
Abstract
Background:
Anaemia is prevalent after intensive care unit (ICU) discharge as a consequence of factors such as blood sampling, concurrent inflammation affecting erythropoiesis, and the use of restrictive ICU red blood cell (RBC) transfusion practice during inpatient stay. ICU survivors experience poor health-related quality of life (HRQoL). Prevalent symptoms include fatigue and weakness, to which anaemia may contribute. There are no trials exploring the effectiveness of treating anaemia with RBC transfusions post-ICU discharge.
Methods and analysis:
The ABC post-ICU trial is a multicentre prospective, parallel group, randomised trial, with embedded moderation and mediation analysis. Participants are adult ICU survivors with anaemia (haemoglobin (Hb) ⩽94 g/L) fit for ICU discharge. Patients are randomised to usual care (default Hb transfusion trigger <70 g/L, target 70–90 g/L) or single-unit RBC transfusions to achieve Hb range 100–120 g/L. The intervention is from randomisation to hospital discharge. Primary outcome is the physical component summary score (PCS) of the 36-item short form (SF-36) health survey, which measures HRQoL, assessed 90 days post-randomisation. Secondary outcomes at 90 days include: hospital length of stay, mortality, fatigue score, activities of daily living, and Mental Component Scale score (MCS) SF-36. Outcomes are also measured at 30 and 180 days. Safety outcomes include: new infections, transfusion-related adverse events, and major adverse cardiac events. Analysis includes a moderation analysis based on baseline recalled PCS SF-36, comorbidity burden, mobility, and systemic inflammation (C-reactive protein (CRP) concentration). A mediation analysis based on 30 days blood samples will explore whether anaemia severity (Hb) or persisting inflammation (CRP) mediates intervention effects. A health-economic analysis over 180 days will be conducted. The sample size is 346, providing 90% power to detect a difference in PCS SF-36 of 5 points, assuming >70% completed SF-36 follow-up.
ClinicalTrials.gov:
NCT04591574.
Keywords
Strengths and limitations
This trial measures the effect of a more liberal blood transfusion strategy for anaemic ICU survivors on outcomes prioritised by patients, such as health-related quality of life, fatigue severity, and ability to undertake activities of daily living.
The trial has statistical power to detect a minimally clinically important difference in the physical component of quality of life using a validated instrument (SF-36).
The trial includes pre-planned moderation analyses to explore whether some groups of patients benefit more or less from the intervention, and mediation analyses to explore factors which may partially explain how the intervention works.
A limitation is that the trial is not blinded from researchers or participants, because this is difficult for blood transfusion interventions.
An anticipated limitation may be follow-up completion for the primary outcome, which is known to be challenging in this patient population. The sample size is inflated to account for this.
Introduction
Approximately 80% of patients requiring organ support in an intensive care unit (ICU) survive their critical illness. Many survivors subsequently experience poor health as a result of physical, mental, and cognitive impairments collectively called a ‘post intensive care syndrome (PICS)’. 1 New disabilities are often superimposed on pre-existing health impairments, and it is estimated that more than 50% of ICU survivors have pre-existing multiple long term conditions (MLTCs). 2
Fatigue is the most prevalent symptom reported by ICU survivors, and is often severe and debilitating.3–5 Impairment of physical function is common, predominantly affecting patients in the first 2–3 months following ICU discharge. Disability, as described by the World Health Organisation (WHO) International Classification of Functioning, Disability and Health Framework, is common, and includes impaired body function (muscle strength, walking ability), activity (ability to carry out Activities of Daily Living (ADLs)), and participation (including health-related quality of life (HRQoL), and return to work).6,7 ICU survivors have excess long-term mortality compared to other hospitalised patients, and utilise substantially higher healthcare resources, especially during the first 3–6 months following ICU discharge. 8 Patients typically spend a further 3 weeks in the acute hospital after ICU discharge, and 25%–30% experience unplanned emergency readmission to hospital within 3 months of hospital discharge.2,9
Anaemia as a potential mechanism of delayed recovery and poor health after critical illness
Anaemia is highly prevalent during critical illness, because of bleeding, blood sampling, and impaired erythrogenesis, and is frequently present in survivors.10,11 A key uncertainty is the degree to which anaemia is a risk-modifiable factor for the longer-term physical, mental, and cognitive impairments that characterise PICS. Although many red blood cell (RBC) transfusion threshold trials have been conducted in different clinical populations, including critical illness, uncertainty remains about the association between anaemia and HRQoL, and whether anaemia treatments improve outcomes other than mortality.12,13 For example, recent trials in acutely brain injured patients suggest more liberal blood transfusion strategies may improve patient-centred outcomes such as HRQoL and functional status.14,15 Other populations in whom restrictive RBC transfusions may not be preferable to liberal transfusions include patients with acute and chronic cardiovascular disease,16,17 and older patients 18 in whom mortality and complications may occur more frequently. In previous ICU-based trials deaths mostly occurred before ICU discharge rather than among ICU survivors. Longer-term disability and HRQoL were rarely reported, and the evidence for these longer-term outcomes is weak. 12 This could be important among ICU survivors, because cardiovascular disease is present in up to 25% of ICU patients, pre-existing MLTCs are prevalent, and mean age is 60–65 years.9,19 Anaemia also persists in many patients following ICU discharge, and is prevalent at hospital discharge. 11 Most clinicians do not investigate the aetiology of post-ICU anaemia or actively treat it. 20 There is a need for further research on the impact of anaemia treatments on outcomes prioritised by patients, such as HRQoL, fatigue severity, and ability to undertaken activities of daily living.
In ICU, impaired erythrogenesis has multifactorial pathophysiology, but systemic inflammation is central by impairing iron absorption and availability through hepcidin upregulation, inhibiting erythropoietin release, and direct inhibition of marrow RBC production.10,21 Persisting systemic inflammation is prevalent after ICU discharge 22 and is associated with poor functional recovery 23 and HRQoL. This could occur via direct effects (e.g. neuro-muscular dysfunction) or indirect effects (e.g. persisting anaemia).
Existing evidence
A large observational literature demonstrates the high disability burden suffered by ICU survivors, especially during the first 3–6 months, but often lasting many years. This includes physical impairments, reduced Activities of Daily Living, and mental and cognitive disability.1,6,24
We found no studies specifically exploring the impact of transfusion strategies after ICU discharge on health status among general ICU population survivors. Two ICU based transfusion trials measured longer-term HRQoL: a trial in septic shock patients, 25 and a trial in older patients requiring prolonged mechanical ventilation. 26 The intervention in both trials occurred during ICU, and ICU mortality was high. Survivor cohorts were small, confirmed markedly reduced HRQoL, but found no differences in HRQoL. These studies had limited statistical power, and did not explore whether baseline health status or other factors influenced any treatment effects. This is relevant, because pre-existing health is an important determinant of longer-term HRQoL in heterogeneous ICU populations.27,28 Recent clinical transfusion guidelines for critical illness do not make recommendations specific to ICU survivors.29,30 No rehabilitation trials have considered anaemia treatment either as the main intervention or as part of the intervention. 31
Alternative approaches to treating post-ICU anaemia include blood conservation strategies, iron therapy, and erythropoietin therapy either individually or in combination. Evidence for these interventions is inconclusive, but several trials are ongoing.32–35 These approaches may also treat anaemia, but the degree and rate of correction may differ from RBC transfusion. Both approaches require further investigation.
Trial hypothesis and objectives
We hypothesise that in patients discharged from intensive care with moderate to severe anaemia, treatment of anaemia using blood transfusions will improve quality of life, disability, and other measures of well-being.
Primary objective
To determine whether treating anaemia from the time of ICU discharge using blood transfusions (target haemoglobin (Hb) 100–120 g/L) results in an improvement in self-reported physical HRQoL 90 days after intensive care discharge, compared with current usual care which recommends transfusions when Hb is less than 70 g/L to achieve a target Hb 70–90 g/L.
Secondary objectives
To determine the effect of treating anaemia using blood transfusions on fatigue, disability, and death rate compared with current usual care.
To determine the safety of treating anaemia with blood transfusions, compared with current usual care.
To determine which patients may benefit most from treating anaemia with blood transfusions.
To determine whether more active treatment of anaemia using blood transfusions is cost-effective.
Methods and analysis
Overall design
A prospective, parallel group, open, unblinded, randomised controlled intervention trial, with embedded moderation and mediation analyses. This publication summarises protocol version 9.0, 14th March 2025.
Trial registration and sponsor
ClinicalTrials.gov Identifier: NCT04591574; Sponsor, ACCORD (https://www.accord.scot/).
Personalised medicine questions
We aim to explore personalised medicine in relation to the use of blood transfusions in ICU survivors. We will explore baseline patient characteristics that may moderate the effects of RBC transfusions and factors during the intervention that may mediate effectiveness of the intervention on the primary outcome, if an effect is found.
Moderation variables: we will explore the importance of four baseline characteristics on moderating treatment effects, namely: (1) baseline co-morbidity score (Functional Comorbidity Index, FCI), 36 (2) recalled (pre-ICU admission) physical HRQoL (Physical Component Summary (PCS) of the 36-item short form (SF-36) health survey), 37 (3) mobility at ICU discharge (ICU mobility scale 38 ) and/or (4) baseline severity of systemic inflammation (C Reactive Protein (CRP) concentration at randomisation).
Mediation variables: we will explore the importance of two potential mediators of intervention effects based on variables at 30 days post-randomisation, namely: (1) severity of persisting inflammation (CRP concentration), and (2) persisting anaemia (Hb concentration).
Cost effectiveness questions
We will estimate cost per Quality-Adjusted Life Year (QALY) and cost per life-year over the study follow-up period. Depending on the results of the short-term outcomes, linkage of routinely collected healthcare data may be undertaken after completing the trial to estimate long-term cost-effectiveness.
Patient and public involvement
We consulted with ICU patients and their families, who helped develop our research. This group strongly supported exploring interventions that might reduce fatigue and disabilities, and agreed that anaemia is an important consideration. Patients felt HRQoL was a key patient-centred outcomes and prioritised fatigue as a key symptom. The Trial Steering Committee (TSC) includes a patient member. ‘How can the physical consequences of critical illness be prevented and what is the best way to support recovery from these after intensive care?’ is a James Lind Alliance priority setting partnership ‘top 10’ question for critical care research.
Inclusion and exclusion criteria
Inclusion and exclusion criteria are shown in Table 1. For inclusion criteria, we chose a Hb threshold of ⩽94 g/L based on the mid-point of the ‘moderate’ anaemia range defined by the World Health Organisation at the time of trial design. 39 For exclusion criteria, we excluded patients with primary neurological ICU-admission diagnoses because these are likely to dominate the primary outcome. Patients recovering from cardiac surgery, post-variceal haemorrhage with chronic liver disease, those with pre-existing dialysis-dependent renal failure, and patients with chronic transfusion-dependent haematological disorders are excluded because best practice evidence is stronger in these groups.
Inclusion and exclusion criteria.

Trial outcomes
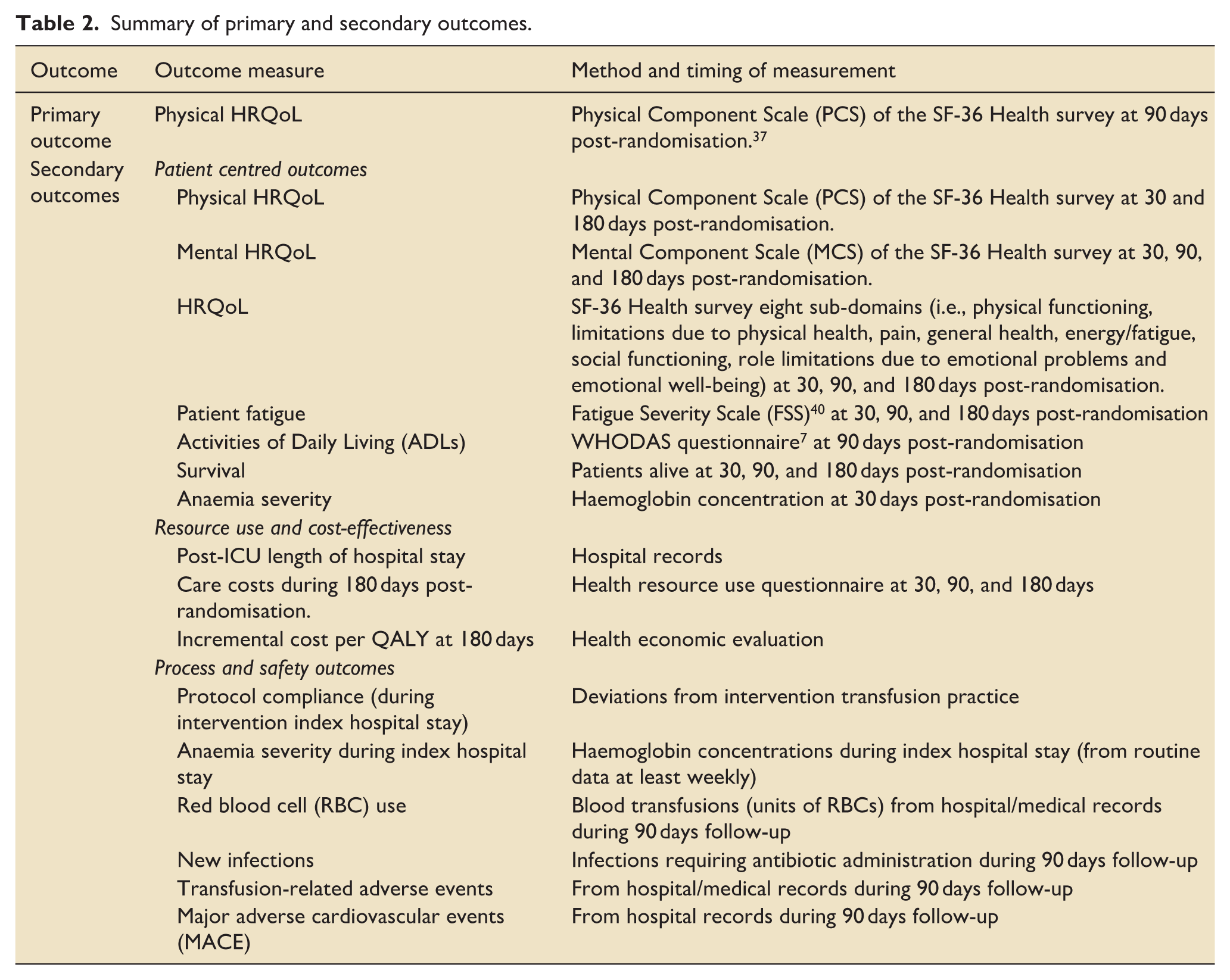
Primary and secondary outcomes are shown in Table 2.
Summary of primary and secondary outcomes.
Patient screening and consent
Patient screening is at the time they are considered ready for ICU discharge by clinical teams. Members of the responsible clinical team are asked to confirm suitability prior to any approach for consent by research staff. The eligibility window is from the time the patient meets inclusion criteria for up to 7 days following the decision by the treating clinician that the patient is ready for ICU discharge, as long as the patient remains in the acute hospital.
Wherever possible, patients are approached when they have mental capacity. However, when the clinical and/or research teams consider that they lack mental capacity approaches appropriate to the local legal arrangements for Adults with Incapacity are used, including a surrogate decision maker (SDM) where appropriate. Any patients in whom consent is from a SDM are approached for consent to remain in the trial once they regain capacity.
Randomisation
Patients are randomised using a web-based randomisation service managed by the Edinburgh Clinical Trials Unit (ECTU) to ensure allocation concealment. Allocation is in a 1:1 ratio, stratified by centre in permuted random blocks. The randomisation sequence was created using computer-generated pseudo random numbers by an ECTU member of staff not involved in clinical care.
Intervention
Duration of intervention
The intervention period lasts from the time of enrolment until discharge from the acute hospital, and ends when one of the following occurs: patient is discharged home from hospital; patient dies during the acute hospital stay; patient is discharged to a rehabilitation hospital or facility; or, patient is discharged to an intermediate or long-term care facility.
If a participant is discharged to another acute care hospital from the enrolling hospital as part of the same hospitalisation episode, the patient remains in the trial and treatment allocation is maintained wherever possible until discharge from that acute hospital.
Assessment and management during intervention period
Measurement of Hb during the intervention period is at the discretion of clinical teams, but is requested by research teams if not routinely available for 7 days.
For both study groups, the Hb is re-checked after each unit of RBCs administered within 24 h unless patients are bleeding consistent with best practice. (NICE Blood Transfusion Quality Statement 3: www.nice.org.uk/guidance/qs138/chapter/Quality-statements).
Transfusion prescription and patient monitoring during transfusion
Transfusions are commenced as soon as possible after a triggering Hb result and within a maximum of 48 h. Transfusions that do not occur or occur after 48 h are reported as protocol deviations. Bedside prescription checking and monitoring of patients during the transfusion period follow locally agreed protocols and procedures. This may include prescription of additional medication, such as diuretics, at the discretion of the clinical team. If Hb is not re-checked within 48 h this is also reported as a deviation. In the usual care group, administration of a blood transfusion at a trigger Hb ⩾70 g/L without the responsible clinician providing a reason for modifying the transfusion trigger is also recorded as a deviation.
Use of alternatives to blood transfusion
Patients in both study groups can receive alternatives to blood transfusion if directed by the clinical teams. However, these should be consistent with current NICE guidance concerning alternatives to blood transfusion (www.nice.org.uk/guidance/ng24).
Other treatment and interventions during the intervention period
All other treatments and therapies provided to patients in both study groups are at the discretion of the clinical teams.
Data collection
Data collection is summarised in Table 3.
Data collection during the trial.
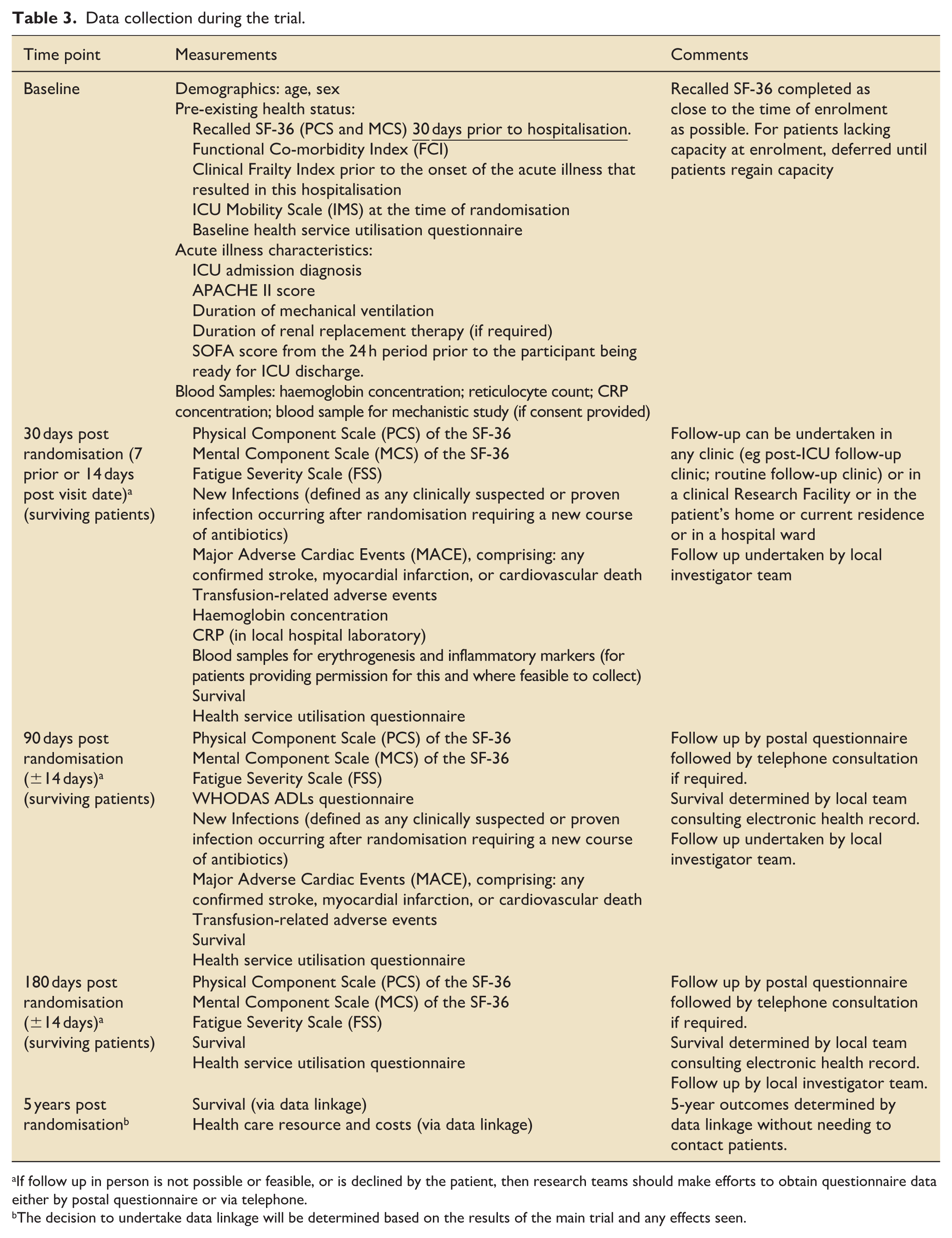
If follow up in person is not possible or feasible, or is declined by the patient, then research teams should make efforts to obtain questionnaire data either by postal questionnaire or via telephone.
The decision to undertake data linkage will be determined based on the results of the main trial and any effects seen.
Sample size
Based on previous research2,41 we estimated mean PCS SF-36 at 90 days will be significantly reduced at 35–40 (normal population average value 50), with standard deviation (SD) of 10–15 points. We defined a minimum clinically important difference as 5 points of the PCS element of the SF-36 at 90 days (smaller differences are of unlikely importance to patients). 42 This represents a ‘medium’ effect size of 0.42. Assuming the mean (SD) PCS SF-36 will be 38 (SD 12) with restrictive (usual) care, to detect an improvement from RBC transfusion to a mean of 43 at 90% power (two-sided P value 0.05) would require 244 patients.
In this population there is a risk of loss to follow-up for SF-36 (other than death), which in most previous trials has been between 10% and 30%. At trial outset we inflated the sample size allowing for a 20% loss to follow-up at 90 days. This required a sample size of 305 (randomised 1:1 between the two groups). Adjusting for pre-specified baseline covariates is expected to further increase study power and/or protect against extra variability.
In May 2024, follow-up completion for the primary outcome was 75% despite active attempts to maximise this. As part of the strategy to ensure internal validity and adequate study power we increased the sample size to ensure complete follow-up for at least 244 surviving patients. At this time, rates of deaths during follow-up were 12/182 patients (7%). A conservative approach was agreed, assuming loss to follow-up might be as high as 30%. This required an inflation in sample size from 305 to 346 patients to ensure at least 90% power for a difference in the SF-36 PCS of 5 points. We also decided to use change from baseline SF-36 PCS as the primary outcome, adjusting for the baseline value in the analysis, and use imputation for missing outcome data in survivors to maximise inclusion of information in the primary analysis (see below). The final sample size is 346 patients.
Analysis
All details of the statistical approaches are specified in the Statistical Analysis Plan (SAP; Supplemental Appendix 1).
Primary outcome
The primary analysis is an intention-to-treat (ITT) comparison of change from baseline PCS SF-36 at 90 days post-randomisation between randomised groups, using a mixed effects linear model, with site as a random effect, and adjustment for age, Functional Comorbidity Index, pre-ICU recalled PCS SF-36, ICU length of stay, ICU mobility scale, and baseline inflammation (assessed by CRP concentration). Given that 20%–30% of randomised patients are expected to have missing SF-36 data, we will use multiple imputation by chained equations (MICE), employing the passive approach model (or an alternative appropriate model) under the missing at random assumption. We will also conduct sensitivity type analyses to investigate the robustness of the findings to the assumptions behind this model.
In addition to the full ITT approach for the primary endpoint, (i.e. the real-world effect of the intervention amongst all those offered the intervention) we will also estimate the treatment efficacy (the idealised effect achieved by those fully compliant with their randomised treatment and all aspects of the protocol).
Moderation analyses of PCS SF-36 scale
We will conduct pre-specified moderation analyses on the primary endpoint. These will fit interaction terms for the four baseline parameters and treatment group, namely: (a) Co-morbidity score (FCI); (b) pre-ICU recalled SF-36; (c) ICU mobility scale; and (d) baseline level of systemic inflammation (CRP concentration). We will first explore the nature of the potential interaction by fitting each baseline parameter as a continuous interaction with treatment, and if warranted, look at suitable categorisation (e.g. above vs below the median) for ease of clinical interpretation.
Mediation analysis of PCS SF-36 scale
We will conduct two mediation analyses on the principal analysis of PCS SF-36 model. These will explore how these factors may mediate the intervention effect. First, we will examine the role of Hb at 30 days post-randomisation, as a measure of anaemia severity and persisting impaired erythropoiesis. Specifically, we will assess whether the degree of anaemia correction at 30 days, according to group allocation, relates to the endpoint. Second, we will explore whether CRP concentration at 30 days post-randomisation, as an indicator of persistent inflammation within each group, mediates the impact of RBC transfusions on the endpoint. This is relevant because RBC transfusions may be pro- or anti-inflammatory, superimposed on the persisting inflammation that often occurs in the post-ICU period.
We also plan to measure a range of biomarkers that may provide additional information about the mechanisms by which the intervention influences the primary outcome, based on blood sampling at baseline and 30 days. These will include effects of the intervention on iron status, marrow erythrogenesis (red blood cell production), inflammation, and immune suppression. These exploratory analyses will be underpinned by a Directed Acyclic Graph (DAG) and pre-specified analysis plan (Supplemental Appendix 2).
Secondary outcomes
Secondary outcomes will be analysed using models appropriate for the type of outcome (e.g. for continuous outcomes as per the primary endpoint/principal analysis; mortality using a Cox proportional hazards model, binary outcomes with adjusted logistic regressions).
Health economic evaluation
The cost-effectiveness of the intervention compared with ‘usual care’ will take the format of a within-trial cost-effectiveness analysis and use a cost-utility analysis framework. The primary health economic outcome measure will be the incremental cost per QALY at 180 days, and a further analysis will assess the cost per Life Year at 180 days. The estimation of QALYs will rely on the SF-6D, derived from the SF-36. 43 If appropriate based on trial findings, the impact of the intervention on the downstream costs, health outcomes and cost effectiveness will be assessed over a 5-year time horizon. The perspective for analysis will be that of the UK National Health Service and Personal Social Services as per NICE guidance (https://www.nice.org.uk/process/pmg36), with a secondary analysis taking a wider societal perspective (e.g. including indirect costs in terms of the costs of lost productivity).
A detailed Health Economic Analysis Plan (HEAP) will be finalised before database lock.
Study within a trial (SWAT): sub-study to explore reasons that patients decline consent to participate
During the trial conduct it has become apparent that consent rates among eligible patients is lower than expected, at around 40% of patients approached for consent. The reason for this unexpected low agreement to participate is uncertain, but important to the validity of this and future trials in ICU survivor populations. With the advice and support of patient collaborators and the Trial Steering Committee, we developed a provisional logic model hypothesising possible reasons for this unexpectedly low consent rate (Supplemental Appendix 3). We designed a mixed methods study including a patient focus group, survey of research staff, and interviews with research staff as a SWAT to increase understanding of patient decision-making in relation to the trial. This is summarised in Supplemental Appendix 3.
Ethics and trial oversight
The trial sponsor is ACCORD (https://www.accord.scot/). Trial oversight is by an independent Trial Steering Committee and Data and Safety Monitoring Committee. Ethical approval is from the Scotland a Research Ethics Committee (REC; Scotland; 19/SS/0105) and the Wales REC4 (England, Wales; 19/WA/0276).
Role of funder and sponsor
The funder had no role in trial design. The sponsor reviewed the trial protocol and ethical submission and approved it (including amendments) prior to submission. Neither will have any role in analysis, interpretation, or the decision to submit for publication.
Current status
Recruitment started in September 2020 at one centre (Edinburgh), but the trial was impacted by the COVID19 pandemic, and wider roll out was postponed. Further site set-up occurred from October 2022. Total sites opened during the trial is 19, across England (15 sites), Scotland (3 sites), and Wales (1 site). Recruitment is currently 313 (April 10th 2025), and is expected to complete by June 30th 2025.
Supplemental Material
sj-docx-1-inc-10.1177_17511437251374884 – Supplemental material for Anaemia management with red blood cell transfusion to improve post-intensive care disability: Protocol for the ABC post-ICU randomised controlled trial
Supplemental material, sj-docx-1-inc-10.1177_17511437251374884 for Anaemia management with red blood cell transfusion to improve post-intensive care disability: Protocol for the ABC post-ICU randomised controlled trial by Timothy S. Walsh, Lydia Emerson, Jo Singleton, Rachel Locherty, David Hope, Stephanie Cholbi, Annabel Giddings, Alix Macdonald, Nazir Lone, Annemarie B. Docherty, Gillian Mead, Simon J. Stanworth, Alexander Drakesmith, Noemi B. A. Roy, Peter Hall, Aileen R. Neilson, Roz Pollock, Aryelly Rodriguez, John Norrie, Christopher J. Weir, Akshay Shah and David Griffith in Journal of the Intensive Care Society
Footnotes
Ethical considerations
Scotland A Research Ethics Committee (REC; Scotland; 19/SS/0105); Wales REC4 (England, Wales; 19/WA/0276).
Author contributions
Conception and acquiring funding: Walsh; Docherty; Drakesmith; Griffith; Hall; Lone; Mead; Neilson; Norrie; Stanworth; Roy.
Trial design: Walsh; Docherty; Drakesmith; Emerson; Cholbi; Giddings; Griffith; Hall; Hope; Locherty; Lone; Macdonald; Mead; Neilson; Norrie; Pollock; Rodriguez; Shah; Singleton; Stanworth; Roy; Weir.
Statistical design and analysis: Walsh; Griffith; Lone; Norrie; Rodriguez; Weir.
Trial oversight: Walsh; Docherty; Drakesmith; Emerson; Cholbi; Giddings; Griffith; Hall; Hope; Locherty; Lone; Macdonald; Mead; Neilson; Norrie; Pollock; Rodriguez; Shah; Singleton; Stanworth; Roy; Weir.
Health economic evaluation: Hall; Neilson.
All authors reviewed the final manuscript and approved it prior to submission.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the J P Moulton Foundation. The trial is being delivered across the United Kingdom with infrastructure support from the National Institute of Health and Care Research (NIHR) Clinical Research Network.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Akshay Shah has received consultancy fees from Pharmacosmos (UK) outside of the submitted work.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.