
Abstract
Background
Anaemia is common in patients who survive critical illness and is associated with high levels of fatigue and poor quality of life. In non-critically ill patients, treating anaemia with intravenous iron has resulted in meaningful improvements in quality of life, but uncertainties regarding the benefits, risks, timing and optimal route of iron therapy in survivors of critical illness remain.
Methods / Design
INtravenous Iron to Treat Anaemia following CriTical care (INTACT) is an open-label, feasibility, parallel group, randomised controlled trial with 1:1 randomisation to either intravenous iron (1000 mg ferric carboxymaltose) or usual medical care. The primary objective is to assess the feasibility of a future, multicentre randomised controlled trial. Participants will be followed up for up to 90 days post-randomisation. The primary outcome measures, which will be used to determine feasibility, are recruitment and randomisation rates, protocol adherence and completeness of follow-up. Secondary outcome measures include collecting clinical, laboratory, health-related quality of life and safety data to inform the power calculations of a future definitive trial.
Conclusion
Improving recovery from critical illness is a recognised research priority. Whether or not correcting anaemia, with intravenous iron, improves health-related quality of life and recovery requires further investigation. If so, it has the potential to become a rapidly translatable intervention. Prior to embarking on a phase III multicentre trial, a carefully designed and implemented feasibility trial is essential.
Introduction
Background
Anaemia is a very common problem in intensive care units (ICUs) affecting 60–80% of patients and is associated with adverse outcomes.1,2 Anaemia persists until hospital discharge and in some studies up to six months following ICU discharge, and is associated with fatigue and poor health-related quality of life (HRQoL).3–5
Iron-restricted erythropoiesis resulting from anaemia of inflammation is extremely common in ICU patients and may occur through absolute iron deficiency, functional iron deficiency or iron sequestration. Dysregulated iron homeostasis is associated with adverse outcomes in ICU patients. 6 Our understanding of iron homeostasis has been improved by the discovery of hepcidin – the master regulator of body iron status. Hepcidin is a small peptide produced by the liver and acts by inhibiting the function of ferroportin, the sole cellular exporter of iron. Hepcidin blocks duodenal iron absorption and results in iron trapping within macrophages and hepatocytes subsequently decreasing the availability of iron for erythropoiesis. 7 Current markers of iron status (e.g. ferritin, transferrin saturation) have limited utility in critically ill patients as the relationship between these markers and iron status is heavily confounded by inflammation. Hepcidin may be a better marker of iron status in critically ill patients, even in the presence of inflammation,8,9 and recent work supports the hypothesis that critically ill patients have potentially iron-responsive anaemia which may not be detectable with currently available tests. One study suggested that 35% of ICU survivors developed evidence of iron deficiency six months after ICU stay. 4
In other contexts, intravenous (IV) iron therapy has been shown to be effective in treating anaemia with subsequent improvements in quality of life.10–13 IV iron is able to bypass hepcidin-induced restriction of iron. 14 Despite this, safety concerns regarding the risk of infection caused IV iron infusion remain. Iron is essential for bacterial growth and exogenous iron administration has been shown to worsen shock and lung injury in canine experimental models of pneumonia. 15 Randomised controlled trials (RCTs) have not always included infection as a pre-defined end point and therefore the risk in critically ill patients remains uncertain.12,16,17
Rationale for a trial
ICU survivors experience poor HRQoL, reduced physical function and fatigue.18–20 Improving recovery from critical illness has been identified as a key research priority by the National Institute for Health Research (NIHR) critical care portfolio and the James Lind Alliance. Evidence from observational studies suggests that untreated anaemia may, in part, be contributing to these symptoms.
Attempts to treat anaemia during acute critical illness have so far been unsuccessful. RCTs of iron supplementation in ICU patients have failed to demonstrate any evidence of benefit.17,21 This could partly be explained by limitations in trial design as studies were underpowered and there was wide variation in patient populations and iron administration regimes. 17 Biological reasons to explain this lack of benefit could be due a hyporeactive bone marrow, secondary to the acute inflammatory response, that may not respond to exogenous iron administration. The peak effect of iron may also not be observed until 2–3 weeks after administration.14,17,22,23 This suggests that any relative risks and benefits of iron probably depend on the timing of administration.
We hypothesise that treating anaemia when patients are recovering from critical illness, when the iron and erythropoietic profiles are more favourable, may be a more suitable time point for intervention, than during the acute phase of critical illness. A large, multicentre RCT is needed to answer this question but there are uncertainties regarding recruitment and follow-up rates, variability in haemoglobin concentrations in the post-ICU period and choice of optimal outcome measures. 18 We have therefore designed the INtravenous Iron to Treat Anaemia following CriTical care (INTACT) feasibility RCT, with the primary aim of assessing recruitment, randomisation and follow-up rates to inform the design of a future adequately powered multicentre trial.
Objectives
Primary objective
To assess the feasibility of a future large multicentre RCT of IV iron to treat anaemia in patients recovering from critical illness in intensive care.
Secondary objective
To collect clinical, laboratory and HRQoL data to inform the power calculations and health economic evaluations of any future definitive trial.
Trial design
INTACT is an open-label, feasibility, parallel group, RCT with 1:1 randomisation to either intervention or usual medical care. The trial is being co-ordinated by the Oxford Clinical Trials Research Unit (OCTRU) and sponsored by the University of Oxford. The funding is provided by the NIHR as part of a Doctoral Research Fellowship (DRF-2017-10-094). The trial is being conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice (GCP).
Methods
Participants, interventions and outcomes
Study setting
This feasibility RCT is being undertaken in three general adult UK ICUs (John Radcliffe Hospital, Oxford; Edinburgh Royal Infirmary, Edinburgh; Royal Berkshire Hospital, Reading), which have a proven track record in ICU research.
Eligibility criteria
Written informed consent is obtained from participants prior to any study procedures taking place. Patients eligible for the study must comply with all of the following before randomisation:
Requirement for ICU (Level 2 or 3 support) for ≥ 24 h and now deemed medically stable for discharge from ICU by the attending physician Last measured laboratory Hb ≤100 g l−1 Planned palliative care Planned home ventilation Primary neurological diagnosis with little/no rehabilitation potential Known hypersensitivity to iron Immunosuppressive therapy for organ transplant IV iron or erythropoietin treatment in the previous four weeks Pregnancy (breastfeeding is not an exclusion criterion) Personal or family history of iron overload disorders such as haemachromatosis or previously documented ferritin >1200 ng ml−1 and/or transferrin saturation > 50% History of severe asthma, eczema or atopic allergy Known severe chronic liver disease (e.g. Child-Pugh C or above) Haemodialysis-dependent chronic kidney disease Acute uncontrolled infection – as judged by the treating clinician (e.g. ongoing bacteraemia or non-resolving sepsis) or patient expected to be on non-prophylactic antibiotics for greater than 14 days.
Patients are excluded if they meet any of the following criteria:
The inclusion and exclusion criteria are designed to include participants who reflect a general population of survivors of critical illness with moderate to severe anaemia, who may benefit from the intervention, and exclude patients who are unlikely to benefit or at increased risk of adverse events (AEs) from IV iron.
Intervention
In the intervention arm, 1000 mg of ferric carboxymaltose is diluted in 100 ml of 0.9% saline and administered as an IV infusion over a minimum of 15 min. The dosing of ferric carboxymaltose is based on the maximum dose that can safely be given according the Summary of Product Characteristics within a reasonable time period. Participants are closely monitored for signs of hypersensitivity during the infusion and for at least 30 min after the treatment. The intervention can be administered at any time between ICU and hospital discharge. For participants who weigh less than 50 kg, the dose would be adjusted to 20 mg kg−1.
Usual medical care
Usual medical care consists of routine ward-based care including observation with monitoring and transfusion when required. Current guidelines advocate adopting a restrictive transfusion policy, with the exception of certain groups of patients such as those with ischaemic heart disease. 24 It is not routine practice to administer IV iron in patients recovering from critical illness at present. Any decision to commence iron therapy in this group would be at the discretion of the treating physician and independent of the study and would be recorded in the case report forms (CRFs).
Participant timeline
This is highlighted in Figure 1.
Study flow chart.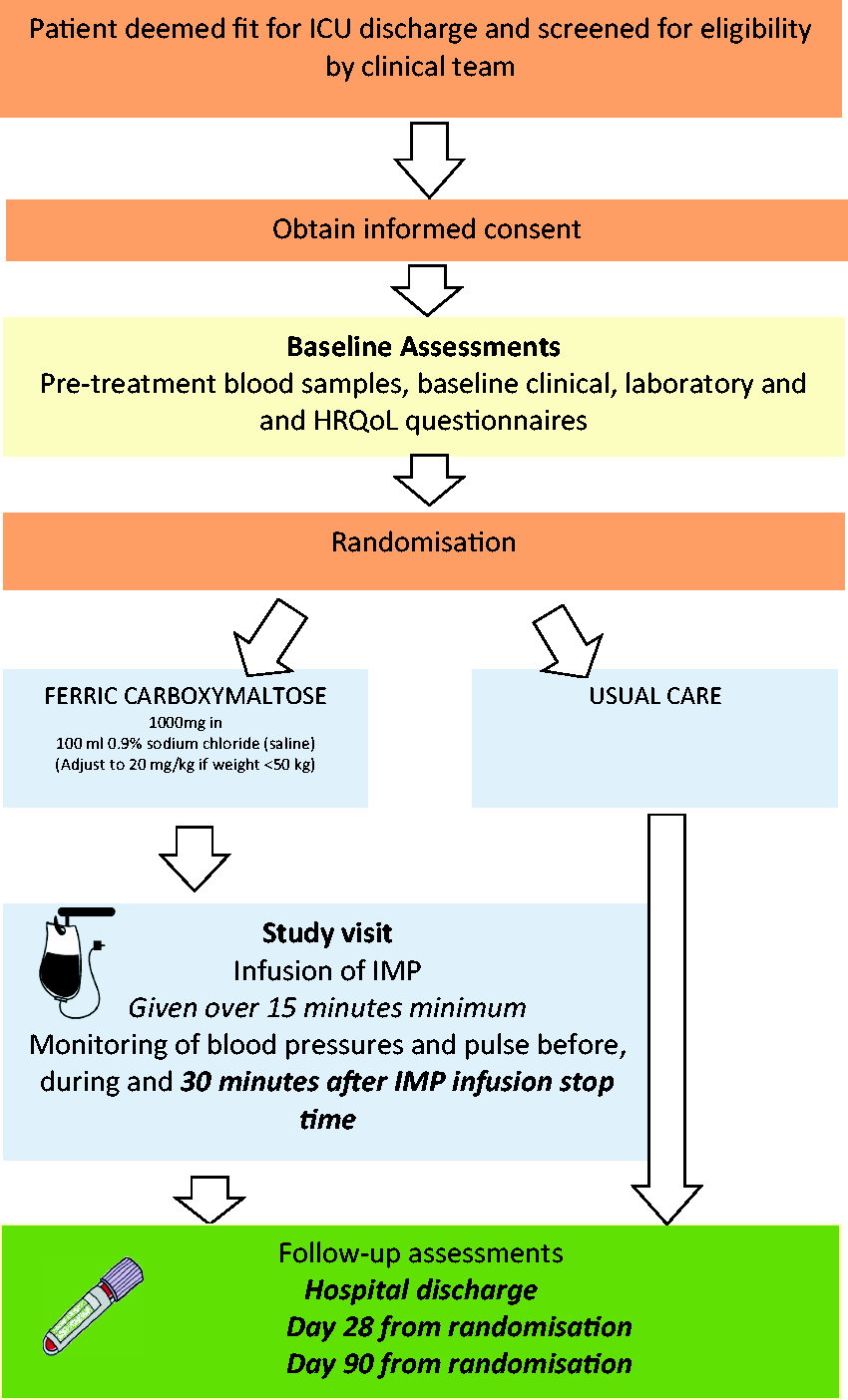
Outcome measures
Primary outcome measures:
Recruitment and randomisation rates overall and by centre Protocol adherence, defined as ‘number of participants allocated to the intervention who go on to the receive the study drug’ Completion rates of HRQoL questionnaires Nosocomial infection Mortality Hospital length of stay Changes in laboratory haematological (e.g. Hb) and iron profiles (e.g. ferritin) from baseline to 28 and 90 days post-randomisation Changes in Multidimensional Fatigue Inventory-20 (MFI-20), Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) and EuroQol-5D-5L (EQ-5D-5L) questionnaire scores from baseline to 28 and 90 days post-randomisation Healthcare resource use
Secondary outcome measures:
Sample size
As this is a feasibility study not aiming to assess treatment effects, we have not undertaken a formal power calculation. At the John Radcliffe Hospital (which will be the main recruitment centre for this trial) approximately 800 patients are admitted to the ICU each year. Of these 120 patients are elective post-operative admissions who would be expected to stay less than 48 h and of the remaining 680 patients, assuming unit mortality of 14%, 584 would be expected to survive ICU. Based on pre-trial work and previous studies, 50% (292 patients) would leave ICU with a Hb of ≤100 g l−1. 25 Assuming 30–40% enrolment, we expect to enrol on average 100 participants per year (approximately 2 participants per week) at this site. From the three sites it is anticipated that 130 patients will be recruited over a 12-month recruitment period.
Assignment of interventions
Randomisation
Eligible participants are randomised on a 1:1 ratio to receive either the intervention or usual medical care using minimisation on a secure web-based system controlled and managed by the OCTRU, stratified on anaemia severity (Hb < 80 g l−1 vs. 80–100 g l−1) and participant ICU length of stay (<7 days vs. ≥ 7 days).
Blinding
We have opted not to use a placebo study drug. This is because ferric carboxymaltose is a challenging substance to blind due to its rusty brown colour.
Data collection, management and analysis
Data collection
Data are collected using paper CRFs and then transferred to a validated electronic data entry system. Clinical data are collected at baseline (pre-randomisation) and hospital discharge. Laboratory and HRQoL data are collected at baseline, and 28- and 90-days post-randomisation. This is outlined in the study schematic in Figure 2 and detailed in Table 1. Laboratory data will include serial measurements of hepcidin throughout the study period.
CONSORT pilot and feasibility trials flow diagram schematic. Timing of visits and data collection. APACHEII: Acute Physiology and Chronic Health Evaluation II; EPO: erythropoietin; EQ-5D-5L, EuroQol-5 Dimension-5 Level; FACIT-F: Functional Assessment of Chronic Illness Therapy-Fatigue; FBC: full blood count; Hb: haemoglobin; HRQoL: health-related quality of life; ICU: intensive care unit; LFTs: liver function tests; MFI-20: Multidimensional Fatigue Inventory-20; sTfR: soluble transferrin receptor; U&Es: urea and electrolytes. Includes ferritin, serum iron, transferrin and transferrin saturation (%).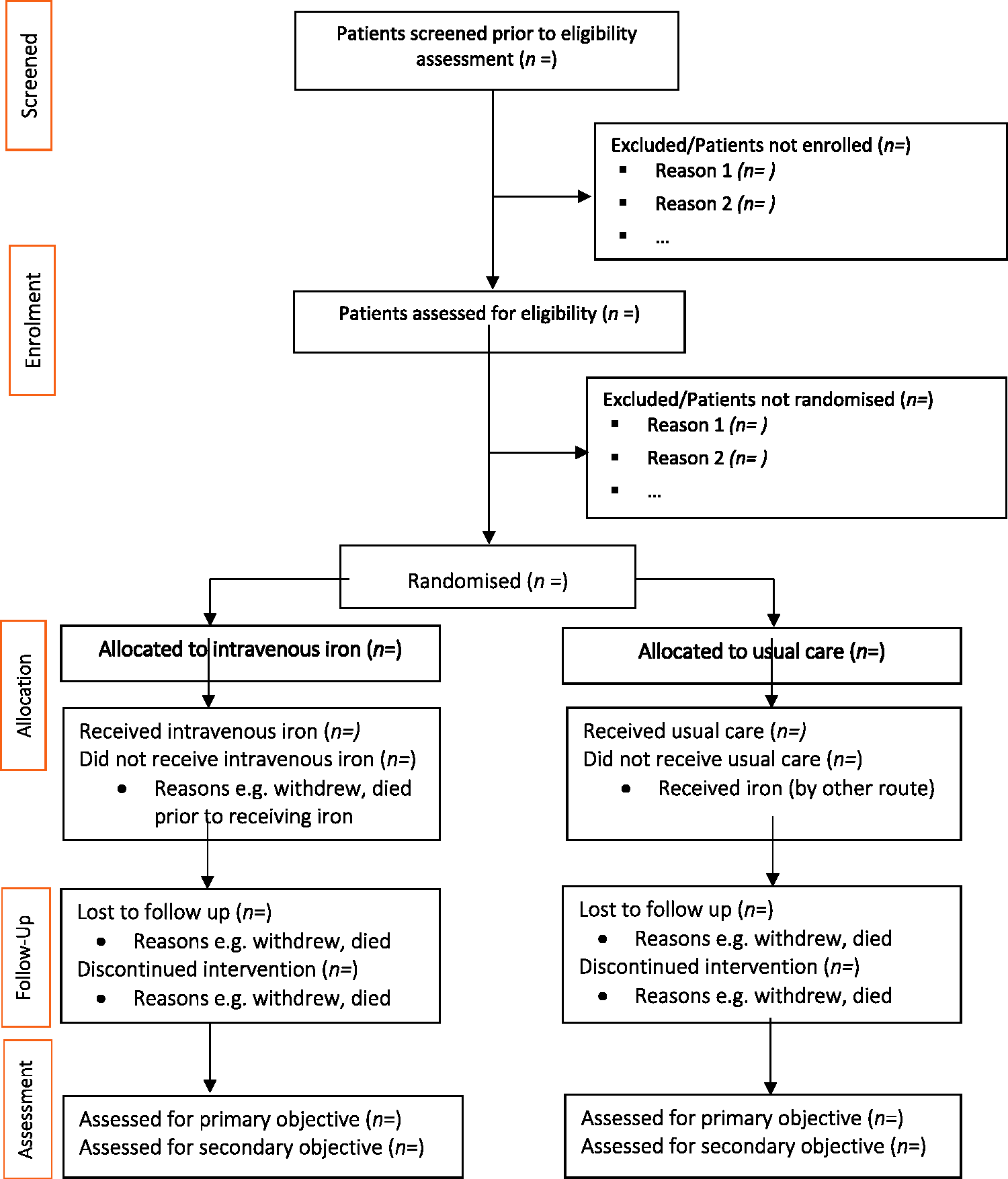

The EQ-5D-5L is a generic preference-based measure of health that is recommended by NICE for economic valuations. The MFI-20 is a 20-item self-reporting score designed and validated to measure fatigue across five domains – general fatigue, physical fatigue, mental fatigue, reduced motivation and reduced activity. 26 Scores can range from 4 to 20, with higher values indicative of increased fatigue levels. Its use in ICU survivors has been described previously. 4 The FACIT-F is a subscale of a questionnaire developed to assess anaemia-related symptoms in patients with cancer. 27 It consists of 13 items referring to the previous seven days. The final scores range from 0 to 52 with higher scores representing less fatigue. It has been validated to assess fatigue in ICU survivors. 28 Approximately 15–20 min will be required to complete all three questionnaires. Participants may ask for help completing the questionnaires from relatives, bedside nurses or a member of the research team. This is felt to be reasonable as problems with concentration and attention are not uncommon in critically ill patients.
Retention
Participation retention in ICU trials is challenging.28–30 To minimise loss to follow-up we will invite participants to our weekly ICU follow-up clinic with full reimbursement of travel costs. If participants are unable to attend, a member of the research team will undertake a home visit with appropriate safe guards in place. Telephone follow-up is offered for participants who live outside a reasonable geographic follow-up area to collect HRQoL data.
Data management
A Data Management Plan has been produced for the study which includes references to confidentiality, access and security arrangements and is available on request from the INTACT study office. In brief, staff at participating centres collect data on paper CRFs, which is then be entered into a password-protected online database (OpenClinica®), located on a secure University server. The database has validation on data fields. Paper CRFs are stored in the Investigator Site File under secure conditions. Paper copies of the consent forms will also be logged into the trial database and then scanned and stored electronically in the e-Trial Master File.
Statistical analysis
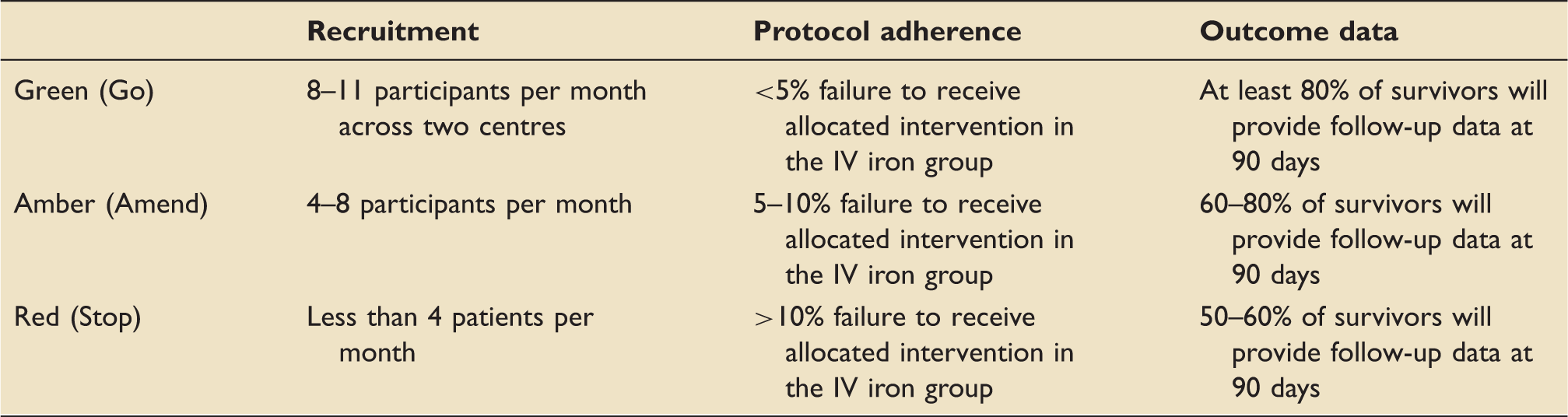
Results will be reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) extension to randomised pilot and feasibility trials. 31 Recruitment and follow-up rates are the main drivers for the feasibility design on the basis that unless reasonable rates can be achieved no formal trial will be possible. Recruitment rate will be calculated by the number of participants randomised as a proportion of total number of eligible participants. Rates will be estimated based on data collected and a 95% confidence interval determined for these measures.
If the estimated recruitment and follow-up rates are such that a multicentre definitive trial is possible, then no formal analysis will be undertaken and data from the feasibility study will be locked and carried over into the definitive trial. No formal analysis of treatment efficacy will be undertaken in this case.
‘Traffic light’ system to determine progression to a definitive trial.
Descriptive statistics will be used to describe the demographic and clinical characteristics between the two groups at baseline. Clinical outcome data will be analysed depending on the type of variables: for continuous variables, the difference in the means and corresponding 95% confidence intervals and analysis of covariance, adjusting for baseline values will be used for comparing the two groups. For categorical variables, the number and percentage of participants in each category will be reported and chi-squared tests will be used for comparing the two treatment groups. If the variables are not normally distributed, alternative non-parametric tests will be used for analysis. Analyses will be conducted on the intention-to-treat (ITT; analysed as randomised) and per-protocol (as ITT but with protocol violators excluded) populations. As this is a small study, treatment effects are likely to have wide confidence intervals and consequently inferences will be tentative and reported as such. Tests will be two-sided and considered to provide evidence for a significant difference if p values are less than 0.05.
Missing data
Reasons for missing data, loss to follow-up and participant withdrawals will be carefully considered, reported and patterns of ‘missingness’ will be explored. Missing data will be minimised by collecting the minimum amount of data required and data queries will be resolved prior to analysis. We have stated that the analysis will be on ITT basis and a per-protocol basis if the study is not considered feasible. There will not be a formal statistical analysis plan and therefore deviations from it are not anticipated. Any deviations from the analysis presented will be reported in the final study report.
Monitoring and safety
Data monitoring
A trial monitoring plan has been developed and will comply with the principles of GCP. As this is a small study with a short duration, there is no formal Data Monitoring Committee. Instead, there is a Trial Oversight Group (TOG), which includes independent clinicians experienced in the fields of clinical trials, intensive care and haematology along with an independent statistician. The TOG will work closely with the Trial Management Group (TMG) and provide overall supervision of the study to ensure it is being conducted in accordance with GCP. A charter containing the details of the members, roles and responsibilities and planned method of functioning has been created and is available on request from the INTACT study office.
Safety
Participants surviving ICU are at a high risk of AEs and serious adverse events (SAEs) secondary to their underlying disease process and/or expected complications of their critical illness. Therefore, only SAEs that might reasonably be a consequence of participation in the trial and are judged by the investigator to not be due to the underlying disease or expected complications of their critical illness will be reported. Ferric carboxymaltose is a drug with a well-known safety profile. The following rare events, if serious, must be reported on the trial specific SAE form:
Anaphylactic/anaphylactoid reactions Loss of consciousness/syncope Bronchospasm Angioedema and facial oedema Severe anaemia (Hb < 80 g l−1) at 28 and/or 90 day follow-up
All SAEs will be submitted to the INTACT trial office within 24 h of the investigator becoming aware.
Auditing
OCTRU will conduct a one-off audit of the study in accordance with the trial monitoring plan. Access to data will also be granted to authorised representatives from the Sponsor, host institution and the regulatory bodies to permit trial-related monitoring, audits and inspections. A trial monitoring plan is available on request from the INTACT study office.
Ethics and dissemination
Regulatory and ethics approval
This is a feasibility study of an investigational medicinal product which is broadly being used within its licensing indication. According to the Medicines and Healthcare products Regulatory Agency (MHRA), this trial has been categorised as type A (no higher than the risk of standard care) as ferric carboxymaltose is indicated for the treatment of iron deficiency when oral preparations are likely to be ineffective or cannot be used. Clinical Trial Authorisation from the MHRA was received on 17 July 2018.
A favourable ethical opinion was obtained from South Central – Berkshire B Research Ethics Committee on 7 July 2018 (18/SC/0308). Health Research Authority approval was obtained on 20 July 2018. A favourable opinion for a substantial amendment was obtained on 18 April 2019 (see Appendix 1).
Consent
Any participants who are deemed potentially suitable will be asked by a member of the clinical team if a member of research team could come along and discuss a research study that they may be eligible for. The clinical team will also provide the participant with a participant information leaflet. All eligible participants who agree verbally to be approached by the research team will be allocated a screening number and entered into the screening log, which will be kept in the investigator site file. The screening log will be completed and maintained by an appropriately trained member of the research team. Where a participant agrees to take part in the study, a copy of the consent form will also be filed in the medical notes. The participant must personally sign and date the latest approved version of the informed consent form before any study specific procedures are performed.
Confidentiality
The research team will ensure that the participants' anonymity is maintained. The participants will be identified only by a participant code on all study documents and the study CRF database, with the exception of the personal identifiers database, which will be held separately to the CRF database. The personal identifiers database will be used for the sole purpose of arranging study follow-up visits. All documents will be stored securely and only accessible by study staff and authorised personnel.
Dissemination policy
The TMG will form the basis of the writing committee and advise on the nature of the publications. The study will be reported in accordance with the CONSORT guidelines and the pilot and feasibility extension. 31 The study findings will be presented at national and international meetings and the results will be published in a high quality, peer reviewed, open-access (via Pubmed) journal. Ongoing study updates will also be provided on the OCTRU website (intact.octru.ox.ac.uk).
Discussion
The problem of anaemia after critical illness is increasingly being recognised. With guidelines advocating restrictive transfusion practices, it is likely that even more ICU survivors will be discharged from hospital with anaemia. No large clinical trial has been undertaken to address the correction of anaemia by the use of IV iron in ICU survivors. INTACT is an open-label, feasibility, RCT designed to answer whether a future large RCT can be undertaken.
The trial design is pragmatic and aims to deliver the intervention at any point between ICU and hospital discharge. The current post-ICU median (IQR) length of stay in our institution is 10 (6–23) days. In order to maximise retention and minimise participant burden, we will offer reimbursement of any travel expenses incurred, and conduct home visits as necessary. This approach was successfully used in a recent RCT evaluating the effect of a physical rehabilitation package in patients recovering from critical care where there was only a 10% loss to follow-up at three months. 33
A limitation of our study is the lack of a placebo infusion. The rusty brown colour of ferric carboxymaltose makes it a challenging substance to blind. This could potentially result in bias, particularly for HRQoL measures. However, a recent large RCT of post-operative IV iron in patients undergoing major elective surgery also used a usual care control group and observed no differences in Short Form-36 (SF-36) scores at 12 weeks post-treatment. 34 As part of our study we will be asking participants at the 90-day follow-up whether or not they remember which group they were randomised to. We will review the decision regarding a placebo infusion for any future RCT.
In conclusion, INTACT is designed to determine whether or not a large RCT of treating anaemia with IV iron in patients recovering from intensive care is feasible. If so, it has the potential to rapidly become a translatable intervention. The trial opened to recruitment in September 2018 and is anticipated to close in December 2019.
Footnotes
Acknowledgements
We would like to thank all the local research team members (Oxford University Hospitals – Archana Bashyal, Paula Hutton, Christie James; Edinburgh Royal Infirmary – Jo Singleton, Nicola Rea, Sarah Clark, Lucy Barclay, Kate Priestley, David Hope, Corrienne McCulloch) involved in the day-to-day running of the study so far and the trial coordinating staff at OCTRU.
Authors' contributions
AS is the chief investigator, protocol author and grant holder. IM and SJD participated in the statistical analysis design of the study and in drafting and revising this manuscript. All authors assisted in the design of this study. All authors have read and approved the final manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is being funded by an NIHR Doctoral Research Fellowship (NIHR-DRF-2017-10-094) awarded to the chief investigator. There is no external industry involvement and all study drugs are being procured through local NHS commissioning arrangements. The University of Oxford is the Sponsor of the study.
Appendix
Appendix 1: History of protocol amendments.
Amendment No.
Protocol version No.
Date issued
Details of changes made
1
2.0
13 Jul 2018
Addition of pregnancy testing, specification of timeframe (24 h) for notification of SAE to CI, and mention of heart rate and blood pressure monitoring before, during and after drug administration
2
3.0
06 Mar 2019
Revision of inclusion and exclusion criteria, including definition of active infection. Greater clarity on recruitment process. Dose adjustment for participants weighing less than 50 kg. Addition of telephone follow-up. Addition of Royal Berkshire Hospital as a recruitment site.