for stimulation was approximately 2 ng/100 µg dust, 16 strongly implicating that the organic dust effects are not primarily induced by LPS.
The current study was performed using neutrophils isolated from peripheral blood. It is important to consider the possibility that neutrophils from lungs and circulation may behave differently, yet COPD is increasingly recognised as a systemic disease 4 and it is not entirely unreasonable to assume that peripheral blood neutrophils display characteristics similar to lung neutrophils. In the present study, we detected differences in neutrophil reactivity between smokers with and without COPD, indicating that systemic engagement increases as the destructive process in the lung progresses.
In conclusion, we have shown that neutrophils are capable producers of chemokines such as CXCL-8 and CCL-3 and that the regulation of these chemokines involves TNF-α. Moreover, we found a diminished effect of TNF-α inhibition on LPS-induced CXCL-8 production in the COPD group, indicating that TNF-α regulation of CXCL-8-release in COPD may be altered. It is possible that this is indicative of other mechanisms taking priority over TNF-α in COPD.
Footnotes
Acknowledgements
The authors would like to thank Dr Anita Simhag, Dr Maria Skedinger and co-workers at Huddinge Hospital for valuable technical assistance. The study was supported by grants from Swedish Heart and Lung Foundation [grant nos 20080442 and 2008437], Karolinska Institutet, AstraZeneca Sweden, Swedish Medical Research Council [grant no. K2010-57X-20368-04-3], King Gustaf V's and Queen Victoria's Foundation [grant no. 2465/07] and Stockholm City County Council [grant nos 20080154 and 20060150].
Conflicts of interest
None of the authors has any potential financial conflict of interest related to this article.