
Abstract
Rotavirus (RV), a leading cause of diarrhea, primarily infects intestinal epithelial cells (IEC). Rotavirus-infected IEC produce IFN-β and express hundreds of IFN-dependent genes. We thus hypothesized that type 1 IFN plays a key role in helping IEC limit RV replication and/or protect against cell death. To test this hypothesis, we examined IEC (HT29 cells) infected with RV (MOI 1) ± neutralizing antibodies to IFN-α/β via microscopy and SDS-PAGE immunoblotting. We hypothesized that neutralization of IFN would be clearly detrimental to RV-infected IEC. Rather, we observed that blockade of IFN function rescued IEC from the apoptotic cell death that otherwise would have occurred 24-48 h following exposure to RV. This resistance to cell death correlated with reduced levels of viral replication at early time points (< 8 h) following infection and eventuated in reduced production of virions. The reduction in RV replication that resulted from IFN neutralization correlated with, and could be recapitulated by, blockade of IFN-induced protein kinase R (PKR) activation, suggesting involvement of this kinase. Interestingly, pharmacologic blockade of caspase activity ablated RV-induced apoptosis and dramatically increased viral protein synthesis, suggesting that IFN-induced apoptosis helps to control RV infection. These results suggest non-mutually exclusive possibilities that IFN signaling is usurped by RV to promote early replication and induction of cell death may be a means by which IFN signaling possibly clears RV from the intestine.
Introduction
Rotaviruses (RVs) are the leading cause of severe dehydrating diarrhea in young children worldwide, resulting in up to 100 million cases and more than 600,000 deaths annually,1,2 Part of the Reoviridae family of dsRNA viruses, RVs primarily infect epithelial cells of the small intestine, causing self-limiting illness that is typically cleared within seven days.1,3,4 While RV infection is associated with B and T cell responses that help assure complete viral clearance and protect against re-infection 3 , mice lacking adaptive immunity maintain considerable control, and sometimes complete clearance, of the virus, indicating that innate immunity is important for controlling this viral pathogen.5,6 Given that intestinal epithelial cells (IEC) are the main cell type targeted by RVs in vivo and the first line of defense against pathogens in the gut in general,7,8 it seems likely that IEC play a role in mediating innate immunity to RVs. Like most viral infections of cells, RV-infected IEC produce type 1 IFN. Moreover, a substantial portion of the overall gene expression elicited in RV-infected IEC was shown to be dependent on type 1 IFN signaling, in that induction was reduced by neutralizing antibodies to type 1 IFN. 9 Additionally, Type 1 IFN levels have also been shown to increase in RV-infected humans and animals. 10
That RV-induced epithelial gene expression exhibits substantial dependence upon type 1 IFN is in accordance with the broad role played by IFN in anti-viral innate immunity. 11 Type 1 IFN, produced in an autocrine or paracrine manner, activates the JAK/signal transducers and activators of transcription (STAT) signaling pathways that induce expression of hundreds of anti-viral genes, also known as interferon-stimulated genes (ISGs), which combinatorially function to limit viral replication and spread in infected and neighboring uninfected cells.11–14 One of these ISGs, interferon regulatory factor 7 (IRF7), is a transcription factor that serves to promote further type 1 IFN production. 12 Another ISG, protein kinase R (PKR), serves as a sensor of viral dsRNA and may facilitate viral clearance via promotion of apoptosis.11,15 Such broad anti-viral action of type 1 IFNs are in accordance with observations that interference with type 1 IFN signaling greatly impairs the ability of mice to clear several classes of viruses, 16 particularly in mice lacking adaptive immunity. 17 In contrast, mice lacking type 1 IFN exhibit relatively normal clearance of RV, even upon a Rag -/- background, 18 making the role of RV-induced type 1 signaling unclear.18,19 On the other hand, pre-treatment of mice with type 1 IFN can reduce diarrhea in vivo20,21 and limit RV infection in vitro. 22 Although studies in RV-infected mice show that loss of type 1 IFN receptors does not alter diarrhea or rate of viral clearance, 19 STAT1 deficiency correlates with increased viral shedding in feces during RV infection. 16 The fact that RV has adopted strategies for thwarting type 1 IFN responses and thus increasing infectivity, such as by encoding NSP1 proteins that dampen type 1 IFN production23,24 or preventing STAT1 accumulation in the nucleus, 25 also supports the notion that type 1 IFNs pose a significant threat to RVs.
We recently reported that the structural components of RVs induce type 1 IFN in IEC, suggesting that RV-induced type 1 IFN responses are activated via PRR-mediated pathways with a likely role for recognition of viral nucleic acids. 9 Here, we sought to determine the role of RV-induced type 1 IFN in affecting the outcome of the RV-IEC interaction. We observed that neutralizing the type 1 IFN response resulted in a dramatic impairment of anti-viral signaling. Surprisingly, such ablation of anti-viral signaling reduced viral replication and prevented RV-induced IEC cell apoptosis. Pharmacologic blockade of caspase activity prevented RV-induced cell death and markedly enhanced RV replication. Taken together, these results suggest that RV exploits type 1 IFN to promote an initial burst of replication but IFN signaling is also beneficial to the host in that it promotes apoptosis that may limit the infection.
Materials and methods
Antibodies and reagents
Rabbit and guinea-pig anti-RV sera were provided as a kind gift from Jon Gentsch at the Center for Disease Control (CDC). Antibodies to total and phosphorylated STAT1 and PKR were obtained from Cell Signaling Technology (Beverly, MA, USA). Total IRF 7 antibodies were purchased from Santa Cruz Technology (Santa Cruz, CA, USA). Human IFN-α/β and antibodies to human IFN-α/β (anti-IFN α/β) were obtained from the National Institute of Allergy and Infectious Disease (NIAID) Reference Reagent Laboratory through ATTC (Manassas, VA, USA). β-Actin antibodies were obtained from Sigma-Aldrich (St. Louis, MO, USA), respectively. Cleaved poly (ADP-ribose) polymerase (PARP) antibody was obtained from Cell Signaling. 2-Aminopurine (2AP) and Z-VAD-FMK were purchased from Sigma and R&D Systems (Minneapolis, MN, USA), respectively.
Cell culture and rotavirus propagation
Model intestinal epithelia (HT29) were cultured as previously described on standard tissue-culture plastics 26 or Lab Tek Chamber slides (Nalge Nunc International, Rochester, NY, USA). Rhesus rotavirus (RRV) was propagated in MA104 cells and titered as previously described.27,28
Cell infection with rotavirus and type 1 IFN antibodies
Before infection, RV was diluted in serum-free medium (SFM) to a multiplicity of infection (MOI) 1 and incubated with 10 µg/ml trypsin (Mediatech, Inc., Manassas, VA, USA, #25-054-CI) for 30 min in a 37οC water bath. Control samples were treated with an equivalent amount of trypsin diluted in SFM (Mock) or SFM alone. Where indicated, neutralizing antibodies to human anti-IFN-α/β (1:100) were added to some preparations of trypsinized RV before infection. Cell monolayers were washed several times with SFM and inoculated with virus alone or virus plus anti-IFN-α/β for 1 h at 37οC/5% CO2 to allow for adsorption. Following adsorption, cells were washed again several times with SFM and then incubated with 2 µg/ml trypsin in SFM or SFM-only for 0–48 hours post-infection (hpi). Cells stimulated with RV in the presence of anti-IFN-α/β were treated with 2 µg/ml trypsin in SFM plus the same concentration of type 1 IFN antibodies used during viral adsorption. At various time points from 0–48 hpi, supernatants were collected and stored at −20οC for ELISA. Cells were washed several times with PBS and resuspended in radioimmunoprecipitation assay II buffer (20 m
Cell stimulation with type 1 IFNs alone or in the presence of type 1 IFN antibodies
Before stimulation, type 1 IFNs (α/β) were diluted in SFM to a concentration of 200 IU/ml. Where indicated, type 1 IFN antibodies (anti-IFN-α, anti-IFN-β) were added to type 1 IFN preparations (1:100). Confluent cells were washed three times in SFM and treated with type 1 IFNs (200 IU/ml) or type 1 IFNs plus anti-IFN-α/β for 0–48 h. At the indicated time points, cells were washed three times with PBS and resuspended in RIPA II buffer for Western blotting or TRIzol for quantitative real-time PCR (qRT-PCR) analysis as described above.
Cell infection with RV and 2mM 2AP
Trypsinized RV was diluted in SFM alone (MOI 1) or SFM containing 2 m
Cell treatment with rotavirus and Z-VAD
Confluent HT29 cells were washed three times with SFM and pre-treated with 20 µM Z-VAD-FMK (pan-caspase inhibitor) for 2 h. Following pre-treatment, cells were washed with SFM and infected with RV (MOI 5) or RV + Z-VAD (20 µM) for 1 h/37°C/5% CO2. Control samples were treated with an equivalent amount of trypsin diluted in SFM (Mock). Cells were washed with SFM and incubated with 2 µg/ml trypsin or 2 µg/ml trypsin containing Z-VAD (20 µM) for 48 hpi. Cells were analyzed via microscopy and Western blotting, as described elsewhere, for viral protein synthesis or apoptosis. Cell supernatants were assessed for number of dead cells via hemocytometer.
Western blotting
Cells were grown to confluence and stimulated with indicated stimuli as described above. At various time points from 0–48 hpi, cells were washed three times in PBS, lysed in RIPA II buffer, as described above, and cleared by centrifugation (10 min at 15000 g, 4οC). Total protein concentrations were estimated for lysates by BioRad Protein Assay (Bio-Rad, Hercules, CA, USA). Equal amounts of protein were assayed for anti-viral and apoptotic markers (IRF7, STAT1 and PKR, cleaved PARP) and viral proteins by 12% SDS-PAGE immunoblotting and membranes were stripped and probed for β-actin (control). Immunoblots were visualized with the ECL system (Amersham Biosciences, Piscataway, NJ, USA). Densitometric analyses were used to measure relative intensities of bands on immunoblots and were performed using the Scion Image beta analysis program (Scion Corporation).
Enzyme-linked immunosorbent assay
Confluent cells were treated with indicated stimuli as described above. At various time points from 0–48 hpi, cell-free supernatants were collected and analyzed for viral protein levels via double antibody sandwich ELISA. The assays were performed using anti-rhesus RV polyclonal antibodies from rabbits and guinea-pigs. Briefly, microtiter plates were coated for 18 h at 20°C with rabbit anti-RV, washed several times with 0.05% Tween/ PBS, and blocked for 1h at 20oC with 1% BSA/PBS. Next, plates were washed again, as before, and incubated for 1 h at 20°C with standards and samples that were diluted in PBS. Standards were prepared from rhesus RV that was propagated and titrated in MA104 cells as described previously.27,28 Samples were prepared from supernatants that were diluted in PBS. Following incubation, guinea-pig anti-RRV was added to the plates for 1 h at 20°C. Next, plates were washed and treated with HRP-conjugated donkey anti-guinea pig antibody (Jackson Immunoresearch, Westgrove, PA, USA) for 1 h at 20°C. After several washes, tetramethylbenzidine (TMB) substrate and STOP solution (KPL, Gaithersburg, MD, USA) were added to the plates. Absorbance readings were taken at 450 nm on a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Microscopy
Confluent cells grown on 8-well Lab-Tek chamber slides (Nalge Nunc International, Rochester, NY, USA) were treated with the indicated stimuli as described above. Cell morphology was observed under an inverted microscope (magnification 20-100x). For immunofluorescent microscopy, unless indicated otherwise, stimulated cells were fixed in ice-cold ethanol (95% EtOH) for 10 min at 20°C, washed three times in PBS-0.01% Tween and permeabilized in 0.1% Triton-X for 8 min at 20°C. Cells were washed three times in PBS-0.01%, followed by incubation in blocking buffer (3% BSA/PBS) for 1 h at 20°C and primary antibody (rabbit anti-RV, 1:10,000) in blocking buffer O/N at 4°C. Cells were washed three times with PBS-0.01% Tween, incubated with secondary antibody (anti-rabbit conjugated to FITC, 1:50; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) in blocking buffer for 1 h at 37οC in a humidified chamber, and washed three times with PBS-0.01% Tween. Cells were counterstained with 4',6-diamidino-2-phenylindole (DAPI) (1:1000/PBS) for 10 min at 20°C in the dark and washed three times as described previously. Stained cells were mounted on slides with fluorescent anti-fade medium (VectaShield; Burlingame, CA, USA) and viewed under a fluorescent microscope. To quantitate fluorescence levels per image, threshold analysis was performed using ImageJ v1.36b software (http://rsb.info.nih.gov/ij/).
TUNEL assay
Confluent cells grown on 8-well Lab-Tek chamber slides (Nalge Nunc International) were treated with the indicated stimuli. Cells were fixed in 4% paraformaldehyde for 1 h at 20°C, permeabilized in 0.1% Triton-X/ 0.1% sodium citrate for 2 min on ice, and labeled by an InSitu Cell Death Detection Kit (Roche Diagnostics, Indianapolis, IN, USA), using TUNEL according to manufacturer’s guidelines. Following labeling, samples were counterstained for nuclei with SYTO83 (Invitrogen, Carlsbad, CA, USA) diluted in PBS (1:5000) for 15 min at 20°C in the dark, mounted onto slides with fluorescent anti-fade medium as described above, and viewed under a Zeiss LSM510 laser scanning confocal microscope (Zeiss Microimaging Inc., Thornwood, NY, USA).
Results
Rotavirus-induced type I IFNs elicit IEC anti-viral signaling and apoptosis
Infection of IECs with RV results in a substantial remodeling of IEC gene expression with significant induction of over 1000 genes.9,29 The majority of such RV-induced gene expression in IECs is dependent upon type 1 IFN in that it was blocked by neutralizing antibodies to IFN-α and IFN-β.
9
Here, we observed that such neutralization of type 1 IFN also dramatically reduced RV-induced phosphorylation of STAT-1 and PKR and synthesis of IRF7 that followed RV infection in IECs (Figure 1A), in further accordance with the notion that type 1 IFN plays a key role in the IEC response to RV.9,22,29 To determine the relative roles of IFN-β or IFN-α in the RV-induced IEC response, we examined these signaling events in the presence of selective antibody to IFN-α or IFN-β. We observed that the effect of adding both antibodies together on anti-viral signaling was largely mimicked by antibody to IFN-β, while antibody to IFN-α was without significant effect (Figure 1B). The failure of anti-IFN-α to block RV-induced responses did not reflect inability of IECs to respond to IFN-α nor the ability of the antibody to neutralize its target, as IECs responded to recombinant IFN-α and the response was completely neutralized by anti-IFN-α (Figure 1C). Rather, the inability of anti-IFN-α to affect RV-induced signaling seemed to simply reflect the lack of a role for IEC IFN-α in this in vitro infection as qRT-PCR showed only very slight expression of the IFN-α gene basally or in response to RV infection (data not shown). Conversely, the blockade of RV-induced signaling by antibody to IFN-β likely shows a role for RV-induced IFN-β expression rather than a non-specific action of the antibody, as this antibody effectively neutralized the action of IFN-β and did not have a marked effect on activity of recombinant IFN-α (Figure 1D). Thus, IFN-β mediates a significant portion of RV-induced signaling in IECs.
Roravirus-induced type I IFN, particularly IFN-β, elicits epithelial anti-viral gene expression. Intestinal epithelial (HT29) cell monolayers were infected with RV (MOI 1), RV plus both type 1 IFN antibodies (anti-IFN-α/β), RV plus either type 1 IFN antibody (anti-IFN-α or anti-IFN-β), and control samples that received equivalent amounts of trypsin diluted in serum-free medium (SFM) (Mock) or SFM alone (0) (A, B). In parallel experiments, confluent HT29 cells were treated with type 1 IFNs (IFN-α or IFN-β) alone or in the presence of either anti-IFN-α or anti-IFN-β (C, D). At various time points from 0-48 hpi, cell lysates were collected and analyzed for expression of viral proteins (VP6) and anti-viral markers via Western blot analysis. Data shown are results from a single experiment and representative of three separate experiments that gave similar results.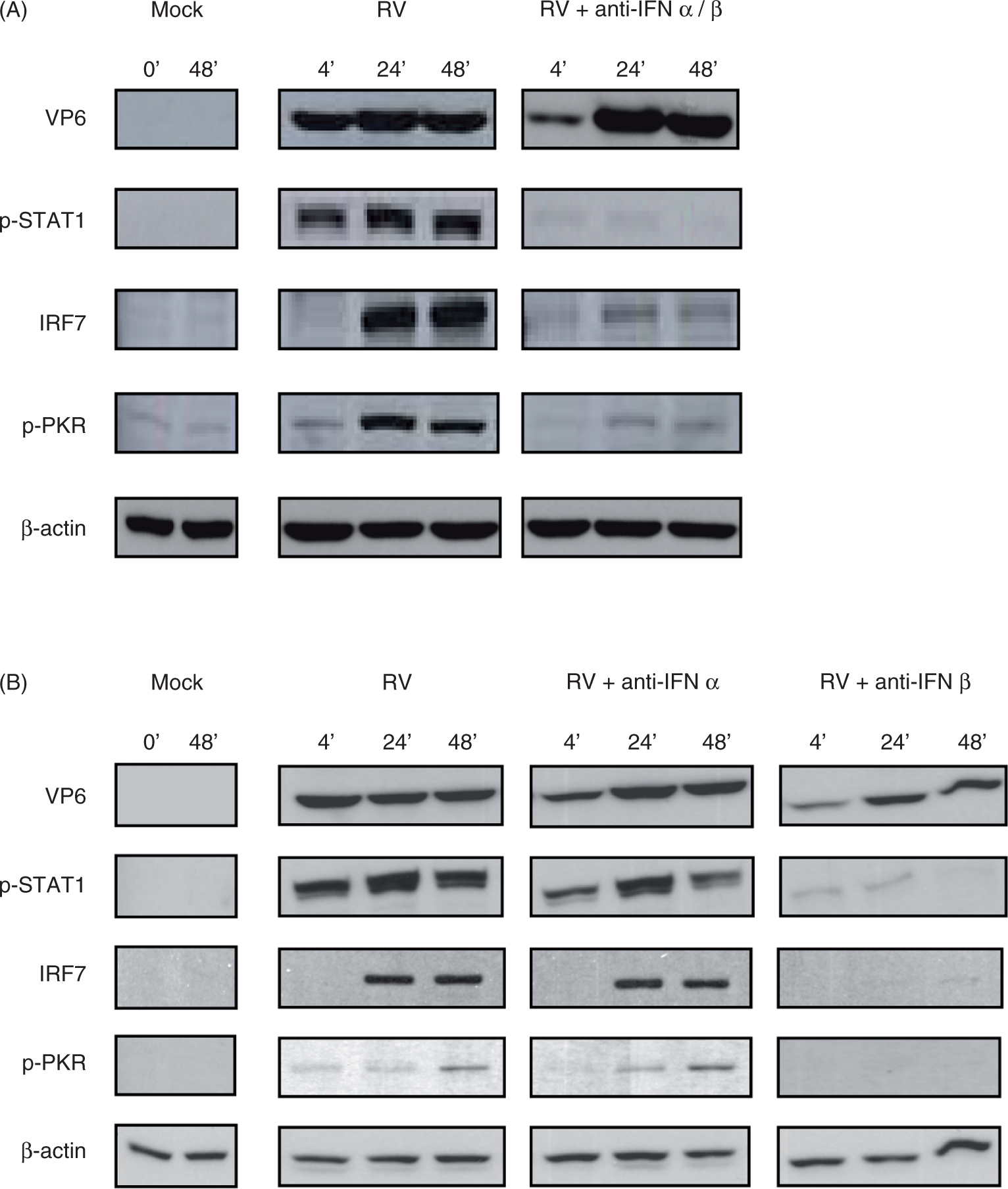
In light of the prominent role of type 1 IFN in mediating RV-induced signaling and gene expression in IECs, we expected that suppression of type 1 IFN activity might result in increased viral replication and/or impair the ability of IECs to withstand the cytotoxic effects of the virus, which, under the conditions used here, normally become apparent around 24-48 h following initiation of infection (Figure 2). Neutralization of type 1 IFN did not cause a significant increase in RV levels as assessed by levels of the viral protein VP6 (Figure 1 A, B). Rather, the most striking effect of IFN neutralization was that it prevented the loss of cells from the culture plate that otherwise occurred by 48 hpi in response to RV (Figure 2A). Closer examination of these cells under an inverted light microscope revealed that RV-infected epithelia exhibited altered shape, membrane fusion, and cell lysis that was also absent when type 1 IFN was neutralized. It has been observed that one means by which type 1 IFN impedes viral infection is via induction of apoptosis in infected and neighboring uninfected cells.
11
Thus, we sought to determine the extent to which the effects of IFN neutralization in preventing IEC loss correlated with effects on IEC apoptosis. Again, IECs were infected with RV alone or RV + anti-IFN-α/β and then examined at 0-48 hpi for evidence of apoptosis. First, we measured levels of cleaved PARP, a downstream substrate of the caspase-3 signaling pathway, that was selected as a marker for apoptosis after verification in separate control experiments that it could be readily detected in IECs treated with staurosporine, a potent inducer of apoptosis
30
(data not shown). Rotavirus infection induced PARP cleavage that was prevented by type 1 IFN neutralization (Figure 2B). Next, we assessed apoptosis via TUNEL assay. Similarly, apoptosis of RV-infected IECs also appeared to occur in an IFN-dependent manner (Figure 2C) that was evident at 24 and 48 hpi. Together, these results support the notion that RV-induced changes in cell morphology are part of an apoptotic process and suggest that type 1 IFN promotes IEC apoptosis in response to RV infection.
Rotavirus-induced type I IFN induces apoptosis in intestinal epithelia. Intestinal epithelial (HT29) cell monolayers were infected with RV (MOI 1) and RV plus type 1 IFN antibodies (anti-IFN-α/β). Control samples received equivalent amounts of trypsin diluted in SFM (Mock) or SFM alone (0). At 48 hpi, cell morphology was observed under an inverted microscope (100x) for evidence of cell death (A). From 0-48 hpi, cell lysates were collected and analyzed for expression of cleaved PARP, an apoptotic marker, via Western blot analysis (B). At 24-48 hpi, cells were fixed, permeabilized, and stained for DNA fragmentation, another marker for apoptosis, via TUNEL assay (C). Fluorescent microscope pictures of cells stained for apoptosis (green, indicated by yellow arrows) and nuclei (red) are shown as merged images (63X) in which at least three fields per specimen were examined (% TUNEL-positive cells for mock, RV, and RV + anti-IFN-treated cells were 1, 10, and 0 at 24 hpi, and 1, 12, and 0 at 48 hpi) . Data shown are results from a single experiment and representative of three separate experiments that gave similar results.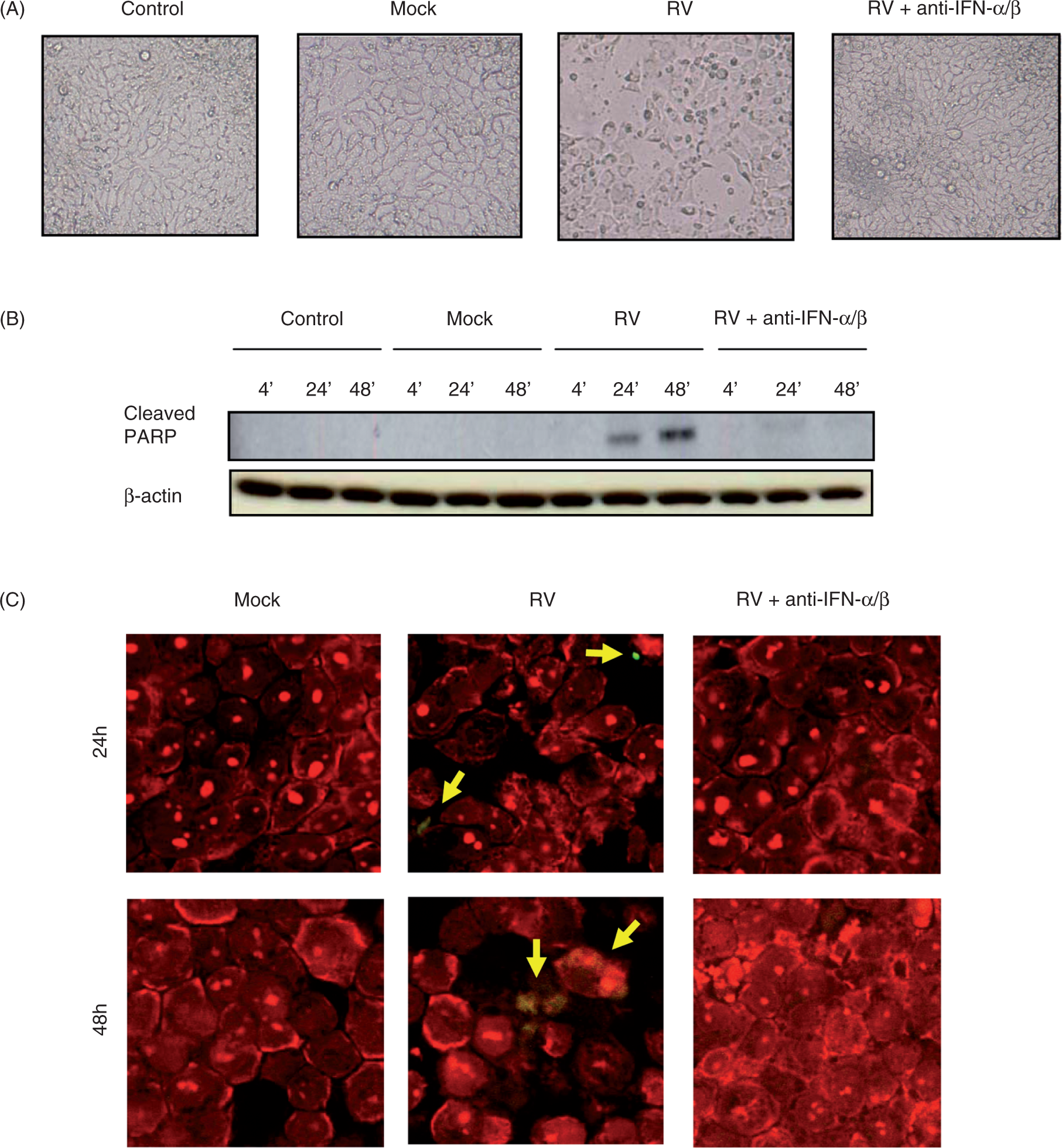
Rotavirus exploits epithelial type 1 IFN responses to promote viral replication and spread
In some viral infections, type 1 IFN does not alter replication in infected cells per se but rather reduces total viral loads by preventing the spread of viral infection from infected to uninfected cells.23,31 One means by which type 1 IFN may prevent viral spread is by promoting apoptosis thus preventing viruses from using cellular machinery for assembly and release.
11
To better understand the role of type 1 IFN in such a context, we next examined the consequences of blocking IFN activity in RV-infected IECs using methods that allowed us to assess the relative amount of cells that contained virus. Specifically, IECs were exposed to RV in the absence or presence of anti-IFN-α/β for 0-24 hpi and the presence of RV was assayed via immuno-fluorescence microscopy. This technique afforded detection of a small but easily observable population of RV-infected cells 4 h following exposure to the virus (Figure 3A). The number of infected cells increased markedly by 8 hpi, and increased slightly further by 24 hpi. Contrary to our original hypothesis that neutralization of IFN would increase viral spread, we observed that blocking type 1 IFN signaling markedly reduced the number of infected cells at 4 and 8 hpi; however, greater numbers of infected cells could be seen by 24 hpi. These findings were further supported by quantitation of fluorescence via NIH image J analysis.
1
The reduced level of viral proteins observed via immunofluorescence at 4 hpi was also seen in Western blot of cell lysates (Figure 3B) generated in parallel and was in accordance with results of Figure 1. The lack of a consistent increase in viral protein synthesis observed via Western blotting at 24-48 hpi could possibly be due to a difference in number of infected cells vs. total level of viral antigens, or, reflect that the lysates were subjected to greater dilution before analysis in order to normalize the protein levels of the samples (as cell loss was greatly reduced by neutralization of type 1 IFN, as described in Figure 2). We also assessed levels of viral antigens in cell-free supernatants that were released by RV-infected epithelia by Western blot and ELISA, which was done without normalizing level of total protein (Figure 3B and Table 2, respectively). Together, these results indicate that blockade of type 1 IFN signaling reduces viral replication, particularly during the early stages of infection. In addition, lack of type 1 IFN activity also correlates with reduced IEC apoptosis and consequently greater numbers of surviving infected cells.
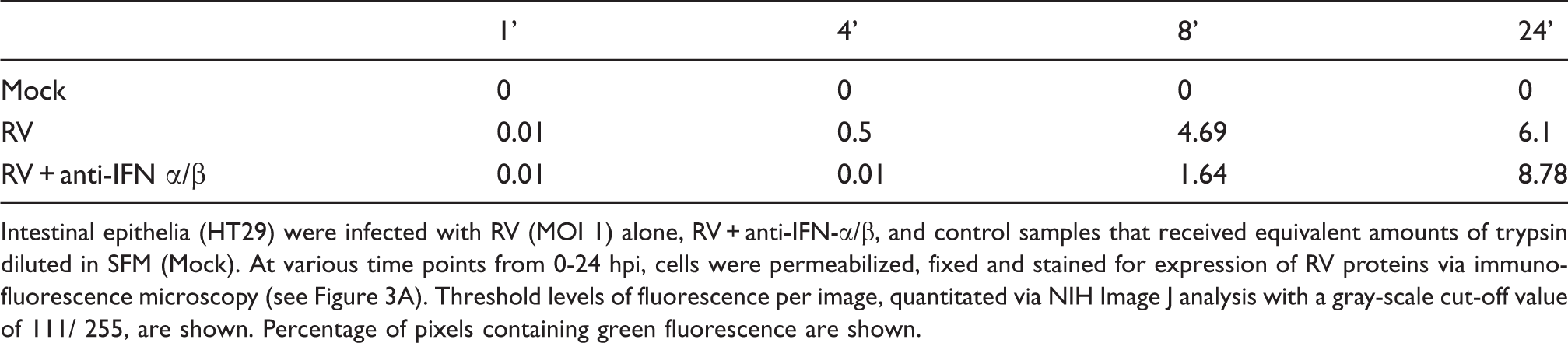
Rotavirus-induced type I IFNs promote, not impair, early viral protein synthesis and cell-to-cell spread. Intestinal epithelial (HT29) cell monolayers were infected with RV (MOI 1), RV plus type 1 IFN antibodies (anti-IFN-α/β), and control samples that received equivalent amounts of trypsin diluted in SFM (Mock). For up to 24 hpi, cells were permeabilized, fixed and stained for viral protein expression (green) and nuclei (blue), and viewed under a fluorescent microscope, magnification 20X (A). Fluorescence per image was quantitated using National Institutes of Health (NIH) Image J analysis (see Table 1). In separate experiments, cells were mock-treated and infected with RV or RV + anti-IFN-α/β for 0-48 hpi, and supernatants or lysates were collected and analyzed via Western blot analysis for viral protein expression (VP2) (B). Viral protein concentration in the supernatants was quantitated by ELISA (see Table 2). Data shown are results from a single experiment and representative of three separate experiments that gave similar results. Percent threshold of fluorescence in RV-infected cells versus RV-infected cells with anti-IFN α/β Intestinal epithelia (HT29) were infected with RV (MOI 1) alone, RV + anti-IFN-α/β, and control samples that received equivalent amounts of trypsin diluted in SFM (Mock). At various time points from 0-24 hpi, cells were permeabilized, fixed and stained for expression of RV proteins via immunofluorescence microscopy (see Figure 3A). Threshold levels of fluorescence per image, quantitated via NIH Image J analysis with a gray-scale cut-off value of 111/ 255, are shown. Percentage of pixels containing green fluorescence are shown. Viral levels in supernatants of RV-infected cells versus RV-infected cells with anti-IFN antibodies Intestinal epithelia (HT29) were infected with RV (MOI 1) alone, RV + anti-IFN-α/β, and control samples that received equivalent amounts of trypsin diluted in SFM (Mock). At various time points from 0-48 hpi, supernatants were collected and assayed for levels of RV proteins (pfu/ml) via ELISA. Mock samples had no detectable levels and are not shown.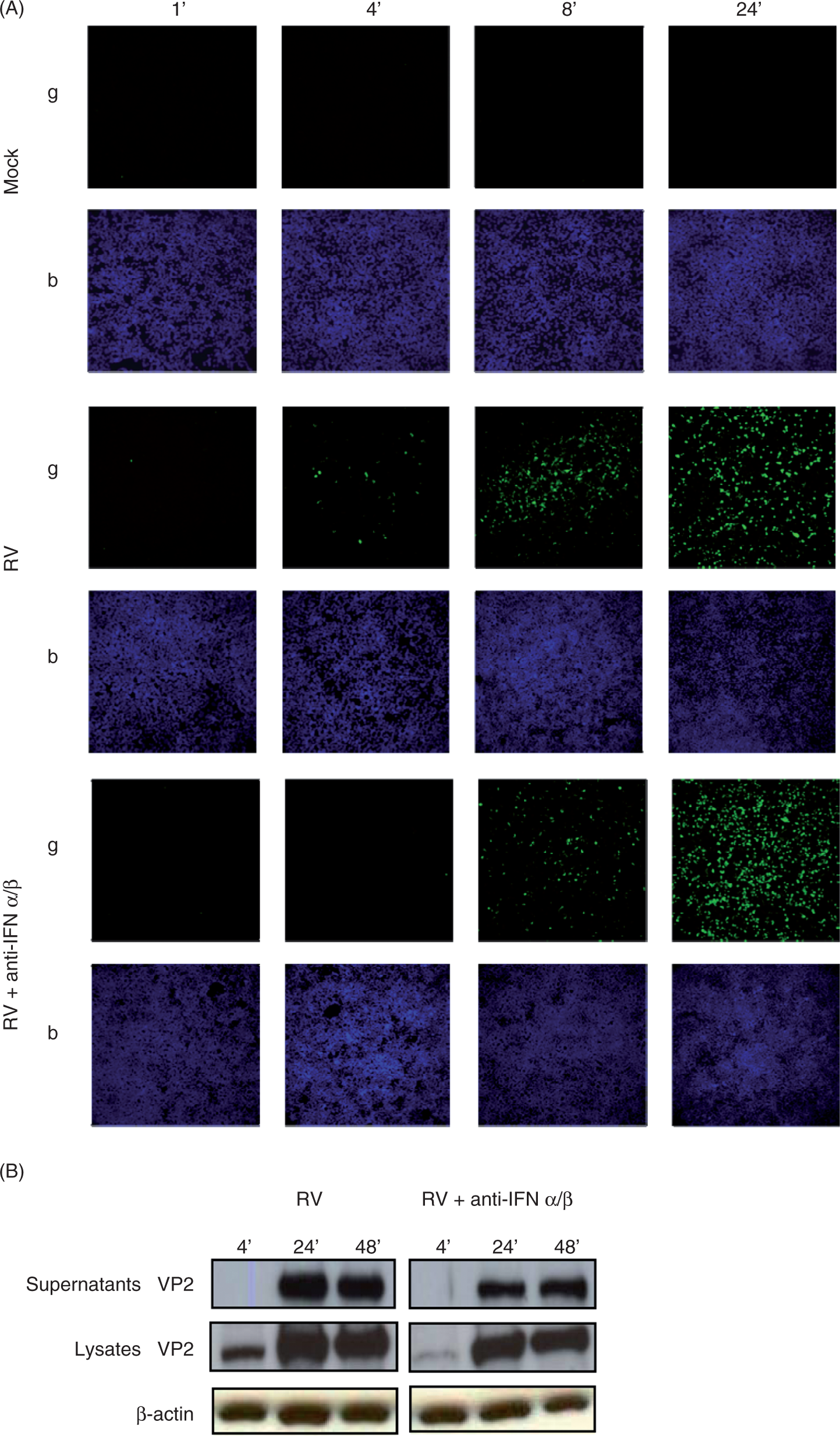

We next began to investigate the mechanism by which blockade of type 1 IFN reduced RV replication in IECs. In considering candidate mechanisms, we noted that one of the kinases whose activation was IFN-dependent, namely PKR, has been shown to correlate with increased infectivity of reoviruses, which belong to the same Reoviridae family of dsRNA viruses as rotavirus32,33 Thus, we investigated whether RV might, similar to reovirus, exploit type 1 IFN signaling to promote PKR-dependent replication. Epithelial cells were infected with RV or RV + 2AP, a PKR inhibitor, and subsequently examined for spread of viral infection via immunofluorescence microscopy. The PKR inhibition reduced the number of RV-infected cells at all time points assayed (Figure 4 and Table 3). To verify that the PKR inhibitor had not simply blocked all signaling in IEC per se, we verified that an event previously shown to be independent of PKR, namely flagellin-induced IL-8 secretion,
34
was not blocked by PKR inhibition (data not shown). These results support the notion that activation of PKR may be one means by which induction of type 1 IFN signaling is exploited by RV.
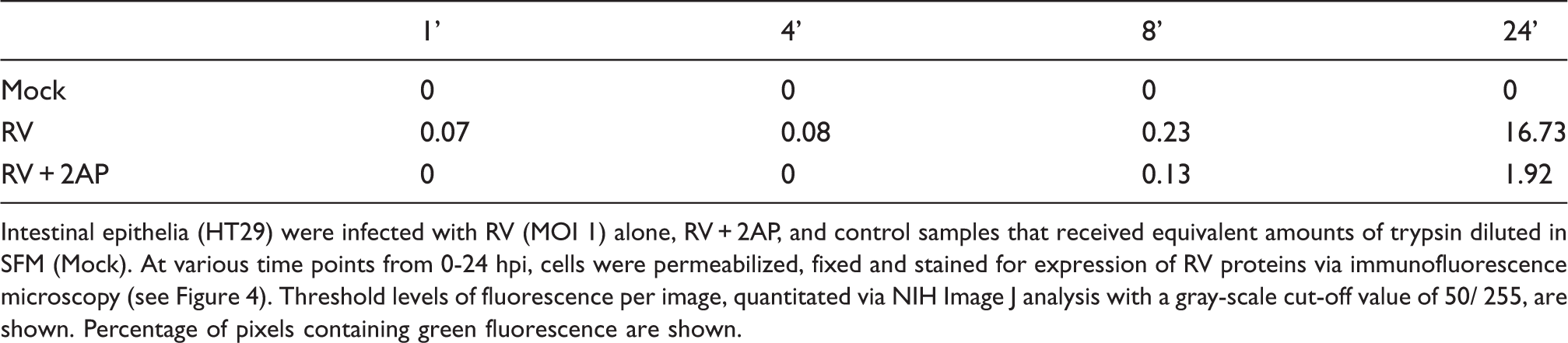
Rotaviral spread is PKR-dependent. Intestinal epithelial (HT29) cell monolayers were infected with RV (MOI 1), RV plus PKR inhibitor 2AP, and control samples that received equivalent amounts of trypsin diluted in SFM (Mock). For up to 24 hpi, cells were permeabilized, fixed and analyzed under a fluorescent microscope for viral protein expression (green) and nuclei (blue) at magnification 20X. Fluorescence per image was quantitated using NIH Image J analysis (see Table 3). Data shown are results of a single experiment and representative of three separate experiments that gave similar results. Percent threshold of fluorescence in RV-infected cells vs. RV-infected cells treated with 2AP Intestinal epithelia (HT29) were infected with RV (MOI 1) alone, RV + 2AP, and control samples that received equivalent amounts of trypsin diluted in SFM (Mock). At various time points from 0-24 hpi, cells were permeabilized, fixed and stained for expression of RV proteins via immunofluorescence microscopy (see Figure 4). Threshold levels of fluorescence per image, quantitated via NIH Image J analysis with a gray-scale cut-off value of 50/ 255, are shown. Percentage of pixels containing green fluorescence are shown.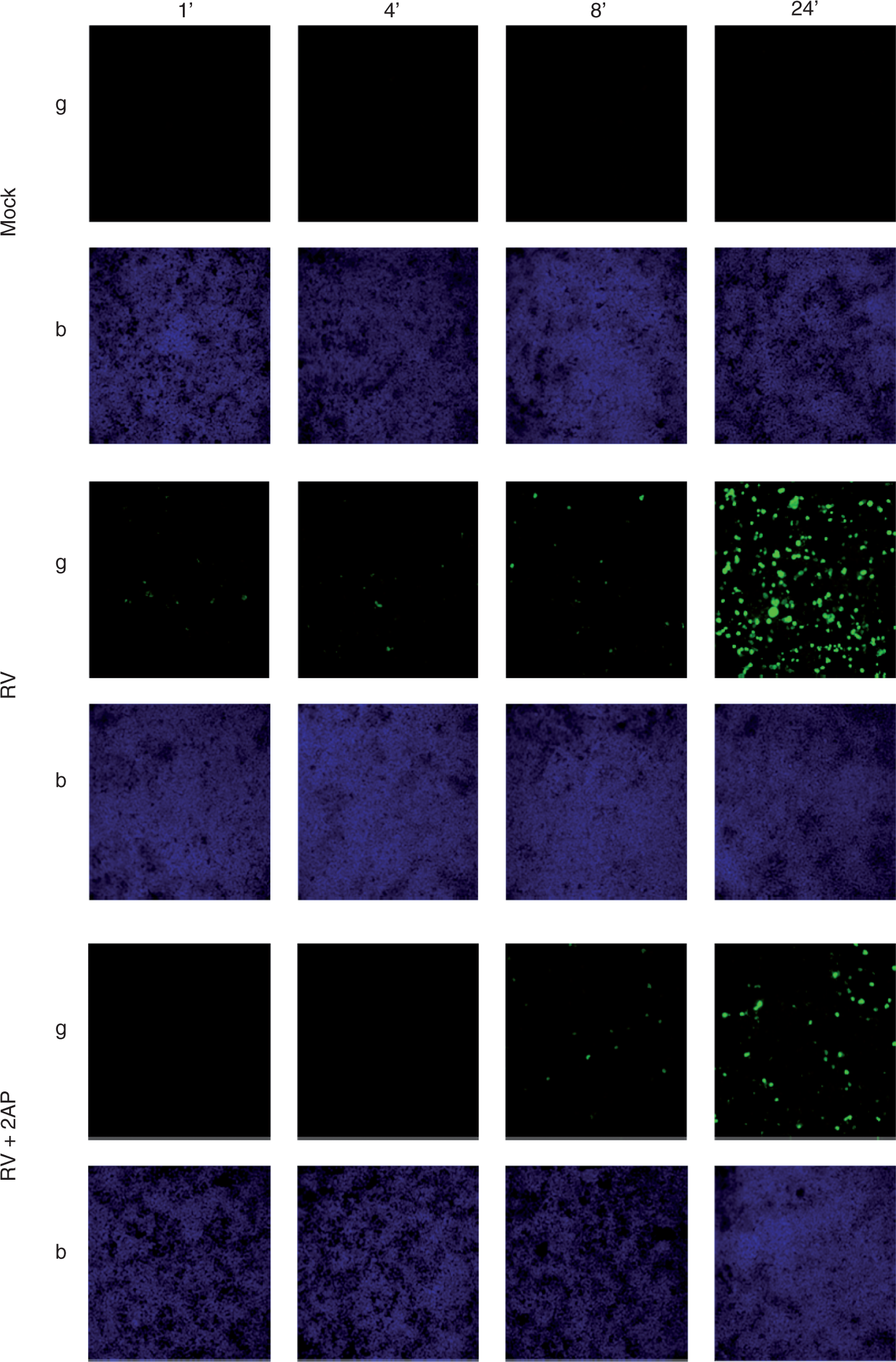
Blockade of caspase activity rescues RV-induced cell death and enhances viral replication
Next, to better understand if the cell death seen in response to RV infection was driven by caspase-mediated apoptosis or represented cell lysis driven by viral replication, we blocked caspase activity with the pan-caspase inhibitor Z-VAD-FMK. We found that at a 20 µM concentration, this compound was not toxic to our cells and was an effective inhibitor of caspase activity in that it completely blocked cleavage of basal and RV-induced cleavage of PARP (Figure 5A). The inhibition of PARP cleavage correlated with a complete elimination of RV-induced cell death. Specifically, microscopic examination of RV-infected cells revealed loss of cells from what had been a confluent monolayer while, in contrast, Z-VAD treated RV-infected cells were at least as confluent as uninfected cells (Figure 5B). In accordance, the supernatants of RV-infected cells contained about four times as many dead floating cells as mock-infected or RV-infected cells that had been treated with Z-VAD. These results further support the notion that RV-induced cell death, which we observed was dependent upon IFN-β, is an apoptotic phenomenon rather than a more generalized virus-induced cytopathic effect. Moreover, preventing RV-induced apoptosis via caspase blockade resulted in a dramatic increase in viral replication (Figure 5A). Thus, while RV may take advantage of IFN-induced signaling at early time points following infection, later induction of IFN-β mediated apoptosis may also benefit the host by limiting viral infection.
Caspase inhibition prevents RV-induced cell death and enhances viral replication. Intestinal epithelial (HT29) cell monolayers were infected with RV (MOI 5), RV plus 20 µM Z-VAD-FMK, and control samples that received equivalent amounts of trypsin diluted in SFM (Mock). Cell morphology was assessed at 0-48 hpi by microscopy (20X). Cell lysates were collected at 48 hpi and assessed for cleaved poly (ADP-ribose) polymerase (PARP), VP6, and β-actin via SDS-PAGE immunoblotting (A). Images in (B) show loss of cells visible at 48 hpi (representative of at least three fields per specimen). Data shown are results of a single experiment and representative of three separate experiments that gave similar results.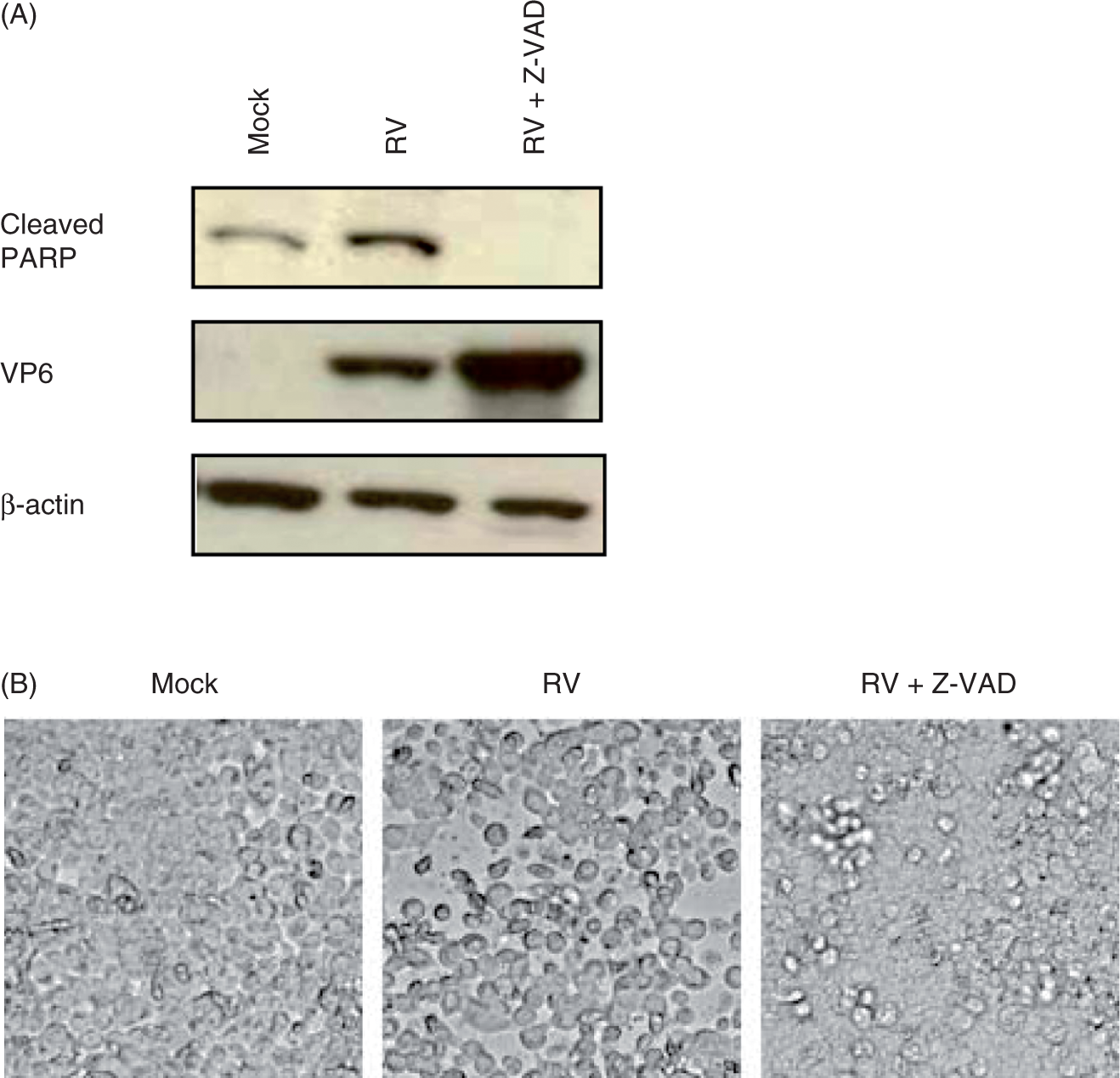
Discussion
Like many cell types infected with viruses, IECs generate type 1 IFN upon infection with RV. Such IFN induction is responsible for induction of over 500 genes in RV-infected IECs. 9 Herein, we observed that, in RV-infected IECs, type 1 IFN, in particular IFN-β, also plays a predominant role in activating some of the phosphorylation events commonly associated with viral infection. Yet, we are not able to completely rule out the possibility that IFN-α plays a minor role in IECs in that IFN-α neutralization resulted in a slight reduction of early RV replication (densitometric quantitation of VP6 levels at 4 hpi in from series of experiments illustrated in Figure 1B indicated blockade of IFN-α reduced VP6 levels by an average of 22% and also appeared to ameliorate early PKR activation). Nonetheless, the predominant role of IFN-β rather than IFN-α in our in vitro system is in accordance with previous work that IFN-α is primarily produced by pDCs, whereas IFN-β is more broadly produced by many cell types including IECs.35,36 However, we observed that IECs were highly responsive to exogenously administered IFN-α and responded in a manner that was indistinguishable from their response to IFN-β. Moreover, this response was not dependent upon IFN-β, which may perhaps have been produced in response to IFN-α. Thus, it is quite possible that IFN-α will play a key role in the innate immune response to RV infection in vivo, in which pDC may be recruited the gut at some point during the infection. Thus, future studies are required to determine if IFN-α has a significant role in modulating RV infection in vivo.
Based on the presumption that all immune responses should be considered beneficial unless proven otherwise, we expected that blockade of IEC type 1 IFN signaling in vitro would enhance RV infection in a manner that would suggest an obvious role for such signaling in anti-viral immunity in vivo. In contrast, neutralization of type 1 IFN primarily modulated RV infection in a way that seemed consistent with the possibility that RV exploits type 1 IFN to promote its replication and cause a pathologic response in the host. Specifically, we observed that blockade of type 1 IFN markedly attenuated the rate of RV replication particularly in the first 8 h following inoculation, suggesting that the type 1 IFN response promotes RV replication. Additionally, we observed that RV-induced type 1 IFN promoted cell death. As for other viruses that cause acute infections, replication rates in vitro often correlate with virulence in vivo, and that RV-induced cell death is thought to play a role in causing clinical manifestations of RV infection,37,38 these results suggest that the type 1 IFN response may be considerably detrimental to RV-infected hosts.
The notion that some viruses have evolved mechanisms to take advantage of IFN signaling has been suggested previously, with Smith et al.32,33 in particular, finding that the IFN-associated PKR activity promoted replication of reovirus, which shares considerable similarity with RV. Our observation that pharmacologic inhibition of PKR suppressed RV replication suggests that RV may exploit IFN signaling in a manner similar to that used by reovirus. Although activation of PKR is known to block protein translation, which would be expected to impede viral replication, recent work by Rojas et al. has shown that RV can obviate the block in translation normally caused by PKR-mediated eIF2-α phosphorylation.39 While taking advantage of signaling induced by type 1 IFN may, thus, be a strategy used by a variety of viruses, there are many more reports of viruses interfering with type 1 IFN signaling in a variety of ways, 40 likely reflecting the broad ability of type 1 IFN to suppress viral infection. Indeed, elegant in vitro studies by Patton and colleagues demonstrate that RV employs this strategy in that one of its non-structural proteins, namely NSP1, suppresses type 1 IFN production via degradation of IFN-inducing IRF transcription factors.23,24 The absence of NSP1-mediated IRF degradation was associated with reduced viral spread, 23 suggesting that inability to suppress IFN signaling might impair RV fitness in vivo. Our observation that a relatively small amount of UV-irradiated rotavirus induced greater IFN signaling than live virus present in IECs 24 h following infection speaks of the ability of RV to suppress IFN signaling. Nonetheless, it should be noted that RV suppression of IFN signaling is not absolute, as RV infection still results in detectable activation of IFN and numerous IFN-activated genes. 9 . Thus, one possibility of reconciling our findings with those of Patton et al., is that a small amount of type 1 IFN signaling, perhaps just enough to activate a threshold level of PKR activation, provides the optimal environment for RV and, thus, any alterations in IFN signaling (i.e. increase or decrease) may reduce RV infectivity.
Another potential way of interpreting our findings is to view RV-induced cell death as a means of innate immunity that is effective in vivo, albeit harder to appreciate in vitro. Under normal conditions in vivo, apoptotic epithelial cells shed into the gut lumen in a manner that preserves gut barrier function.41,42 Thus, one can envisage that using this process may be a safe, efficient means of eliminating RV-infected cells and, thus, IFN promotion of apoptosis may, in fact, be of benefit to the host. In this context, one could view the loss of RV-infected IECs from the cell culture plate observed herein as a means of viral clearance, as in vivo these cells would be flushed out of the intestine in the fecal stream. The high regenerative capacity of the intestine would likely allow for a considerable level of viral clearance by this mechanism before loss of barrier function and subsequent inflammation would ensue, which is in accordance with observations that RV infection is not associated with histopathologic inflammation. 3 However, such a mechanism of RV clearance, if indeed operative in vivo, may not only provide a benefit to the host but may also be likely to aid RV in its dissemination to new hosts, presumably via fecal-to-oral route. 43 Thus, RV-induced IFN-mediated apoptosis may, in fact, be mutually beneficial to both RV and the host, in accordance with the notion that ancient pathogens have co-evolved with their hosts.
In considering the relative importance of various in vitro observations discussed herein, we note that, in contrast to the case for most other viral infections, loss of the type 1 IFN receptor and subsequent ablation of all type 1 IFN responses does not have a dramatic alteration on the course of infection.18,19 Our favored interpretation of this observation is that, overall, the type 1 IFN response is utilized by both RV and the host to promote viral replication and clearance, respectively. Thus, the net result of eliminating type 1 IFN signaling is rather modest, although it seems to modulate local dynamics of the infectious process. In this scenario, it may be possible to modulate the course of infection by RV and other viruses by better understanding and subsequently more precise manipulation of type 1 IFN signaling and innate immunity in general.
Footnotes
Acknowledgements
We thank Kathryn Knoop, Jennifer Hood, Benyue Zhang and Susan Voss for their technical assistance. We also thank Mary Estes (Baylor University) and Jon Gentsch (Center for Disease Control and Prevention) for helpful advice and reagents. This work was supported by NIH grant AI083420 to AG, AI080656 and a Digestive Disease Research and Development Center (DDRDC) grant DK064399, and a Digestive Diseases Research Core Center (DDRCC) grant DK66338.