
Abstract
Background:
Between April 2022 and March 2023, 43.8% (88,259) patients admitted to Intensive Care Units (ICU) in the United Kingdom (UK) required breathing support through a ventilator, the majority require sedation. Unfortunately, mechanical ventilation is associated with high mortality and morbidity, and sedative agents currently used have significant side effects including hypotension and delirium. They are also implicated in long-term psychological sequelae such as major depression and posttraumatic stress disorder. Ketamine has been utilised in anaesthesia for over 50 years and has an excellent safety profile. The diverse properties of ketamine are the focus of much research currently, including its properties as a potent antidepressant. Ketamine has not been fully investigated in the context of ICU, and there are gaps in the evidence that warrant further investigation through a large randomised controlled trial. Preparatory work for such a study includes refining study designs, identifying key clinical and patient centred outcomes and exploring barriers to implementation, which is the focus of this work.
Methods:
SHOCK-ICU is a single centre, non-randomised, feasibility study assessing the feasibility of continuous ketamine infusions for the provision of sedation for 30 patients undergoing mechanical ventilation on the ICU.
Data will be collected at baseline, daily until >48 h without mechanical ventilation, ICU discharge, and 90-days from enrolment. Data collection will include trial aspects such as expected recruitment, refusal, and follow-up rates, ability to collect data, and exploratory assessment of clinical efficacy markers.
Primary outcome:
The primary outcome is study feasibility; this will be assessed using pre-defined progression criteria that will aid design of future ketamine sedation studies.
Keywords
Introduction
Background and rationale
Mechanical ventilation is a common intervention in the Intensive Care Unit (ICU), with approximately 43.8% (88,259) of all patients admitted to ICU across the United Kingdom (UK) between April 2022 and March 2023 requiring breathing support on a ventilator. 1 Worldwide estimates from 2010 were that 20 million patients worldwide require invasive mechanical ventilation each year. 2 For most patients, sedation is a requirement for mechanical ventilation. Optimising sedation is a fundamental aspect in managing critically ill patients. Agent selection is a balance of risks and benefits. Most traditional sedatives (including propofol, benzodiazepines and alpha-2-agonists) are associated with multiple significant and potentially detrimental adverse effects; commonly hypotension, bradycardia and prolonged mechanical ventilation.3 –7
A significant consequence of sedatives, in particular benzodiazepines, is the increased risk of delirium in ICU.8,9 ICU delirium causes significant distress for both patients and their relatives, increases the work burden for ICU staff, and puts patients at significant risk of serious complications, for example, accidental removal of endotracheal tubes, tracheostomies, and venous catheters, post-traumatic stress disorder (PTSD), and increases mortality.10,11
Ketamine is a water and lipid soluble N-methyl D-aspartic acid (NMDA) receptor antagonist that has been used since the 1970s to provide cataleptic, amnesic, analgesic and dose dependant anaesthetic effects. 12 Owing to its ability to stimulate the sympathetic nervous system, preserving heart rate and blood pressure, whist avoiding respiratory suppression, ketamine has become increasingly popular as an anaesthetic agent for emergency surgical procedures in hypotensive patients.13,14
Although having been available in clinical practice for 50 years, and becoming licensed for major depressive disorder (MDD) treatment in 2019, 15 ketamine by continuous infusion remains a rarely used sedative to facilitate mechanical ventilation in UK practice. 16 A review of the literature revealed a paucity of high-quality evidence with very few well-designed prospective studies. 17 Despite the lack of well-designed, well-powered studies, the reported findings suggest a range of potential patient benefits, including improved sedation and pain scores, reduced concomitant sedative infusions, reduced opioid requirement and haemodynamic stability.
Given that psychological symptoms such as depression or PTSD following ICU admission are also common, ketamine’s ability to rapidly provide antidepressant effects may be beneficial in post-ICU MDD when used as a sedative on ICU.18 –20 Publications investigating the link between ketamine use on ICU (for analgesia, sedation or otherwise) and depressive symptoms following or during ICU admission currently remain limited to case reports.21,22
A large prospective randomised controlled trial (RCT) is required to provide robust evidence with regards to continuous ketamine sedation. However, as this represents a novel sedation regime and making significant changes to current sedation practices may face potential clinical and cultural barriers to implementation; therefore, careful planning of future studies is required in order to minimise chances of implementation failure. This study will investigate the feasibility of conducting a future multi-centre, randomised trial of ketamine sedation on ICU.
Methods
Trial setting and design
The study is set in adult critical care and is a single-centre, single-arm, prospective, feasibility study of continuous ketamine infusions for primary sedation in patients undergoing mechanical ventilation on the intensive care unit (ICU).
This study is a clinical trial of an investigational medicinal product (CTIMP) and has been approved by the Medicines and Healthcare products Regulatory Agency (MHRA; CTA 18166/0242/001-0001) and the Health Research Authority and Health and Care Research Wales (HRA and HCRW; 22/EE/0186). The study is registered on ISRCTN (ISRCTN13274002). The current version of the protocol (V1.1, 11/07/2024) is available from the ISRCTN website.
Objectives
Primary objective
The primary objective is to establish the feasibility of using continuous ketamine infusions for sedation and the collection of potential key future endpoints to inform a subsequent randomised controlled trial. Trial methods and protocol implementation will be assessed against the pre-defined progression criteria outlined in Table 1. The elements assessed will include the expected rates of eligibility, recruitment, and follow-up, protocol adherence, ability to collect proposed data including clinical endpoints, patient reported outcome measures and health economic data.
Progression criteria.
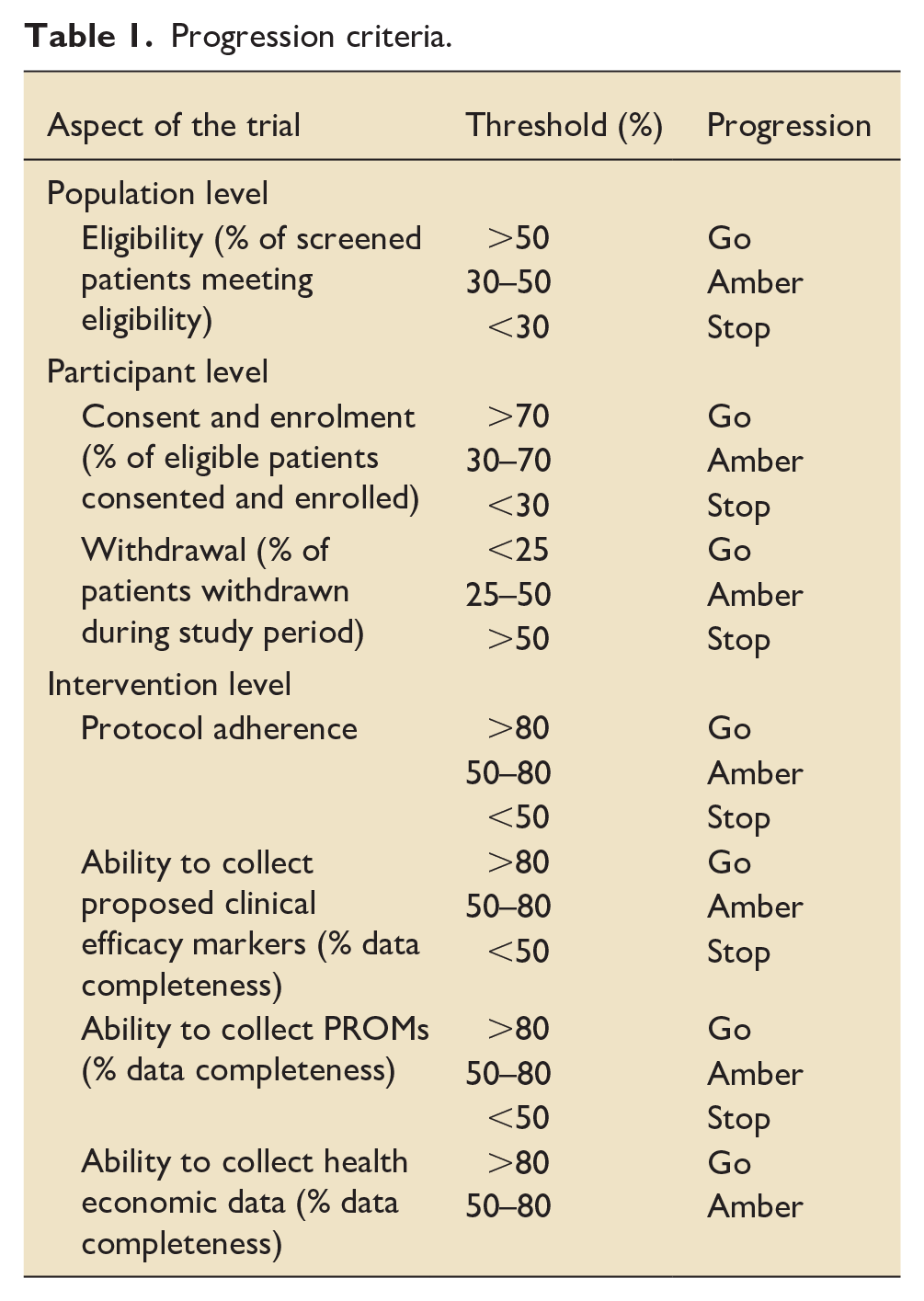
Identification of prospective clinical and patient-centred endpoints as well as early indicators of efficacy and safety will also be assessed. These will be compared to published data in peer reviewed literature as well as local data from the study ICU and published Intensive Care National Audit and Research Centre (ICNARC) records.
This includes exploratory assessments of clinical efficacy markers through monitoring of outcomes and clinical events of interest relating to the investigational medicinal product (IMP) throughout the patient’s ICU stay, hospital stay, and at 90-day follow-up. These include but are not limited to mortality, length of stay, duration of ventilation, patients’ sedative requirements, cardiovascular support requirements, incidence of delirium and incidence of adverse events or adverse reactions. A full list of clinical efficacy and exploratory outcome measurements can be found in the Supplemental Materials.
Patient and public involvement
PPI work has been carried out through previous ICU patient focus groups held in conjunction with the NIHR Biomedical Research Centre (BRC), Leeds. Six focus group participants described their experiences as patients in ICU, revealing a high incidence of negative recalled experiences, particularly with regards to delirium and sedation. Participants were asked to provide their thoughts on the rationale, acceptability and design of the proposed study. All participants felt that the intervention was acceptable, even given the negative reputation of ketamine, and that they would be willing to receive a ketamine-based sedation regime if they were in ICU. This PPI work also helped refine design aspects such as plain English summaries, and considerations when gaining assent from relatives.
The study has also undergone external PPI review from the national ICU charity ‘ICU Steps’ and the ‘National Institute of Academic Anaesthesia Patient, Carer and Public Involvement and Engagement (NIAA PCPIE)’ who confirmed this to be an area of high importance for patients and their relatives, and as having potential to significantly impact patient experience and outcomes.
Participants, interventions and outcomes
Eligibility criteria
The inclusion and exclusion criteria are in keeping with recent ICU sedation studies and were designed using a combination of expert opinion, retrospective review of the trial site patient population, literature review and regulatory requirements (Table 2).
Inclusion and exclusion criteria.

O’Grady jaundice to encephalopathy time intervals: Hyper-acute = <7 days, acute = 8–28 days, sub-acute = 5–12 weeks. 23
These tests should be performed and recorded in the medical notes as part of the standard of care for ICU patients. Any potential participants in this category without liver function tests from the previous 7 days at the time of eligibility screening will be excluded from participation.
Any woman of childbearing potential (as defined by Clinical Trials Facilitation and Coordination Group 24 that is, fertile, following menarche and until becoming post-menopausal unless permanently sterile. Permanent sterilisation includes hysterectomy, bilateral salpingectomy and bilateral oophorectomy) lacking capacity with a possibility of being pregnant should have a pregnancy test performed and recorded in the medical notes as part of the standard of care for ICU patients. Any potential participants in this category without a valid negative pregnancy test at the time of eligibility screening will be excluded from participation.
It is not a requirement to measure intraocular pressure specifically (beyond any clinical reason to outside of the study). Any patient with a documented history of raised intra-ocular pressure or on long-term treatment will be excluded.
Recruitment and screening
Patients requiring mechanical ventilation will be screened using the SHOCK-ICU eligibility checklist as soon after identification as possible to avoid delays in enrolment and initiation of IMP. Screening will continue for up to 48 h post initiation of mechanical ventilation. Periods of mechanical ventilation occurring prior to admission, for example, in operating theatres or emergency department will not count towards the 48 h eligibility time, except mechanical ventilation occurring at an external ICU. Screening may occur multiple times during the 48 h if appropriate.
Informed consent
At the point of enrolment patients will lack the capacity to consent because they will be receiving sedative medications. Assent will be obtained in accordance with UK law either through a personal legal representative (usually the next of kin) or if a personal representative is unavailable, then a professional legal representative will be consulted.
Given the time-critical nature of enrolment and treatment (earlier intervention may correlate with preferable outcomes) consent is required within 2 h of confirmation of eligibility.
Should a participant regain capacity during the study period, they will be asked to provide retrospective consent. This will occur as soon as practically possible upon identification of regaining capacity.
Participants, their legal representative, or their professional representative are free to withdraw from the study without reason at any point. Refusal to participate or withdrawal from the study will not impact any other aspects of care.
The consent process is illustrated in Figure 1 in the Supplemental Materials.
Interventions
Patients will commence intravenous infusion of open-label study drug according to a weight-based dose regimen (see Supplemental Materials). Dosing will be based on actual body weight unless BMI >40 kg/m2, in which case an adjusted body weight (ABW) will be used. 25
Clinical staff will transition patients to achieve sedation with the IMP as quickly as clinically feasible and safe, to replicate routine practice. Alfentanil will be used for analgesia alongside the IMP and titrated using clinical judgement to replicate standard care.
Patients will be titrated to achieve the default sedation target of most awake and comfortable state unless otherwise clinically indicated (Richmond Agitation Sedation Scale −2 to +1).
Duration of ketamine infusions will be determined by the treating clinical team up to a maximum of 90 days. There is no minimum stipulated duration of intervention, and all participants should undergo regular attempts to wean from sedation and mechanical ventilation as appropriate and according to local ICU guidelines and standard of care procedures.
Endpoints
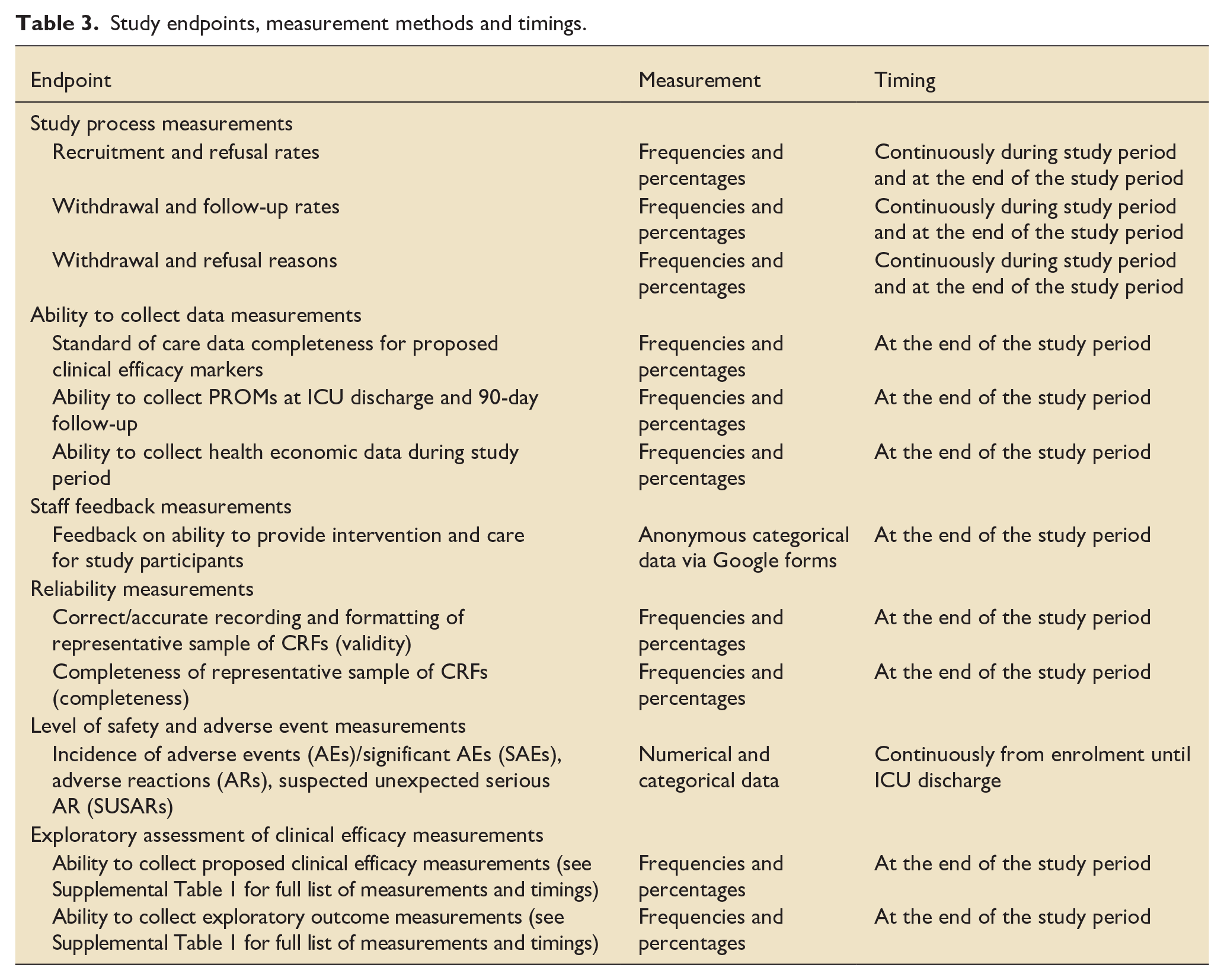
The endpoints used to assess the study objectives are detailed in Table 1. The potentially important clinical efficacy measurements are detailed in Supplemental Table 1. The proposed clinical efficacy measurements are derived from either routinely collected ICU medical and nursing data or based on the scientific premise of the study. These measurements have the potential to become key endpoints or yield key results in subsequent larger RCTs, and therefore it is useful to assess firstly if accurate collection is possible (Table 3).
Study endpoints, measurement methods and timings.
Sample size
As this is a feasibility study, a formal power calculation is not suitable. Estimated sample sizes were tested using Lewis et al.’s method of hypothesis testing of feasibility outcomes based on progression criteria via the authors published application SS-Progress (https://ss-progress.shinyapps.io/ss_progress_app/).26,27
Based on this method, 59 potential participants would need to be screened and 30 participants would need to be enrolled to power assessments of progression criteria at both participant and intervention level with a power of ⩾92%. Therefore, the study aims to enrol 30 participants.
Assignment of intervention
Allocation/Blinding
This is an open-label, non-randomised study.
Data collection, management and analysis
Data collection methods
Data will be collected using SHOCK-ICU case report forms (CRF) at baseline, daily until off mechanical ventilation for >48 h, ICU discharge, and 90-days.
Statistical analysis
Simple descriptive statistics (frequencies, percentages) will be used to assess the study endpoints and used to inform predefined progression criteria for the study protocol.
Incidence of clinical events, including adverse drug reactions, AEs, SAEs and SUSARs will be compared to published data in peer reviewed literature as well as available records from the study ICU and Intensive Care National Audit and Research Centre (ICNARC).
Progression criteria will be assessed according to the predetermined thresholds set out in Table 1.
Monitoring
A trial specific monitoring and reporting plan has been agreed with the sponsor and regulatory bodies, including of AEs, SAEs, ARs, SUSARs and urgent safety measures.
Ethics
Research ethics approval
The trial will be conducted in accordance with the UK Policy Framework for Health and Social Care Research 2018, the Medicines for Human Use (Clinical Trials) Regulations 2004 and subsequent amendments, Data Protection Act 2018 and Guidelines for Good Clinical Practice (GCP). This trial will be carried out under a Clinical Trial Authorisation in accordance with the Medicines for Human Use (Clinical Trials) regulations.
The study been approved by the Medicines and Healthcare products Regulatory Agency (MHRA; CTA 18166/0242/001-0001) and the Health Research Authority and Health and Care Research Wales (HRA and HCRW; 22/EE/0186). The study is registered on ISRCTN (ISRCTN13274002) and the full study protocol is available from the ISRCTN website.
Confidentiality
All research data collected as part of the study will be anonymised and stored securely according to legal requirements. Any personal data collected will be stored separately from clinical data.
Dissemination plans
Data depositing
Archiving will be authorised by the Sponsor following submission of the End of Trial report. Long-term storage and archiving will occur in accordance with MHRA guidance and will be archived using the LTHT approved external archiving service for at least 25 years after completion or discontinuation of the study.
Publications
The results will be submitted for publication in relevant peer reviewed literature and for presentation at meetings. Material will be included in a thesis to be submitted to University of Leeds. Summaries of the trial will be made available to the participants and the investigators.
Supplemental Material
sj-docx-1-inc-10.1177_17511437251327565 – Supplemental material for The Sedative and Haemodynamic effects Of Continuous Ketamine infusions on Intensive Care Unit patients (SHOCK-ICU): Investigating key outcomes, resource utilisation and staff decision-making: Clinical feasibility study protocol
Supplemental material, sj-docx-1-inc-10.1177_17511437251327565 for The Sedative and Haemodynamic effects Of Continuous Ketamine infusions on Intensive Care Unit patients (SHOCK-ICU): Investigating key outcomes, resource utilisation and staff decision-making: Clinical feasibility study protocol by Nicholas D Richards, Simon J Howell, Mark C Bellamy, James Beck, Fiona Tingerides, Ruben Mujica-Mota, Hilary L Bekker, Samuel Relton and Helen Thorp in Journal of the Intensive Care Society
Supplemental Material
sj-docx-2-inc-10.1177_17511437251327565 – Supplemental material for The Sedative and Haemodynamic effects Of Continuous Ketamine infusions on Intensive Care Unit patients (SHOCK-ICU): Investigating key outcomes, resource utilisation and staff decision-making: Clinical feasibility study protocol
Supplemental material, sj-docx-2-inc-10.1177_17511437251327565 for The Sedative and Haemodynamic effects Of Continuous Ketamine infusions on Intensive Care Unit patients (SHOCK-ICU): Investigating key outcomes, resource utilisation and staff decision-making: Clinical feasibility study protocol by Nicholas D Richards, Simon J Howell, Mark C Bellamy, James Beck, Fiona Tingerides, Ruben Mujica-Mota, Hilary L Bekker, Samuel Relton and Helen Thorp in Journal of the Intensive Care Society
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: NR (principal investigator) is in receipt of the Intensive Care Society Road to Research Award to fund activities relating to this study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Principal Investigator research time partly funded through Leeds Doctoral Scholarship (University of Leeds) and is in receipt of the Intensive Care Society Road to Research Award to fund activities relating to this study.
Protocol version
Protocol version: V1.1
Protocol identifier: 2022-CT02
Sponsor
Research & Innovation Centre
St James’s University Hospital
Trial management group
The Trial Management Group (TMG) will comprise of the following persons:
- Dr James Beck, Chief investigator and joint clinical lead
- Professor Mark C Bellamy, Joint clinical lead
- Dr Nicholas D Richards, Principal Investigator
- Professor Simon J Howell, Primary Supervisor
- Dr Ruben Mujica-Mota, Health Economics lead
- Dr Samuel Relton, Statistical lead
- Fiona Tingerides, Critical Care Pharmacy lead
Trial registration
ISRCTN registration number: ISRCTN13274002
IRAS number: 1007276
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.