
Abstract
Respiratory and enteric viral infections cause significant morbidity and mortality world-wide and represent a major socio-economic burden. Many of these viruses have received unprecedented public and media interest in recent years. A popular public misconception is that viruses are a threat to which the human body has only limited defences. However, the majority of primary and secondary exposures to virus are asymptomatic or induce only minor symptoms. The mucosal epithelial surfaces are the main portal of entry for viral pathogens and are centrally involved in the initiation, maintenance and polarisation of the innate and adaptive immune response to infection. This review describes the defences employed by the epithelium of the respiratory and gastrointestinal tracts during viral infections with focus on epithelial modulation of the immune response at the innate/adaptive interface.
Introduction
Respiratory and gastrointestinal viral infections cause significant morbidity and mortality world-wide. In children, the most common causes of death globally are pneumonia (18%) and diarrhoea (15%), 1 with a large proportion of these due to viral infections. In developing countries, respiratory syncytial virus (RSV) accounts for up to 40% of hospital admissions due to childhood lower respiratory tract infections (pneumonia or bronchiolitis). 2 Respiratory syncytial virus infection is estimated to cause up to 200,000 deaths per year, with most of these in developing countries 3 and intestinal rotavirus infection leads to as many as 500,000 deaths per year. 4 Prevention and treatment of these viral infections and their sequelae represent a significant socio-economic burden.5,6 Influenza-related emergency department visits have been estimated to cost US$279 million annually in the US, with a further US$163 million spent on influenza-related hospitalisations. 7
The emergence of new viruses in the human population, the acquisition of human infectivity in some strains of virus previously restricted to animals and the re-assortment of genes within the influenza virus resulting in the generation of a new strain of an old foe has led to unprecedented public and media attention in recent years.8,9 During the most recent viral epidemic, media campaigns have left the public with the perception that viruses are a threat to which the human body has only limited defences. However, the human body can be very efficient at eliminating viruses with minimal inflammation or injury. The majority of primary and secondary exposures to virus are asymptomatic or induce only minor symptoms.
The main portals of entry for viral pathogens are the internal epithelial surfaces of the respiratory and gastrointestinal tracts. The 5-m long gastrointestinal tract is continually exposed to external microbes through ingestion, notwithstanding the resident commensal bacteria which outnumber eukaryotic cells in the body. 10 Similarly the 500-m2 surface area of the respiratory surface is exposed to 10,000 litres of air a day, laden with microbial stimuli including viruses. 11 Situated at these interfaces is the mucosal epithelium, centrally involved in the initiation, maintenance, polarisation and resolution of the virus-induced immune response. A rapid and effective innate response is crucial for limiting virus spread and viral load while adaptive immunity develops. However, initiation of an overly vigorous inflammatory process can be both detrimental to recovery and have protracted consequences such as increased susceptibility to excessive inflammation upon subsequent infection as seen in recurrent viral wheeze following viral childhood bronchiolitis.
This review will compare and contrast the mechanisms employed by epithelial cells at the respiratory and gastrointestinal surfaces, with particular focus on their response to viruses and their ability to modulate innate and adaptive immunity.
The innate and adaptive responses defined
Innate immunity is classically considered to be the immediate response mounted through the recognition of common or conserved residues derived from, or present on, pathogens. Such residues are referred to as pathogen-associated molecular patterns (PAMPs). Recognition of PAMPs is primarily via a restricted number of highly conserved, high-affinity receptors termed pattern recognition receptors (PRRs). These signals can become amplified by damage-associated molecular pattern (DAMPs) molecules such as metabolites and nucleotides released during cellular injury. Importantly, the innate immune response is not confined to ‘classical’ immune cells but also includes structural cells such as the epithelium. By contrast, the adaptive immune response involves B- and T-lymphocytes and has a vast repertoire of receptors generated by gene re-arrangement and hypermutation. Lymphocyte receptors are initially of low affinity and frequency; however, by utilising selection and amplification of rapidly dividing lymphocytes, frequency, affinity (in the case of B-cells) and avidity are improved producing lymphocytes bearing highly specific receptors within 4–5 d after antigen recognition.
Epithelial humoral responses to viruses
Although the epithelium is the first cell layer that viruses encounter in the human body, it is not always the first physical barrier to viral entry. The airways of healthy individuals are coated in a watery periciliary layer approximately 5–10 µm thick. This layer contains an array of antimicrobial products produced largely by the underlying epithelium and facilitates rapid paracrine signalling between epithelial cells in response to viral infection. Following irritation or infection of the respiratory surface, this periciliary layer becomes covered by a mucus layer secreted by submucosal glands and goblet cells, providing a further barrier to microbial entry. 12 This mucus layer is typically restricted to the tracheal and bronchial surfaces; however, inflammation can lead to Clara cells (progenitor/surfactant-producing epithelial cells) in the respiratory bronchioles adopting goblet cell function, extending the coverage of the mucus layer and reducing respiratory efficiency. 13 At the respiratory surface, ciliated epithelium accounts for over 60% of all epithelial cells, with each cell possessing approximately 200 cilia. Ciliated cells drive the mucocilary escalator removing mucus and periciliary layers containing virus and viral antigens from the lung at a rate of approximately 20 mm/min in the large bronchi and 1 mm/min in the bronchioles. 14 Release of cellular nucleotides and signalling molecules as a result of viral infection can markedly increase beat frequency of cilia, the thickness and consistency of the fluid layer and the factors secreted by the epithelium.15,16 These mucus and periciliar layers also act as a pH buffer, which can prevent the entry of some viruses in addition to harbouring epithelial-derived antimicrobial factors.
Residing in the pulmonary fluid layer are alveolar macrophages capable of secreting a broad range of inflammatory cytokines following bacterial and viral stimuli. However, these cells are rendered anti-inflammatory by the underlying epithelium. 17 Transforming growth factor-β tethered to the surface of the epithelium, expression of inhibitory ligands such as CD200 and secretion of a number of soluble factors prevent macrophages from making excessively vigorous, potentially deleterious responses to innocuous antigen. 17
In the gut, hyperpolarised columnar epithelial cells are covered at their apical surface by a 500-nm thick glycocalyx layer on negatively charged microvilli which limits the uptake of antigens whilst still promoting nutrient absorption. 18 This is overlaid by a mucin rich layer; a glycoprotein coat composed of carbohydrate side chains bound to a protein skeleton. 19 Both membrane-bound and secreted mucins consist of a core protein (apomucin) with 18 genes currently known to encode these proteins in humans. 20 These glycoproteins bind competitively to surface receptors acting as a barrier to microbes such as rotavirus 21 but also act as a source of energy for commensal bacteria. 22
Epithelial cells are significant contributors of soluble factors in the humoral innate immune response during viral infection. The principal methods of protection employed by the humoral innate immune response include direct killing of microbes, initiation of the complement cascade to enhance engulfment of pathogens or infected cells by professional phagocytes and the enhancement of cellular surveillance/pathogen recognition. The details of the complement cascade are outwith the scope of this review but are reviewed extensively elsewhere.
23
In brief, this cascade is composed of a classical route which can be regarded as part of the adaptive immune response since it involves antibody, the alternative pathway resulting from complement stabilisation on the surface of pathogens or infected/apoptotic cells, and the lectin pathway resulting from the recognition and binding of lectins or ficolins to PAMPs. Of particular interest with regard to viral infection are the following soluble factors in the humoral innate immune response: collectins, ficolins, pentraxins, nitric oxide and antimicrobial peptides (Figure 1).
Innate, non-cytokine, non-chemokine epithelial responses to viruses in the respiratory and gastrointestinal tract. MBL, mannose binding lectin; Sp-A/D, surfactant protein A/D; CRP, C-reactive protein; HBD1/2, human beta defensins 1/2.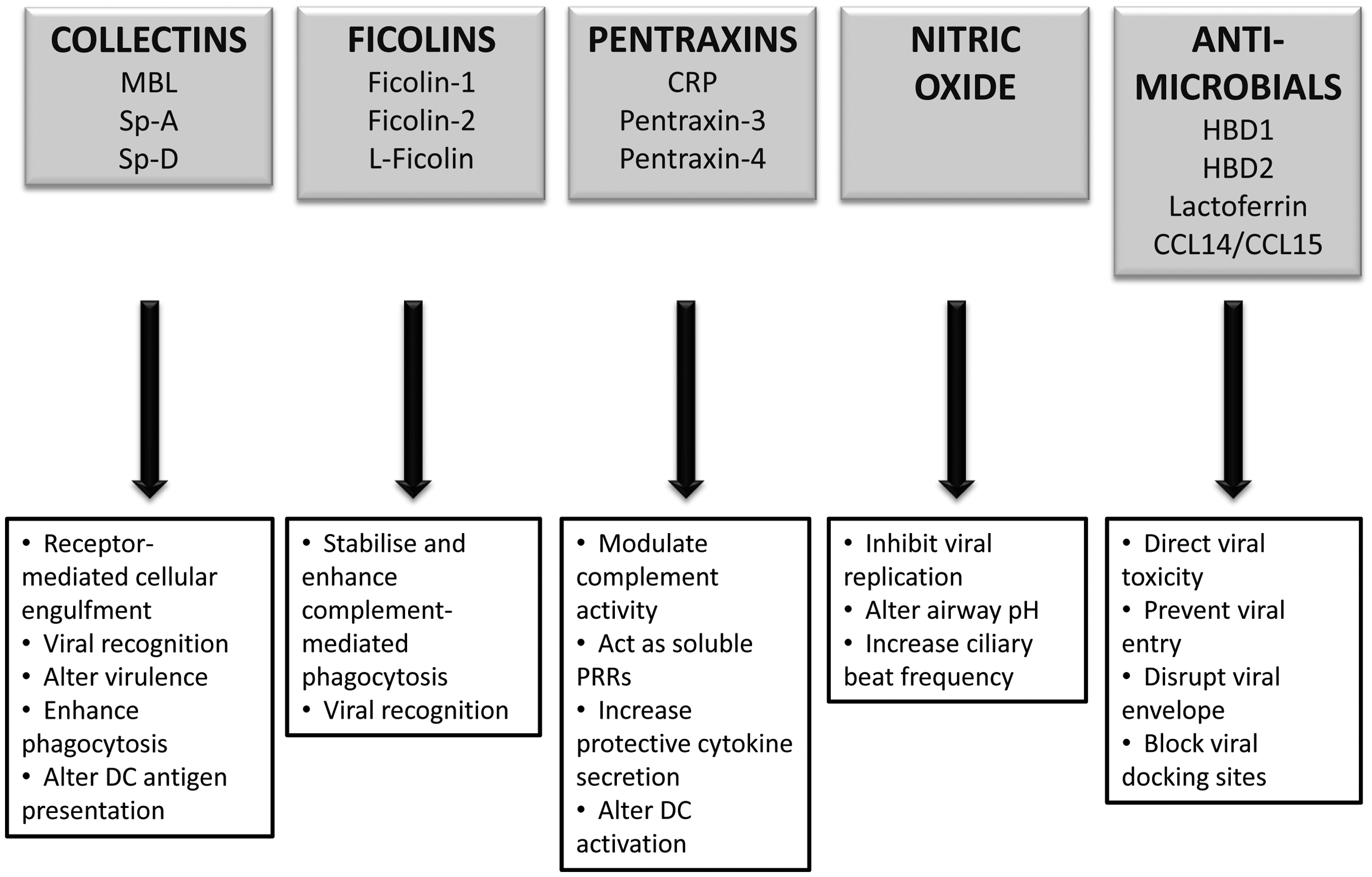
Collectins
The collectins are soluble PRRs containing a collagen motif that can bind to glycoproteins on enveloped viruses such as influenza virus and RSV, as well as non-enveloped viruses such as rhinovirus and rotavirus. Although some species-specific homologues exist, the most conserved collectins include mannose binding lectin (MBL) derived principally from hepatocytes in the liver 24 and the large hydrophilic surfactant proteins A (Sp-A) and D (Sp-D), mainly produced by respiratory epithelium, but also by the epithelium of the digestive tract. Studies in vitro, ex vivo and in animal models would suggest that infection and inflammation in the absence of collectins results in excessive inflammatory cell recruitment, cytokine secretion and excessive oxygen free radical generation. 25 – 28 The surfactants Sp-A and Sp-D are primarily expressed on the luminal surface of pulmonary epithelial cells, specifically by alveolar type II and Clara cells.29,30 Expression of Sp-A and SP-D has also been identified in the gastrointestinal mucosa of rats31,32 and has been shown to be up-regulated in intestinal inflammation and in the presence of bacteria such as Helicobacter pylori. 32 Although there is some evidence suggesting a role for surfactants in animal models of intestinal rotavirus infection, there is currently no work demonstrating their function in human enteric viral infection. 33
The regulation of Sp-A and Sp-D expression is complex and incompletely characterised, with differences in promoter binding sites leading to differential Sp-A and Sp-D responses to interferon γ (IFN-γ), tumor necrosis factor α (TNF-α), transforming growth factor β (TGF-β) and glucocorticoids. 34 Unlike MBL and ficolins, surfactants increase cellular engulfment not by direct complement activation but via a receptor-mediated effect. Although an area of conjecture, one attractive model has been put forward by Gardai et al. 35 and recently reviewed by Haczku. 34 This model suggests that, in the absence of PAMPs, the surfactants are bound to cell receptors by their heads which possess immunosuppressive properties; however, in the presence of PAMPs, they bind with their collagenous tail and initiate pro-inflammatory cytokine release and possibly engulfment.34,35
Surfactants can aid in the recognition of both enveloped and non-enveloped viruses by binding to glycoproteins on their surface. In addition, surfactants can also directly interfere with the entry of viruses into cells, with influenza infection perhaps the best characterised example.36,37 Recent pandemic strains have lacked susceptibility to Sp-D mediated opsonisation and neutralisation, which could have been contributory to their virulence. 38 Once infection has occurred, surfactants can also limit inflammation by enhancing phagocytosis of apoptotic and infected epithelium, although the exact manner in which this occurs is also still a matter of debate. 34 Surfactant gene polymorphism studies in RSV infection of infants indicate that, while susceptibility to infection may not be linked to surfactant function, the severity of response may be. 39 It is, therefore, of interest to note that RSV has also recently been shown to reduce the translation efficiency of Sp-A. 40 Surfactants also appear to modulate adaptive immunity with Sp-A inhibiting and Sp-D augmenting antigen presentation by the cell type which initiates much of the adaptive response, the dendritic cell (DC).41,42 Surfactant protein-A and Sp-D might, therefore, be expected to be counter-regulatory; however, both inhibit the proliferation of mitogen-stimulated T-lymphocytes via IL-2 inhibition, thus the exact manner in which these molecules modulate adaptive immunity has yet to be completely elucidated (reviewed elsewhere34,43).
Ficolins
There is some limited expression of the ficolins, a group of oligomeric lectins, in lung epithelium with little evidence of presence in the gut. Ficolin-3 (also called H-ficolin, Hakata antigen, thermolabile β-2 macroglycoprotein, or thermolabile substance) is produced by type II alveolar epithelial cells and ciliated bronchial epithelial cells in human lung, with ficolin-1 (M-ficolin) only expressed by type II epithelium.44,45 Although much is as yet unknown about the ficolins, they have been shown to stabilise and enhance MBL complement-mediated phagocytosis, 46 with recent work showing evidence of viral recognition by L-ficolin. 47 Thus, ficolins may as yet be revealed to be integral components of the classical and lectin-based complement cascades and are promising new targets for investigation.
Pentraxins
The pentraxins are a superfamily of innate humoral proteins further sub-divided into the short and long forms. Evidence for direct viral interaction with the short pentraxins is limited and primarily relates to enhanced recognition of virus-infected or apoptotic cells or steric hindrance to receptor-mediated viral uptake. 48 The long pentraxins are largely liver-derived and include C-reactive protein (CRP) and serum amyloid P component (SAP). 49 These, more highly conserved, long pentraxins which include the neuronal pentraxins and pentraxins 3 and 4, are produced by a variety of different cell types. These molecules are thought to modulate complement activity and to act as soluble PRRs.49,50 Although expression has been identified at both the enteric and respiratory epithelial surfaces, the majority of work with regard to specific cellular expression is in the respiratory tract; the most studied of these being pentraxin 3. Expression in epithelial cells is up-regulated by inflammatory cytokines 51 and binding of viruses such as human and mouse cytomegalovirus (CMV) and influenza virus by pentraxin 3 can reduce viral entry and increase protective cytokine secretion.48,49 Studies with fungal and viral infections in pentraxin 3 knockout animals indicate that pentraxins also affect DC activation with important implications for the subsequent polarisation of adaptive lymphocyte responses.49,52
Nitric oxide
Epithelial cells of both the respiratory and enteric surfaces produce nitric oxide (NO) during inflammation. Elevated levels have been identified in upper respiratory tract viral infections with NO directly inhibiting the replication of viruses and reducing T helper 1 (Th1) and natural killer (NK) cell chemokine production by the epithelium. 53 An interesting side effect of NO up-regulation is that NO synthase activation in bronchial epithelial cells reduces sodium channel and acid buffering activity. This lowers airway pH and has been shown to reduce the replication of some viruses such as rhinoviruses. 54 Nitric oxide release has also been associated with increased cilia beat frequency leading to more effective viral clearance. 55 Although NO has been shown to be produced in the gut, 56 its role during viral infection is not yet fully understood. It has been demonstrated that NO plays a role in rotavirus-induced diarrhoea in several studies in human and murine models.57,58 However, from these studies, it is not clear whether NO acts to dampen the response to rotavirus infection or to perpetuate the diarrhoeal illness.
Antimicrobial molecules
Human beta defensin (HBD) 1 is constitutively expressed by epithelium but like HBD 2 and HBD 3 it can be up-regulated in response to viral PAMPs and inflammatory cytokines. The HBDs have antiviral activity against the enveloped viruses herpes simplex virus (HSV) 2, influenza A and RSV. Their antiviral mechanisms include direct toxicity and prevention of viral entry, but is primarily thought to arise from disruption of the viral envelope/membrane. 59 (The role of defensins in viral infections was recently reviewed by Ding et al. 60 ) Epithelium can also be coated in gland-, plasma- or neutrophil-derived antimicrobial molecules such as lactoferrin. Lactoferrin has been shown to associate directly with many viruses including CMV, HSV, hepatitis B and C (HBV and HCV) viruses, rotaviruses, and adenovirus but it is predominantly thought to block infection by occupying potential viral docking sites on the epithelium such as glycoaminoglycans. 61 Some constitutively expressed chemokines of the gastrointestinal epithelium such as CCL14/15 also have proven anti-microbial/viral function;62,63 however, the overall role of defensins, cathelicidins and RNases are less understood in the intestine. The more general roles of antimicrobial peptides are reviewed elsewhere.59,64,65
Viral recognition by epithelial cells
The epithelium at the respiratory and enteric surfaces expresses a diverse range of PRRs capable of detecting viruses. Expression varies with epithelial subtype and location, with specific expression patterns described in several excellent PRR-focused reviews.66,67 Epithelial-expressed PRRs are briefly described below and include cell surface expressed C-type lectins (cell surface variants of the secreted collectins), intra- and extracellular toll-like receptors (TLR), the intracellular RNA-dependent protein kinase (PKR), retinoic acid–inducible gene I (RIG-I) like receptors (RLR) and nucleotide binding domain and leucine-rich repeat containing receptors (NLR).
Detection of TLRs would appear to be methodologically variable66,68 and, in many situations, proof of function is not established. Respiratory epithelium appears to be capable of expressing all the main TLR, although expression will vary with epithelial subtype and anatomical position within the respiratory tract. 69 By contrast, evidence of protein expression in the intestine is confined to TLRs 2, 3, 4, 5 and 9 with some studies reporting RNA expression of others. 69
Toll-like receptor 3 recognises double-stranded RNA (dsRNA), such as reovirus, but is also implicated in the recognition of single-stranded RNA (ssRNA) during viral replication of ssRNA viruses such as RSV and west Nile virus. Toll-like receptor 7 recognises ssRNA enriched with guanosine or uridine (HIV, influenza virus). Toll-like receptor 8 is similar to TLR7 and recognises ssRNA. Toll-like receptor 9 recognises single- and double-stranded DNAs and unmethylated deoxycytidylate-phosphate-deoxyguanylate (CpG) motifs more classically associated with bacteria, but also found in viruses such as HSV1. 70 Expression and function of TLRs 7 and 9 have been well characterised in plasmacytoid DCs, but their role in epithelial recognition of viral infection requires further clarification. 71 The TLRs 3, 7, 8 and 9 are localised predominantly within intracellular endosomal–lysosomal compartments in epithelial cells.
Some of the TLRs expressed by respiratory and enteric epithelium, which are more commonly associated with bacterial recognition, also have antiviral function. Toll-like receptor 4, normally associated with recognition of bacterial lipopolysaccharide is important for the recognition of and response to RSV fusion proteins.72,73 Toll-like receptor 2, more typically associated with bacterial lipoprotein responses, induces antiviral responses, mostly in inflammatory monocytes, to a range of viruses including measles, hepatitis C, HSV, HCMV and RSV. 74 – 76 Similarly, release of DAMPs through apoptosis or necrosis as a result of viral infection can also trigger PRR-mediated recognition of viral inflammation.
The major members of the RLR family, RIG-I and melanoma-differentiation-associated gene 5 (MDA5), are localised in the cytosol of epithelial cells. The RIG-I binds RNA bearing a 5’-triphosphate residue, a feature absent from host cytoplasmic RNA due to eukaryotic RNA metabolism. Recognition of both ssRNA and dsRNA would appear to be possible, although the importance of 5’-triphosphate residues in ssRNA continues to be an area of conjecture,77,78 as RIG-I has been reported to recognise uridine- and adenosine-rich regions of RNA, found in the PAMP structure of hepatitis C virus. 79
MDA5 binds long dsRNA structures within the cytosol. The PKR recognises dsDNA or RNA with short stem loops, is induced by IFNs and inhibits translation through the phosphorylation of the alpha subunit of eukaryotic initiation factor 2α (eIF-2α). 80 The most exciting breakthrough in innate viral recognition in recent years was the demonstration that the NLR nucleotide-binding oligomerisation domain 2 (NOD2) acts as a cytoplasmic viral PRR by triggering activation of interferon-regulatory factor 3 (IRF-3) and production of IFN-β. 81 Mutations in the gene encoding NOD2 have been found to lead to susceptibility to Crohn’s disease, a chronic inflammatory condition of the intestinal tract. 82 The discovery that NOD2 acts as a viral PRR may lead to new avenues of research in this complex disease. Although the PRRs utilise varying signalling pathways, each of these receptors has the capability of activating IFNs and, therefore, the transcription of IFN-inducible genes.
Unlike the lower airways, the gut is rich in microbial microflora. Intestinal epithelial cells were thought to be hyporesponsive to PRR ligands, despite receptor expression, via a series of PRR inhibitory pathways or by limiting expression levels of PRRs preventing deleterious inflammation. However, rotavirus and coxsackievirus induce significantly increased mRNA expression of TLR2, TLR3, TLR7 and TLR8 in an intestinal epithelial cell (IEC) line. 83 Modulation of epithelial-derived cytokines such as IL-25 (IL-17E), IL-33 and thymic stromal lymphopoietin (TSLP) by TLR signalling in gut epithelial cells also indicates that PRRs are active in these cells. 84 – 86 Interleukin-25, IL-33 and TSLP are cytokines capable of directing Th2 responses and receptors are present on DCs, macrophages, invariant natural killer T cells, mast cells and T cells. 87 – 91 At the intestinal surface, these cytokines are thought to maintain tonic signalling and regulation of inflammation, whereas at the respiratory surface they have been implicated in the development of asthma.89,92 The identification of TLR-inhibiting receptors such as TIR8 (SIGIRR) in gut epithelium adds greater complexity to an already dynamic process. 93
It is of interest that many viruses utilise cell surface innate immune molecules as a means of entering epithelial cells. The enveloped dsDNA viron of the vaccinia virus, for example, is enriched in phosphatidylserine, a phospholipid used for the macropinocytic uptake of apoptotic bodies. 94 Adenovirus 3, which infects both the respiratory and digestive tracts, enters cells via macropinocytosis induced by binding to CD46, the normal function of which is to prevent injury by complement. 95 Measles virus also utilises CD46, and can also use TLR2 interaction to induce surface expression of CD150 to facilitate viral entry. 76
Interferon-mediated responses to viral infection
The signalling pathways induced by viral recognition and infection are both complex and incompletely characterised; they have been previously extensively reviewed. 96 In general, activation of viral recognition receptors leads to activation of signalling pathways involving, among others, NF-κB, MAPKs and the IFN regulatory factors. As stimulation of the majority of viral recognition receptors results in induction of type I IFNs, we will, therefore, focus on the effects of these cytokines following viral recognition.
The type I IFN response is principally for the control and containment of viral replication within the epithelial surface, prior to the generation of effective adaptive immunity, including virus-specific CD8 T-cells. Although some evidence of constitutive expression has been reported, the majority of type I IFNs are inducible, requiring de novo transcription and translation. The type I IFNs, IFN-α and IFN-β are acid stable and elicit a non-specific antiviral response. Interferon-α and IFN-β differ in some of their biological and physiochemical properties but have many overlapping functional properties and share common receptors. The IFNs can increase cellular synthesis of antiviral sensors such as PKR kinase and antiviral factors such as RNAse-1 and 2,5-oligoadenylate synthase which degrade viral RNA and prevent viral replication in infected and neighbouring cells. The IFNs can also up-regulate the cell surface expression of major histocompatibility complex (MHC) class I. Presentation of virus-specific peptides on MHC class-I to specific cytotoxic T-cells aids in the killing of infected cells in a well characterised process. However, viruses commonly down-regulate MHC class I expression as an immune evasion strategy. 97 NK cells which expresses a range of invariant cell surface receptors, some similar to the collectins described earlier, are capable of recognising and killing these MHC-Ilow infected cells in an IFN regulated process. The IFNs can activate NK cells, increasing their activity 20–100 times. Furthermore, IFNs serve to protect neighbouring uninfected cells from NK cell induced cell death by inducing MHC-I on their cell surface, a natural inhibitor of NK activation.98,99 Interferon-regulated inflammatory cytokines include TNF-α, IL-1 and IL-6. These cytokines activate neighbouring epithelium, alveolar macrophages and DCs, initiating a local inflammatory cytokine cascade with effects ranging from increased ciliary beat frequency to increased recruitment of further immune cells. A multitude of viruses have evolved mechanisms to inhibit IFN responses such as the non-structural proteins (NS) 1 and 2 of RSV, NS 1 produced by influenza virus and the SM protein produced by Epstein–Barr virus. 100 – 103
Although almost all cells can up-regulate type I IFNs in response to viral stimuli plasmacytoid DCs (pDCs) are more readily activated by a broader range of viral stimuli and release a greater amount of IFNs more rapidly than epithelial cells. 104 However, the ratio of pDC to epithelium dictates that by sheer numbers the epithelium can also be considered to be a significant source of type I IFN at the respiratory and gastrointestinal surfaces, and that activation of pDC by epithelium can serve to amplify this IFN signalling.
With the exception of some autoregulatory factors such as the suppressor of the cytokine (SOCS) signalling family of proteins, 105 the result of IFN-regulated gene expression is primarily the up-regulation of inflammatory mediators and much of the pathology associated with viral infection can be attributed to overly vigorous immune responses. Much of this inflammation depends on the activation of the NF-κB signalling pathway and associated signalling factors. It is, therefore, of great therapeutic interest that the epithelial cells of the respiratory surfaces themselves can generate an inhibitor of NF-κB mediated inflammatory pathways through the conversion of 25-hydroxyvitamin D3 to active 1,25-dihydroxyvitamin D3. Furthermore, despite its anti-inflammatory properties, active 1,25-dihydroxyvitamin D3 preserves essential antiviral IFN signalling mechanisms and increases the secretion of antimicrobial peptides. 106
The innate/adaptive crossover
Dendritic cells
To address the role of the epithelium of the respiratory and digestive tracts in the transition from innate to adaptive response, it is necessary to understand its relationship with DCs. Mucosal DCs can be recruited from the circulation as pre-existing DCs or as monocytes that differentiate into DCs. In the respiratory tract, DCs can also be derived from local precursor populations in the lung. 107 Dendritic cells reside in close association with the basolateral surface of the epithelium in both the airway and the gastrointestinal tract and can intercalate their dendrites through the epithelium to sample the apical milieu, a process thought to be triggered by PRRs on the overlying epithelium. 108 A substantial number of sub-classifications of DCs have been proposed and can be both species- and organ system-specific as recently discussed by Geissmann et al. 109 In the context of the present review, human DCs can be categorised into two functional groups, the conventional myeloid DC (mDC) and the pDC.
The CD11c-expressing mDC is phagocytic, produces a multitude of inflammatory cytokines and chemokines and can activate naïve CD4+ T-cells by antigen-specific MHC class-II/T-cell receptor interaction. mDCs can detect a broad selection of pathogens via a range of TLRs and intracellular NLR molecules. 110 A typical migration pattern involves collection of antigen at the epithelial or sub-epithelial surface, activation and migration through the lymphatic system to organised lymphoid tissues and presentation of antigen to naïve T-cells resulting in an adaptive immune response. Viral infections primarily result in the generation of T-cells with Th1 like cytokine profiles, although in some viral infections, such as severe RSV infection, Th2 cell responses my also develop. 111
The pDCs by contrast lack CD11c, but express the IL-3 receptor CD123 and the markers CD303 and CD304. The pDCs have low phagocytic capacity and, as mentioned previously, secrete large amounts of type I IFNs. pDC readily detect viruses via expression of TLR7 and TLR9 but lack many of the PRRs which respond to other microbial stimuli.71,110 Similar to mDCs, pDCs can migrate to organised lymphoid tissues, but do so primarily via the blood, crossing high endothelial venules for entry. 112 However, unlike mDCs, pDCs do not appear to have a significant role in naïve CD4+ T-lymphocyte stimulation, although it has been suggested that pDCs are specialised to cross present antigens rapidly to CD8 T-cells, accelerating the generation of antigen specific anti-viral immune responses. 113
Epithelial–dendritic cell interactions
Epithelial cells have the means to control not only the recruitment and/or local differentiation and activation of DCs via cytokine and chemokine expression, but also to polarise the subsequent T-cell responses. Furthermore, even once DCs are recruited, epithelium can continue to influence the immune response. Activated epithelium with up-regulated MHC class-II can act as accessory cells in local mucosal lymphoproliferation. 114
In the resting state, mDCs take up antigens in the absence of cytokine or PAMP stimulation at the respiratory surfaces, and in the presence of a regulatory cytokine milieu at the enteric surfaces. These mDCs then present antigen to antigen-specific T-cells without expressing the co-stimulatory molecules required for T-cell activation leading to failed, abortive or regulatory T-cell responses. In viral infection, mDCs are activated by PAMPs, cytokines including IFNs derived from epithelium and/or pDCs which can fully activate mDCs. Activated mDCs then go on to initiate effector T-cell responses as outlined above. Type I IFNs can polarise mDCs to induce Th1 lymphocytes for the development of anti-viral T-cell mediated immunity.115,116 Interferon-stimulated mDCs also have enhanced cross presentation ability, leading to increased presentation of exogenous antigens to CD8 T-cells. 117
Epithelial cell secretion of ligands for the chemokine receptors CCR1, CCR2, CCR5, CCR6, CXCR1, CXCR2, CXCR4 can recruit further monocytes and DCs during a viral infection, 118 with type I IFNs directing monocyte differentiation into mDC. 116 Secretion of IL-15 by the epithelium can preferentially polarise recruited monocytes to a pDC phenotype, 119 with antiviral and anti-inflammatory properties.120,121 Furthermore, pDC and epithelial-derived IFN can, in combination with IL-6, drive B-cell differentiation and maturation into antibody secreting plasma cells. 122 Virus-specific antibody can be advantageous in triggering engulfment and complement-mediated destruction of infected cells; however, antibody and complement complexes may also contribute to inflammation and disease as has been suggested in severe RSV infection. 123
The release of adenosine or exogenous degradation of ATP derived from epithelium and macrophages can also recruit monocytes and immature mDCs, which express adenosine receptors, to sites of inflammation and infection. Upon maturation, mDCs down-regulate the chemotactic nucleotide receptors facilitating a movement to the lymph nodes. 124 Exposure of mDCs to adenosine or triphosphate nucleotides can result in a shift from pro-inflammatory to anti-inflammatory cytokine secretion and a bias for the generation of Th2 lymphocytes. 125 – 128 Nucleotides released from epithelium may also reduce IFN production by pDC. 129 Studies utilising CD14+ monocytes differentiated in the presence of airway epithelium suggest that the epithelium programmes mDCs to take up and process more antigen and express TLR3 and TLR4, developing an ‘antimicrobial phenotype’. 130 There is limited evidence regarding the response of intestinal DCs to viral infection. It has been shown, however, that mDCs are able to take up and present virus from infected enterocytes. Following infection with murine reovirus, gut DCs contained structural viral proteins. By contrast, enterocytes stained for both structural and non-structural components. 131 Taken together with the fact that there was co-localisation with apoptotic inclusions and cytokeratin, this suggests that mDCs can obtain viral antigens from apoptotic enterocytes through phagocytosis. 132
Observations made in our laboratory with primary and cell-line respiratory epithelium in co-culture with mDCs and T-cells suggest that uninfected epithelium can polarise T-lymphocytes to a regulatory phenotype and inhibit both mDC-induced and direct T-cell activation. 133 Similar observations have been made with bronchial, squamous and type II like carcinoma cell lines. 134 The inhibitory capacity of lung epithelial cells was lost upon infection with RSV. 133 Thus, it appears that epithelial cells maintain the fine balance of preventing inappropriate immune and inflammatory responses in order to limit injury, whilst facilitating the recruitment and activation of inflammatory cells to remove infection. The potential for epithelial cell regulation in the initiation and exacerbation of allergic conditions in the lung130,135 and of Th2 cell responses in the gut 136 has gained particular attention. Activated epithelium is able to produce factors such as TSLP, which induces Th2 polarising ability in DCs, 135 GM-CSF which induces DC differentiation, and the cytokines IL-33 (an eosinophil activator) and IL-25 (a eosinophil chemo-attractant). 135 By comparison, the work investigating the polarisation of DC and T cell response by epithelium in viral infection is limited 137 and an area that requires further attention, particularly when considering that many urgently required vaccines are against viral pathogens first encountered on mucosal surfaces.
Apoptosis
The ultimate antiviral defence in the epithelial cell’s repertoire is the induction of its own death, which can initiate further signalling processes.138,139 The manner of this cell death is crucial. Lytic release of viral particles and necrosis of infected epithelium will recruit inflammatory cells to a site of infection. Actively released triphosphate nucleotides, as mentioned previously, also act as chemo-attractants to receptor bearing cells. 140 By contrast, apoptosis can be anti-inflammatory with engulfment of apoptotic cells by professional phagocytes or DCs reducing their inflammatory cytokine production and co-stimulatory molecule expression. 141 Therefore, the manner by which an epithelial cell dies during a viral infection may determine the balance between inflammation and resolution.
Conclusions
As we come to understand the complexities and dynamics of the interaction between the epithelium and the cells of the immune response, the epithelium can be viewed as more than just a barrier and considered a component of the innate immune response in its own right. Although major advances have been made to determine the innate immune response to bacteria at the pulmonary and gastrointestinal surfaces, the mechanisms of viral recognition at these sites, especially the gut, is less well understood. In addition, at the innate/adaptive interface, the epithelium is revealed as a potent regulator and an exciting target for the development of future therapeutics.
Footnotes
Acknowledgements
The authors thank Professor Sarah E.M. Howie for critically reading this manuscript. The authors have no conflict of interest to declare and this review received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.