
Abstract
Haemophagocytic lymphohistiocytosis (HLH) is a syndrome of severe immune dysregulation, characterised by extreme inflammation, fever, cytopaenias and organ dysfunction. HLH can be triggered by conditions such as infection, autoimmune disease and malignancy, among others. Both a familial and a secondary form have been described, the latter being increasingly recognised in adult patients with critical illness. HLH is difficult to diagnose, often under-recognised and carries a high mortality. Patients can present in a very similar fashion to sepsis and the two syndromes can co-exist and overlap, yet HLH requires specific immunosuppressive therapy. HLH should be actively excluded in patients with presumed sepsis who either lack a clear focus of infection or who are not responding to energetic infection management. Elevated serum ferritin is a key biomarker that may indicate the need for further investigations for HLH and can guide treatment. Early diagnosis and a multidisciplinary approach to HLH management may save lives.
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is a potentially life-threatening syndrome characterised by dysregulation of immune cells, excessive production of cytokines, extreme inflammation and tissue damage. HLH may be indolent or progress to a self-perpetuating, hyperinflammatory cytokine storm and multiorgan failure. HLH can be triggered by infection, malignancy and rheumatological disease and has a high acute mortality rate of 40%. 1 HLH is almost certainly under-recognised, or only recognised late at least partly due to the protean nature of presenting symptoms and lack of validated diagnostic criteria in adults. Early recognition and treatment of HLH potentially improves outcomes.2–5
The true prevalence of HLH in critical care is unknown. Sepsis, in particular, has a large overlap with HLH in both clinical features and pathophysiology 3 and, crucially, infection may be an HLH trigger. HLH or a related syndrome of sepsis-induced hyperinflammation may be present in a distinct subset of critically ill patients with presumed or confirmed sepsis, particularly those who deteriorate despite intensive infection management. Given the very high mortality, it is important to consider and identify HLH early.6,7 Furthermore, therapeutic approaches involve heavy immunosuppression, a strategy that could appear counter-intuitive in septic patients. 8
This article aims to provide an overview of adult-onset HLH for the general intensive care clinician, to outline pragmatic diagnostic and treatment approaches in light of the existing evidence, and to discuss emerging avenues for better characterisation of this heterogeneous clinical syndrome.
Historical context and terminology
Historically, HLH has been classified into familial HLH (fHLH, a genetic disorder presenting in infancy) and secondary or acquired HLH (sHLH, a secondary immune phenomenon diagnosed in later childhood, adolescence or adult life). However, there is a growing recognition of a complex interplay between inherited, acquired and environmental factors across all forms of HLH, leading to a large variability in the timing and severity of clinical disease expression. A key message for clinicians is that sHLH can occur at any age.
The existing terminology for sHLH is confusing, leading to a growing call for a unified nomenclature across specialties. 8 HLH in the context of autoimmune disease is commonly referred to as macrophage activation syndrome (MAS). 8 However, there are multiple alternative terms used, some of which refer to specific clinical circumstances. These include MALS (MAS-like syndrome), hyperferritinaemic syndrome, viral-associated haemophagocytic syndrome and sepsis–HLH overlap syndrome, among others.3,9–11 Of note, some studies have used broad surrogate definitions such as ‘sepsis with hepatobiliary dysfunction and disseminated intravascular coagulation’.11,12 Given this lack of unified terminology, acquired HLH should perhaps be considered a clinical syndrome rather than a ‘disease’ in itself, with the emphasis on early recognition and treatment. For practical purposes, we use the term sHLH in this review.
Pathogenesis
HLH is caused by a failure of the negative feedback loops that normally control inflammation. Our understanding of the complex pathophysiology of secondary HLH is still evolving; a basic overview of current concepts is therefore provided in Figure 1. The result of this dysregulation is persistent immune activation and a self-perpetuating hyperinflammatory state: the cytokine storm.
8
The clinical sequelae are fever, cytopaenias, end-organ damage (often manifested as multiple organ dysfunction) and, in some cases, haemophagocytosis evident on biopsy of bone marrow or other tissues. It is important to note, however, that despite the syndromic name, haemophagocytosis need not be present and is not required for diagnosis.1,8
Pathogenesis of sHLH. An underlying defect in NK cells and CTLs, on the background of preexisting susceptibility, leads to failure to clear antigen from infected tumour or autoimmune cells and antigen-presenting dendritic cells. This leads to uncontrolled immune stimulation with activation and proliferation of macrophages and CTLs in a self-perpetuating fashion. A massive production of cytokines (cytokine storm) results in hyperinflammation, end-organ damage and haemophagocytosis.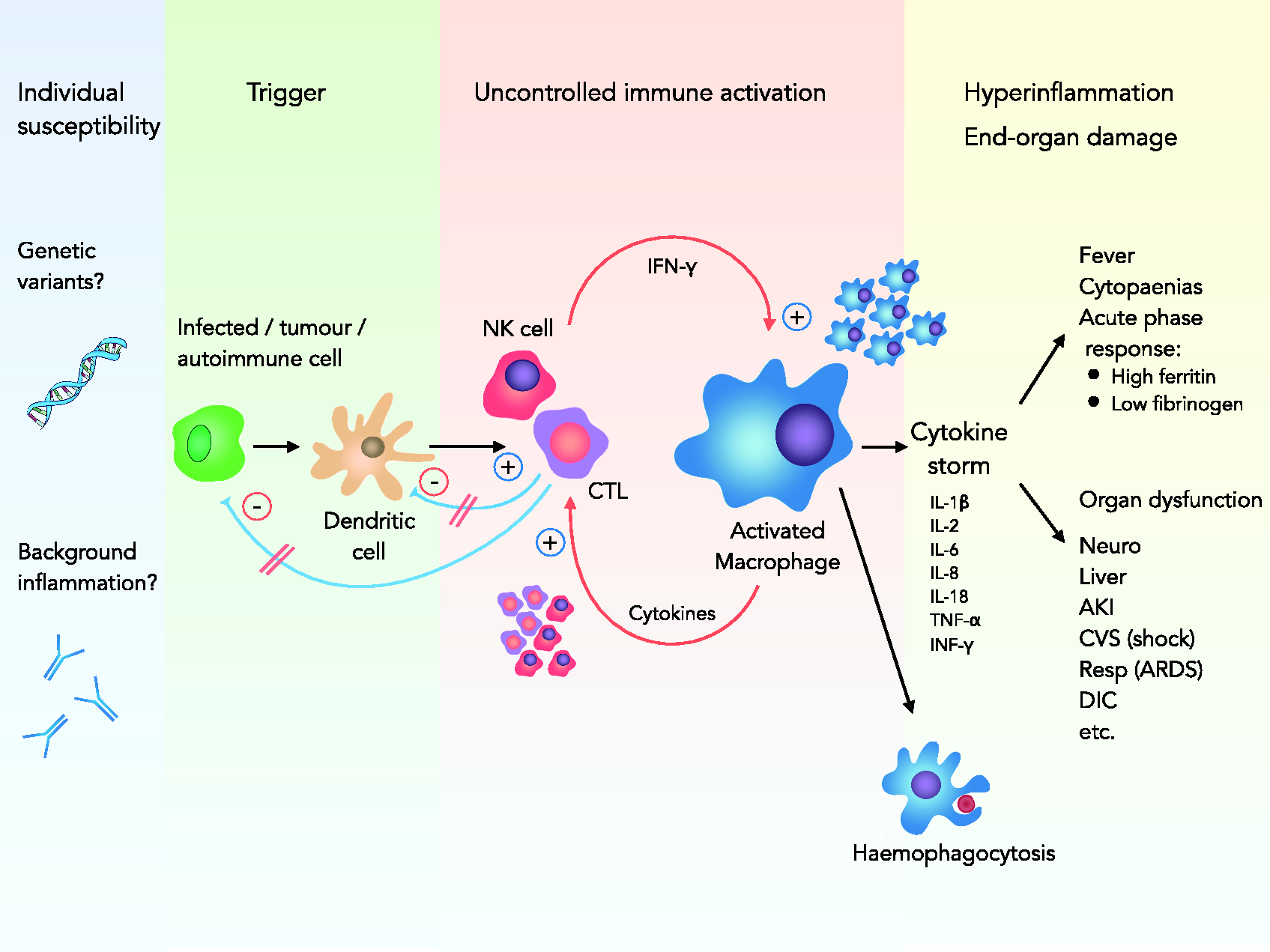
Susceptibility to HLH seems to have a genetic basis for both primary and secondary forms. In fHLH patients, several discrete causative genetic defects involving cytolytic pathways have been identified. In sHLH, the genetic contribution is less well understood to date. However, there is emerging evidence for genetic variants involving similar genes and, for example, low natural killer cell (NK cell) activity has been reported in both sHLH patients and close relatives.13,14
sHLH can be triggered by a large variety of stimuli, including infection, haematological and other malignancy, rheumatological or autoimmune disease, severe burns, chemotherapy, other cancer therapies and stem-cell transplant, among others. 1 However, in a proportion of patients no discrete trigger can be identified at the time.
Infection and sHLH
Infective triggers for sHLH.

CMV: cytomegalovirus; HHV-6: human herpesvirus 6; HIV: human immunodeficiency virus; HSV: herpes simplex virus; sHLH: (secondary) haemophagocytic lymphohistiocytosis; spp: species; VZV: varicella zoster virus.
Among many possible viral triggers, Epstein–Barr virus (EBV) is the predominant agent recognised in the developed world and much of Asia.1,19 However, its pathophysiological role is complex. EBV can be an independent trigger of HLH or precipitate sHLH in conjunction with associated malignancy (EBV-driven lymphoproliferative disorders). In some patients, EBV viraemia simply reflects the activation of lymphocytes containing virus; in this context, EBV can be seen as an innocent bystander. 20
Bacterial infections are reported in around 9% of sHLH cases. 1 Parasitic and fungal triggers are less frequently observed but should be considered in patients with immunosuppression, a relevant travel history or other suggestive clinical context.1,19 Visceral leishmaniasis is of particular importance as it is very effectively treated with liposomal amphotericin, even in the context of sHLH. The majority of recent UK cases were acquired in the Mediterranean littoral including Spain and Italy, with only small numbers from Africa, Asia and South America. 21
Of note, the initial screen for an infective sHLH precipitant is often negative; specialised/repeat sampling is often required.
sHLH in malignancy and associated therapies
sHLH can be a direct consequence of malignancy or a result of cancer treatments such as chemotherapy, haematopoietic stem cell transplantation or novel immune therapies. Most commonly, it is associated with haematological malignancy, in particular lymphomas.22–24 sHLH has also been reported in solid cancers and is probably underdiagnosed in this group. sHLH may also occur after both haematological and solid organ transplantation, and is often associated with opportunistic infections. 1 After haematopoietic stem cell transplantation, sHLH can mimic graft versus host disease, in which case hyperferritinaemia might be the key differentiating diagnostic feature.8,25,26
Most recently, hyperinflammatory clinical syndromes have been described in patients receiving novel targeted immunotherapy, increasingly used in cancer management. Intensivists are likely to see progressively more patients suffering from their unique side effects. Examples of potentially HLH-inducing therapies include monoclonal antibodies, dendritic vaccines, checkpoint inhibitor combinations and chimeric-antigen receptor T-cell (CAR-T) therapies. 22 CAR-T therapy is strongly associated with overproduction of cytokines leading to systemic inflammation and organ dysfunction. The clinical syndrome is termed cytokine release syndrome (CRS) in its initial phase, but may progress to sHLH if severe. While the incidence of CRS is estimated to be very high (74%–100% in the anti-CD19 setting), the risk of developing sHLH in this context is currently less clear. 27
Autoimmune disease and MAS
A large number of rheumatological and autoimmune diseases are associated with sHLH, where the syndrome is termed MAS for historical reasons. The strongest connection exists with systemic juvenile idiopathic arthritis (sJIA), with an estimated sHLH incidence of 10% in paediatric and adolescent sJIA populations, often co-triggered by infection. In adult rheumatological practice, sHLH is most closely associated with adult-onset Still’s disease (on the same disease continuum as sJIA), and with systemic lupus erythematosus. In addition, sHLH has been described complicating rheumatoid arthritis, systemic vasculitides, inflammatory bowel disease and other conditions, where the main suspected triggers are infection and drug therapy.1,8,28
Sepsis and HLH: Overlap and distinguishing features
There is large overlap between the definitions for sHLH and sepsis. The recent consensus definition (‘Sepsis-3’) characterises sepsis as ‘life-threatening organ dysfunction caused by a dysregulated host response to infection’, 29 a description that would be equally applicable to sHLH triggered by infection. Both conditions share similarities in their pathophysiology, cytokine profile and clinical features.3,7 It has to be noted that sepsis may demonstrate a variety of immunological phenotypes, from a predominant inflammatory response to suppressed immune function or a combination of both, and this may be subject to evolution over time.30,31 It has been suggested that sHLH could be implicated in the hyperinflammatory subset of septic patients.10,11,32 This is in keeping with a post-hoc analysis by Shakoory et al. of a randomised controlled trial (RCT) from the 1990s, which suggested a potential mortality benefit from IL-1 receptor blockade with Anakinra in septic ICU patients with hepatobiliary dysfunction and disseminated intravascular coagulopathy (DIC), but not in patients with either alone. 12 However, due to statistical uncertainty (low fragility index), the study should be regarded as hypothesis-generating only at present.
The true incidence of sHLH in sepsis remains unknown. Recently, Kyriazopoulou et al. estimated the incidence of a MALS at around 4% in a large sepsis cohort. 11 MALS was defined by a modified HScore (see further detail below), HPB and DIC or a combination of these, and was associated with a significant rise in early mortality. This further supports the existence of sHLH secondary to bacterial infection, although its true prevalence remains imprecise. Aside from the fact that human immunodeficiency virus (HIV)-positive patients were excluded, no further information was provided on the source of sepsis, microbiology, or comorbidity to clarify the context of MALS. It shall therefore be of interest to see whether Anakinra reduces mortality in an ongoing RCT among a homogenous group of septic patients (based on serum ferritin >4420 ng/mL, lung infections, primary bacteraemia and acute cholangitis but without risk factors from biological treatment, immunodeficiency, stage IV malignancy or autoimmune disease (PROVIDE Trial, ClinicalTrials.gov NCT03332225)).
Differentiating sHLH from sepsis may be difficult. sHLH may be present in the absence of infection but cause an identical clinical presentation to septic shock (a ‘sepsis mimic’). In addition, sHLH can be triggered by infection or predispose to sepsis by virtue of its inherent immune dysfunction.3,10,32 Furthermore, both conditions can co-exist in the context of an underlying disease such as malignancy or an autoimmune condition. From a practical perspective, sHLH could, and probably should, be suspected in all critically ill patients with unexplained fever, cytopaenias and organ dysfunction, particularly those not responding to aggressive sepsis treatment.
Clinical and laboratory features in the context of critical care
Clinical presentation
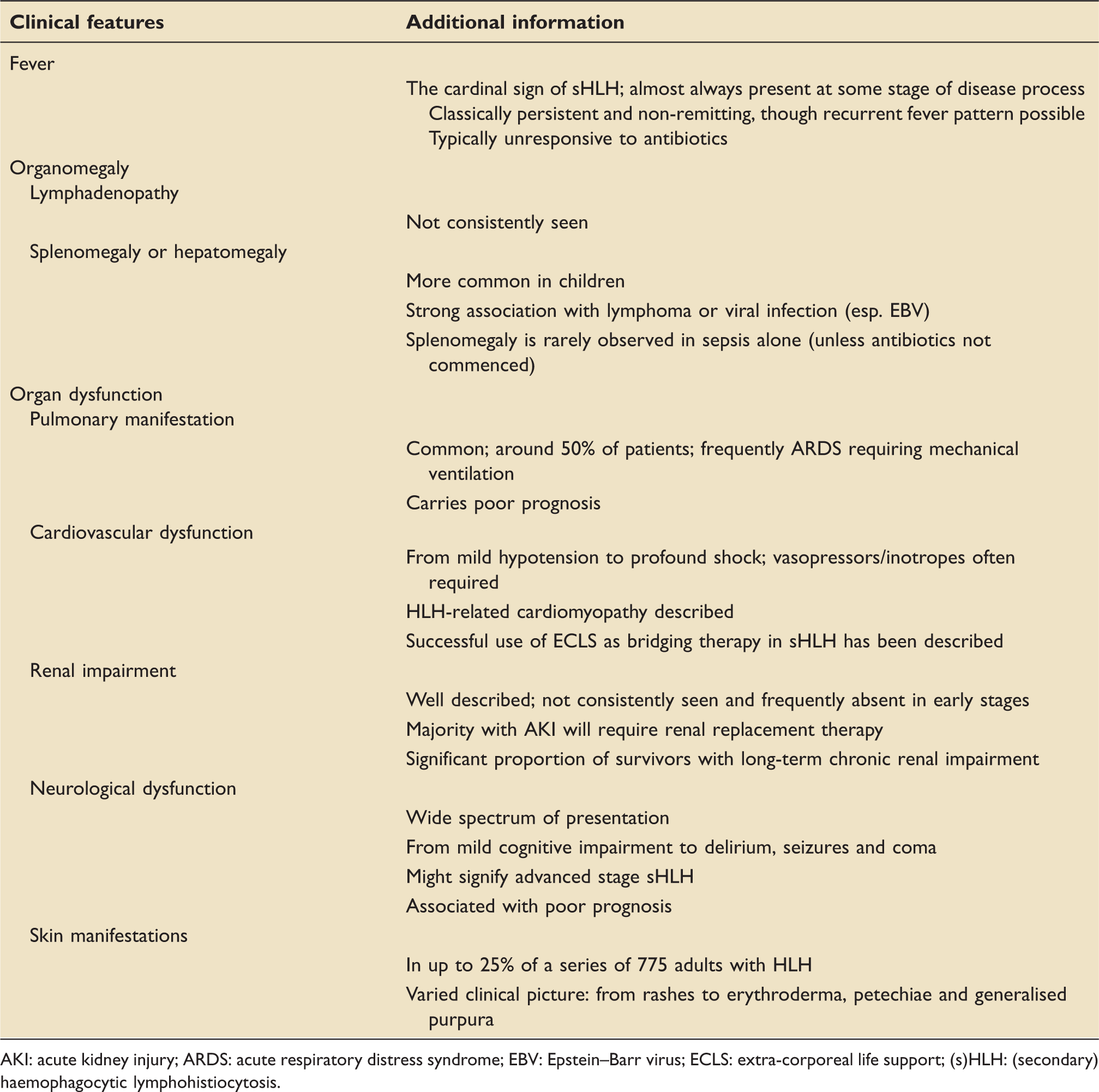
AKI: acute kidney injury; ARDS: acute respiratory distress syndrome; EBV: Epstein–Barr virus; ECLS: extra-corporeal life support; (s)HLH: (secondary) haemophagocytic lymphohistiocytosis.
Laboratory features
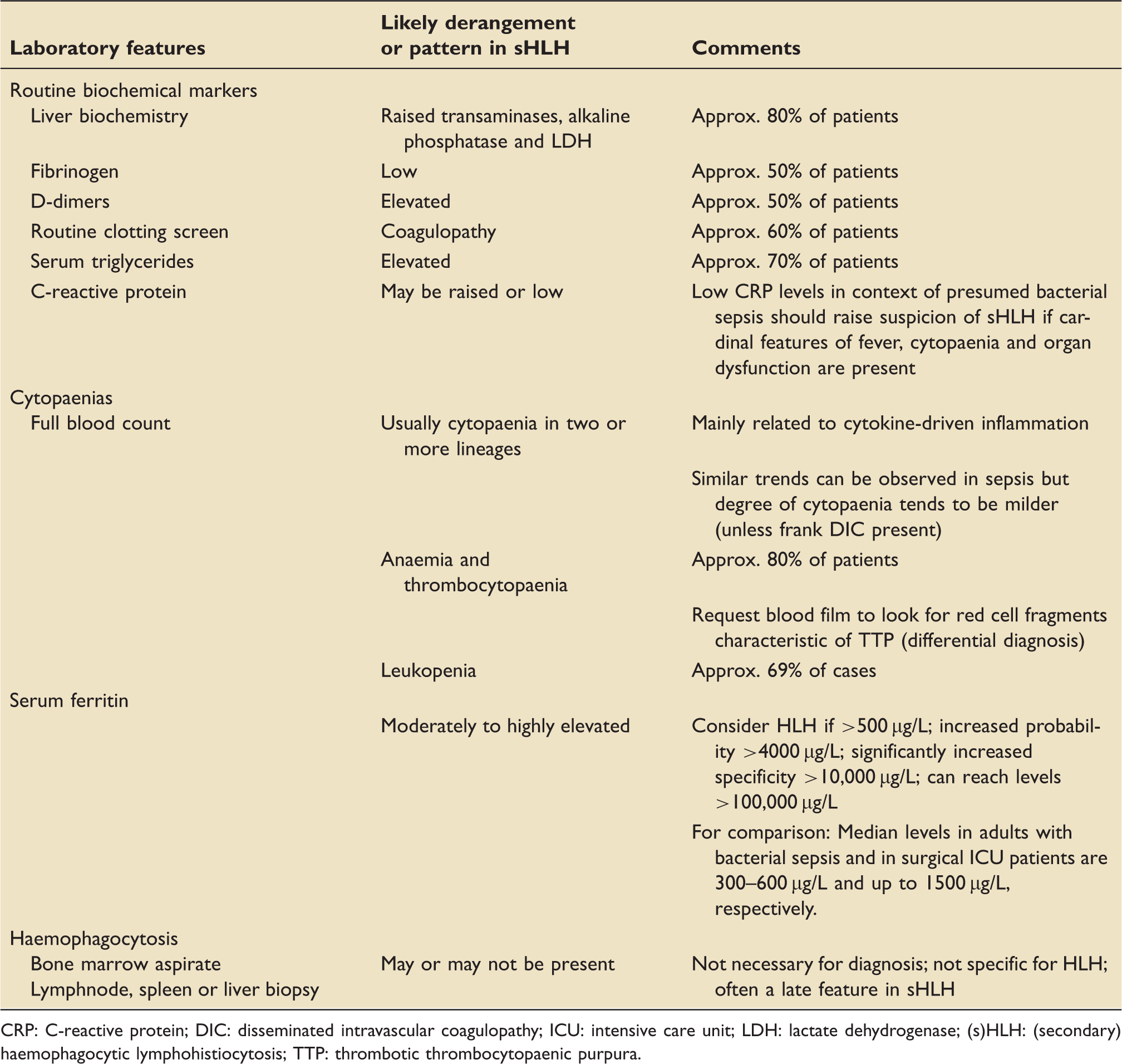
CRP: C-reactive protein; DIC: disseminated intravascular coagulopathy; ICU: intensive care unit; LDH: lactate dehydrogenase; (s)HLH: (secondary) haemophagocytic lymphohistiocytosis; TTP: thrombotic thrombocytopaenic purpura.
Serum ferritin
Ferritin is a protein with numerous functions including iron storage, cell signalling and regulation of immune function. It is used as an inflammatory biomarker, released into serum in response to high levels of cytokines, hormones and oxidative stress. The differential diagnosis for hyperferritinaemia includes iron overload, liver dysfunction, chronic renal failure with secondary siderosis, malignancy, inflammatory disease, infection, sepsis and HLH, among others. Very high levels (>10,000 µg/L) are 96% specific and 90% sensitive for HLH in children. 40 However, marked hyperferritinaemia is less specific for HLH in adults41,42 and must be used in combination with other criteria. Concomitant measurement of glycosylated ferritin may potentially increase the diagnostic specificity of ferritin. 43 Trends in serum ferritin levels are useful in tracking progress, predicting short-term outcome and identifying rebound hyperinflammation.8,44
Haemophagocytosis
Haemophagocytosis refers to a process where macrophages engulf mature or immature blood cells in bone marrow or other reticuloendothelial organs. While this histological finding lends its name to HLH, it is neither pathognomonic nor is it required for HLH diagnosis. 1 Of note, haemophagocytes may be systematically misclassified using the histological method, which lacks specificity and sensitivity as a test of haemophagocytosis. 45 Within critical care, a degree of haemophagocytosis has been observed in 60% of patients with sepsis and thrombocytopaenia 46 and in 65% of autopsies from patients with multiple organ dysfunction. 47 If invasive diagnostic samples are obtained, pathologists should be alerted that HLH is being considered.
Specific immune markers and additional laboratory tests
A variety of potential specific immunological response markers for sHLH have been described, such as soluble interleukin 2 receptor (sIL-2R or sCD25), soluble CD163, NK cell activity and cytokine profiling.2,3,48 Given their prolonged turn-around times and lack of routine availability, none of these specialised immunological assays offer immediate diagnostic benefits in the critical care setting. Specialist advice from immunology/HLH multidisciplinary teams (MDTs) should be sought on which tests to use.
Clinical diagnosis
Diagnostic criteria
HLH-2004 criteria and HScore.
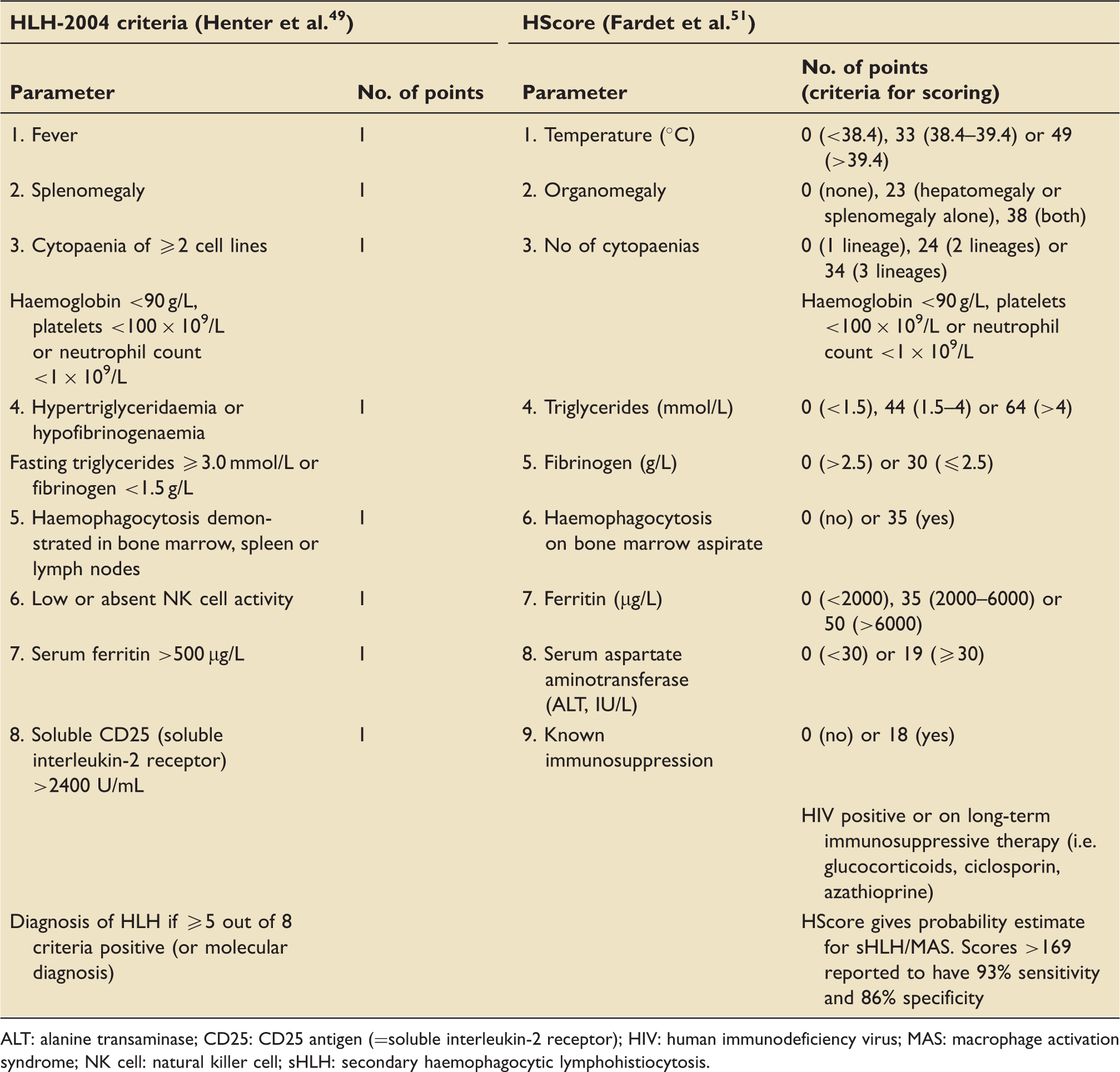
ALT: alanine transaminase; CD25: CD25 antigen (=soluble interleukin-2 receptor); HIV: human immunodeficiency virus; MAS: macrophage activation syndrome; NK cell: natural killer cell; sHLH: secondary haemophagocytic lymphohistiocytosis.
Of note, neither has been validated in a critical care population and both have significant short-comings:
HLH-2004 is the diagnostic framework for fHLH, with diagnosis requiring the presence of five positive criteria from eight categories. Although frequently used for diagnosis in adult sHLH for lack of alternative criteria, HLH-2004 has only been validated in children and encompasses a variety of tests that are impractical in routine intensive care practice. Furthermore, turn-round times for specialised tests are usually slow, leading to concerns that the score does not become positive until very late in the clinical presentation, at which point patients may no longer be salvageable.
49
The HScore is a simple bedside scoring system specifically developed for sHLH. It encompasses nine routine clinical and laboratory parameters, is easy to compute with an online calculator (http://saintantoine.aphp.fr/score/) and provides a probability score for the presence of sHLH. The HScore is based on a single-centre retrospective study with external validation for rheumatological patients.50,51 While it has not been fully validated as a diagnostic tool for sHLH in the critical care population, Kyriazopoulou et al. found a modified HScore to correlate with the presence of MALS and, by proxy, outcome in patients with sepsis.
11
Diagnostic approach in critical care
We suggest the following pragmatic approach to diagnosis of sHLH in critical care patients:
sHLH should be suspected in any unwell patient with unexplained fever, cytopaenias (especially thrombocytopaenia) and organ dysfunction. A serum ferritin level should be checked in these cases and, if elevated >500 µg/L or rapidly rising on serial measurements, should trigger further investigations. Ferritin levels >4000 µg/L in sepsis significantly increase the likelihood of concomitant sHLH,11,52 while levels >10,000 µg/L are highly concerning and may indicate immediate need for treatment. Additional work-up should include clinical assessment, laboratory tests and imaging to establish the probability of sHLH, and to identify potential triggers. Calculation of the HScore may aid in corroborating a clinical suspicion but time-critical treatment should not be withheld on the basis of insufficient HLH-2004 criteria or a low HScore.
A final diagnosis of sHLH hinges on clinical judgement and pattern recognition (see Figure 2). When possible, a dedicated HLH expert opinion should be sought (from a local or regional HLH expert or MDT) to advise on diagnosis and immediate treatment. Unfortunately, sHLH carries an exceedingly high mortality if advanced. Therefore, the risk of missing the diagnosis needs to be balanced against potential side-effects from aggressive immunosuppressive therapy. Early treatment should be considered if there is a reasonable clinical suspicion.
Diagnostic approach for sHLH in critical care.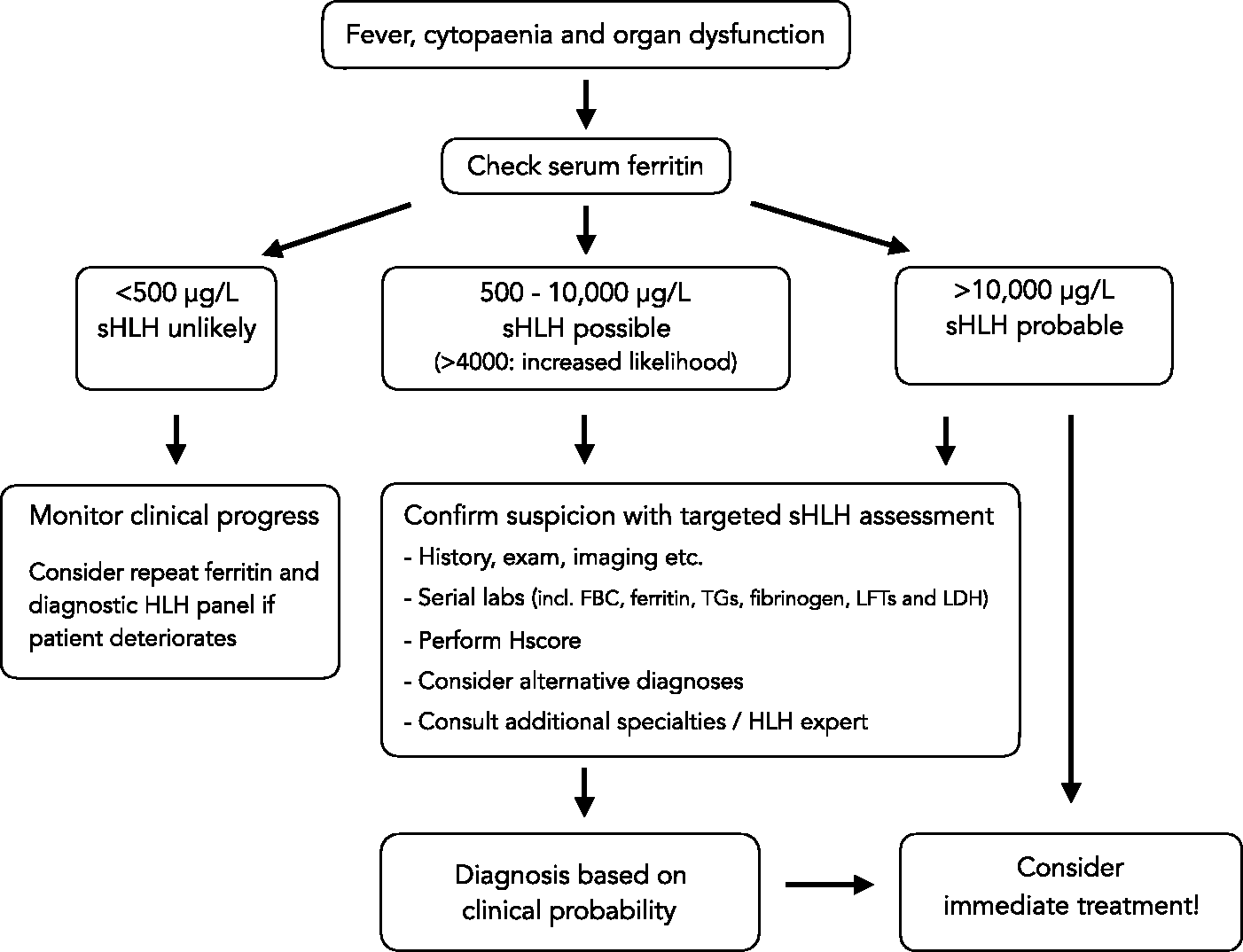
Suggested initial work-up
Diagnostic work-up for sHLH in critical care.

aPTT: activated partial thromboplastin time; ALT: alanine aminotransferase; AST: aspartate aminotransferase; CMV: cytomegalovirus; CSF: cerebrospinal fluid; CT: computed tomography; CXR: chest radiograph; EBV: Epstein–Barr virus; ECG: electrocardiogram; GTT: gamma glutamyl transferase; HIV: human immunodeficiency virus; IVIG: intravenous immunoglobulin; LDH: lactate dehydrogenase; MRI: magnetic resonance imaging; PCP: pneumocystis pneumonitis; PCR: polymerase chain reaction; PET: positron emission tomography; PT: prothrombin time; sHLH: secondary haemophagocytic lymphohistiocytosis; TB: tuberculosis.
Targeted imaging, including ultrasound and cross-sectional radiology are important to establish underlying triggers and alternative causes for the clinical presentation; CT scanning of chest, abdomen and pelvis may be considered for relative ease of availability and the broad diagnostic scope. Specialty consultations from infectious diseases, rheumatology or haematology should be sought as clinically indicated; these may inform further specialised testing (see Tables 3 and 5). Of note, repeat sampling may be required to identify underlying malignancy (such as lymphoma diagnosis masked by steroids) or occult infection (unusual pathogens). Bone marrow examination, additional tissue sampling, positron emission tomography (PET) scanning and other investigations (such as cell-free DNA for lymphoma diagnosis) may be helpful.
Treatment approach in critical care
There are no randomised trials to guide treatment of sHLH in adults. Most treatment regimens have been extrapolated from the intensive immunosuppressive treatment of fHLH. Hence, decision to initiate therapy and choice of agents depend largely on expert opinion and clinical experience. Based on available evidence, we suggest that the general principles for treatment of adult onset sHLH treatment on critical care should revolve around:
Early diagnosis Supportive care and multiple organ support Rapid initiation of specific HLH therapy to control cytokine storm Identification and treatment of any underlying trigger Early involvement of an HLH expert or HLH MDT.
Suggested treatment approach for sHLH in critical care.
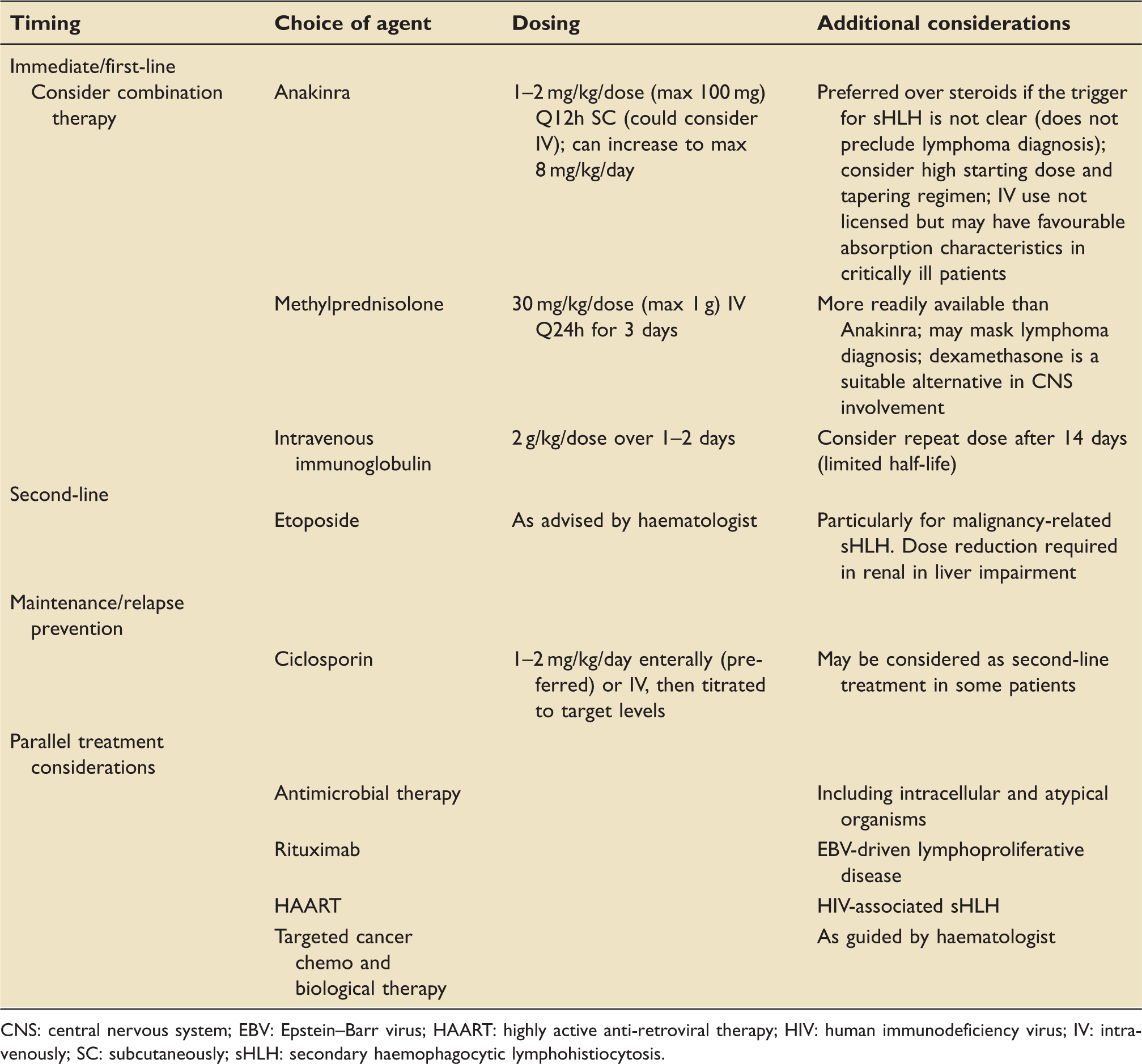
CNS: central nervous system; EBV: Epstein–Barr virus; HAART: highly active anti-retroviral therapy; HIV: human immunodeficiency virus; IV: intravenously; SC: subcutaneously; sHLH: secondary haemophagocytic lymphohistiocytosis.
Steroids are usually the mainstay of initial immunosuppression. If already in use, ‘sepsis-dose’ steroids such as hydrocortisone may be converted to a course of high-dose pulsed methylprednisolone. Dexamethasone is a suitable alternative and may be superior if central nervous system (CNS) involvement is suspected (around one-third of patients).
There are concerns regarding the primary use of steroids when the HLH-driver has not yet been identified. Some lymphomas are initially very steroid-sensitive, so starting treatment with high-dose steroids prior to taking all the required biopsies or performing a PET scan risks masking the lymphoma diagnosis. Conversely, a delay in definitive treatment is again associated with poor outcome. If available, therefore, there is a strong argument for using Anakinra as first-line treatment where diagnostic uncertainty exists. Anakinra is a recombinant IL-1 receptor antagonist and very effective in controlling the initial cytokine storm of sHLH. 8 It is well tolerated, has a favourable side-effect profile and is therefore increasingly being considered as an early treatment modality in sHLH. The financial cost of Anakinra is moderate, yet local availability for emergency use on critical care may be limited. Anakinra is usually given subcutaneously (SC) but can be given intravenously, which might be preferable in unwell ICU patients with unclear SC absorption characteristics. A higher than usual starting dose with a plan for gradual weaning should be considered in very sick patients. Due to its short half-life, Anakinra may be administered in split doses or via continuous infusion.
HLH is a recognised indication for intravenous immunoglobulin. 54 A repeat dose should be considered after 2 weeks due to the limited half-life. Plasma exchange to remove cytokines is an alternative but has little evidence in adults.1,2
Etoposide is a cytotoxic agent with particular activity against macrophages and cytotoxic T cells. It should be considered a third-line treatment for refractory sHLH cases, particularly in EBV-driven disease and malignancy. Caution is advised in severe renal and liver impairment, and dose reductions are advised to mitigate haematological toxicity.1,7,32 Intrathecal methotrexate may be added with progressive CNS involvement.1,8 Ciclosporin has a potential role in relapse prevention but is associated with neurotoxicity, including posterior reversible encephalopathy. This may be difficult to distinguish from HLH with CNS involvement. 8
Aside from routine antibacterial management, specific therapies for intracellular diseases such as tuberculosis, rickettsial disease and leishmaniasis may be crucial. 2
Highly active antiretroviral therapy (HAART) has improved the prognosis for HIV-related HLH. 2 HAART may cure sHLH in the context of primary HIV infection and may ameliorate mild to moderate sHLH in context of cytomegalovirus or human herpesvirus 8 (HHV-8) viraemia by virtue of immune reconstitution. However, it is unlikely to be effective in fulminant AIDS-related sHLH. In the latter context, additional pathology such as HHV-8-associated lymphoproliferative disorder should be considered as this may benefit from immediate anti-cytokine treatment (IL-6 inhibition) and rituximab. 55 Rituximab also plays an important role in EBV-driven lymphoproliferative disorder. 56
Further novel therapies including biological agents are on the horizon but evidence so far is scarce.1,8
Salvage therapy with haematopoietic stem cell transplant (HSCT) may rarely be considered in selected cases of sHLH, who survive the initial critical illness but suffer from relapsing disease, CNS involvement or haematological malignancy.1,2,22 In the UK, sHLH is an accepted indication for HSCT under the ‘transplant for rare diseases’ category, and patients should be discussed with an experienced transplant centre.
Prognosis
sHLH carries a high acute mortality of around 40% for all groups combined. 1 Malignancy-associated HLH has a particularly poor prognosis with acute mortality exceeding 80% and 5-year survival of <15%.8,22 In ICU patients, the published hospital mortality rates range from 52% 7 to 68%. 6 The presence of shock or severe thrombocytopaenia indicates increased risk. 7 Of interest, the maximum serum ferritin elevation concentration is directly correlated with an increased mortality risk, while a rapid fall in serum ferritin levels in response to treatment is associated with a more favourable short-term outcome.8,44
Future perspectives
Prospective research on sHLH in critical care is still in its infancy. The Hellenic Sepsis Study Group is currently conducting a randomised controlled trial (PROVIDE Trial) on the use of Anakinra in patients with high ferritin, sepsis, liver dysfunction and DIC. 57 However, the wider problem remains that sHLH in the context of sepsis essentially constitutes a syndrome within a syndrome. Experience from existing critical care research indicates that large patient heterogeneity often reduces the ability to demonstrate meaningful changes in clinical outcomes within randomised controlled studies.
In addition to research, there is clearly a need for better understanding of sHLH as a clinical phenomenon and for a systematic approach to patient identification, diagnosis and management across different specialties. This is facilitated by local or regional HLH MDTs, which would ideally include representation from rheumatology, haematology, infectious disease and critical care, with additional specialties as needed. Increasingly, HLH MDTs are being established within both paediatric and adult settings. 53 Within the UK, a national cross-specialty collaboration on HLH is being developed (HASC; HLH across specialty collaboration). This draws upon the specialties above with additional clinical input from immunology, genetics, virology, transplant specialists, etc. Future work will focus on establishing a national HLH register in order to capture clinical data and create a comprehensive biobank, following the footsteps of countries such as Germany (hlh-registry.org) and the USA (histiocytesociety.org). The initial aims are to characterise clinical phenotypes of sHLH, identify potential genetic variants in adult sHLH and explore avenues for clinical research.
Conclusion
sHLH is not a disease in itself but rather a clinical syndrome of extreme hyperinflammation, which is triggered by infection, auto-immunity or malignancy, among others. sHLH can lead to rapidly life-threatening organ dysfunction and is likely underdiagnosed in critical care. It should be suspected and actively investigated in any unwell patient with unexplained fever, cytopaenias, progressive organ dysfunction and failure to respond to intensive infection management. Treatment revolves around high-quality supportive care, early anti-inflammatory and immunosuppressive therapy and identification and treatment of any precipitating pathology. In a patient with undifferentiated rapidly progressing organ failure, or if the presumed sepsis diagnosis does not quite ‘feel’ right or respond appropriately, think, could this be HLH?
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs
Kris Bauchmuller https://orcid.org/0000-0001-9648-1328 Rachel Tattersall https://orcid.org/0000-0002-1087-6546 Christopher McNamara https://orcid.org/0000-0002-8581-2790 Mervyn Singer
https://orcid.org/0000-0002-1042-6350 Stephen J Brett
![]()