
Abstract
Immune therapy to ease the burden of sepsis has thus far failed to consistently improve patient outcomes. Advances in cancer immune therapy and awareness that prolonged immune-suppression in sepsis can leave patients vulnerable to secondary infection and death have driven resurgence in the field of sepsis immune-therapy investigation. As we develop and evaluate these novel therapies, we must learn from past experiences where single-mediator targeted immune therapies were blindly delivered to heterogeneous patient cohorts with complex and evolving immune responses. Advances in genomics, proteomics, metabolomics, and point-of-care technology, coupled with a better understanding of sepsis pathogenesis, have meant that personalised immune-therapy is on the horizon. Here, we review the complex immune pathogenesis in sepsis and the contemporary immune therapies that are being investigated to manipulate this response. An outline of the immune biomarkers that may be used to support this approach is also provided.
Introduction
Sepsis is defined as the dysregulated immune response to proven or suspected infection, which if unresolved may progress to its severest form, septic shock. 1 Sepsis remains a leading cause of ICU admission and death throughout the world. In 2006, a major pan-European study found that 37% of ICU patients developed sepsis at some point during their admission, with mortality rates in excess of 50% in those with septic shock. 2 According to data from the United Kingdom Sepsis Trust, an estimated 250,000 cases of sepsis occur per annum in the UK, resulting in 44,000 deaths, at an annual cost of £2 billion to the NHS. 3
Despite advances in our understanding of its pathogenesis, novel therapeutic interventions to significantly reduce the burden of sepsis have remained elusive.4–6 Current treatment options are limited, and it is only through refinements in the way we deliver supportive care that mortality has fallen over the years. These refinements include: early recognition and resuscitation, timely source control, and prompt antibiotic therapy. 7
The lack of new antimicrobials over the last few decades, coupled with widespread use of broad-spectrum antibiotics in both humans and livestock animals, has resulted in the emergence of multi-drug resistance (MDR) bacteria. There is growing concern that we may be entering a ‘post-antibiotic’ era, where not only vulnerable patients but also those undergoing routine procedures could develop untreatable, life-threatening infections. In 2016, a UK government review estimated that by 2050, 10 million lives could be lost annually if solutions are not found for the emergence of anti-microbial resistance. 8 The government has therefore pledged to make addressing this issue a national priority.
A possible solution to this emerging problem would be to find alternative solutions to combat the disease process, by developing novel immune-modulatory therapeutic agents that modulate the harmful host response and help the body to restore homeostasis during sepsis. However, despite attracting much interest over the last 20 years and initial promise in animal models, this approach has yet to translate into convincing success in clinical trials. In some cases, it has even proved to be harmful.4,9 Therefore, before we embark on further efforts in this field, it is incumbent upon us to try to understand why previous attempts have failed and to better understand the complex immune-pathogenesis of sepsis.
In this review, we provide an overview of how the immune system becomes dysregulated in sepsis, and the therapies currently being developed to manipulate these responses. A review of the biomarkers that have been investigated to aid in this approach is also provided.
The immune-pathogenesis of sepsis – The SIRS/CARS paradigm
Due to poor sensitivity and specificity, the term systemic inflammatory response syndrome (SIRS) has fallen away from the latest clinical definition of sepsis, but it remains a useful concept when describing the two opposing immune states that exist during sepsis: the hyper-inflammatory SIRS and the compensatory anti-inflammatory response syndrome (CARS).
The clinical phenotype in early sepsis is classically one of overwhelming systemic inflammation, characterised by fever, tachycardia, hypotension, and respiratory dysfunction. These clinical characteristics have been ascribed to the ‘storm’ of pro-inflammatory cytokines, such as tumour necrosis factor alpha (TNF-α), interleukin (IL)- 6, and IL-1 that were initially thought to be the sole protagonists in early sepsis. 10
It was Bone, 11 who in a seminal paper in 1996 balanced this notion by describing the compensatory anti-inflammatory response syndrome (CARS). He hypothesised that this opposing response modulated the destructive nature of the hyper-inflammatory SIRS response, aiming to restore homeostasis. This general concept is now widely accepted, and has since been further developed and characterised. 12 It is associated with the enhanced release of certain anti-inflammatory mediators such as transforming growth factor β1 (TGF-β1), soluble tumour necrosis factor receptor (sTNFR), and IL-10, which along with other anti-inflammatory cytokines have been associated with poor outcomes. 13
Hotchkiss and others have subsequently refined these concepts by describing a two-phase model of sepsis, whereby early SIRS is followed by a CARS response. 14 However, recent gene-profiling studies have demonstrated that anti-inflammatory mediators are upregulated in the early period of sepsis progression and, moreover, do not support a simple hyper-/hypo-immune model of sepsis.15,16 Cavaillon and colleagues, 17 on the other hand, have argued that these responses are spatially ‘compartmentalised’ or separated within the different tissues of the body, and that the concept of sepsis-induced whole body immune suppression is oversimplified. This hypothesis is based on ex-vivo observations that immune cells from the tissues are fully responsive to stimuli (‘SIRS compartment’), whilst simultaneously those from the bloodstream appear to be hypo-responsive (‘CARS compartment’).4,17 This dichotomy raises the possibility that, via either resident populations or extravasating blood leukocytes (e.g. monocytes and neutrophils), appropriate responses to localised infection can co-exist with systemic hypo-responsiveness. Caution is therefore required in using the immune state of circulating leucocytes to determine the appropriateness of systemic adjunctive immune therapies in sepsis.
Though there may be disagreement about the timing and spatial regulation of sepsis, it is generally agreed that if short-lived and self-limited, CARS is an essential counter-balance to the destructive nature of the acute hyper-inflammatory response, and is tasked with preventing maladaptive multiple-organ inflammation and dysfunction through restricting the propagation of systemic inflammation from its source. 11
Immune suppression at the bedside
With the advent of modern ICU supportive therapies, the likelihood of patients surviving the initial hyperinflammatory phase of their illness, without succumbing to multi-organ failure, has increased. However, immune homeostasis is not always restored in these patients and they may enter a phase of protracted immunosuppression, 12 synonymous with CARS, that some have termed ‘immunoparalysis.’ 18 This subclinical state can last for many months, 19 leaving survivors of the initial insult vulnerable to secondary infection, late organ failure, and death.20,21 Consistent with these findings, a post-mortem study by Boomer et al. 21 of sepsis patients showed evidence of widespread immune depression occurring within major organs, whilst Torgersen et al. 22 found that almost 80% of patients with sepsis had unresolved foci of infection at the time of death.
Gentile et al. 23 recently defined the syndrome that occurs when patients survive their initial sepsis insult, and become chronically critically ill – the ‘persistent inflammation-immunosuppression and catabolism syndrome’ (PICS). PICS elegantly incorporates sepsis-induced immunosuppression and the clinical phenotype that we are often confronted with at the bedside; that of persistent inflammation and organ failure that drives a need for ongoing low-grade supportive therapy, whilst protein catabolism, worsened by suboptimal nutrition, often leads to cachexia and poor wound healing. 24 The authors define PICS in patients that have been admitted to ICU for more than 14 days using surrogate clinical markers of inflammation (CRP > 50 µg/dL), immunosuppression (total lymphocyte count <0.8 × 109/L), and catabolism (weight loss >10% during hospitalisation or BMI < 18, albumin <30 g/dL, creatinine height index <80%). 23 Patients who survive PICS often have poor functional outcomes, a poor quality of life, and a limited chance of long-term survival. 24
Furthermore, the increasing frequency of opportunistic infections with low-virulence organisms that have developed MDR to antibiotics, such as Acenitobacter baumannii, likely reflects the increasing prevalence of PICS in the critically ill, rather than what the media portrays as the emergence of ‘superbugs.’ 25
Mechanisms of immunosuppression in sepsis
In order to understand how we might manipulate the immune response and improve outcome, it is important to understand how the cellular and humoral elements of the opposing immune responses become dysregulated during sepsis.
The immune response to pathogen invasion can be divided into the innate and adaptive arms, both of which consist of humoral and cellular elements. The innate immune response is the first line of defence against pathogens, but it is non-specific and of relatively low potency.
Innate immunity consists of the following components: epithelial barriers like the skin and mucosal membranes, phagocytic leukocytes (neutrophils and monocytes), natural killer cells (cytotoxic leukocytes), circulating plasma proteins, cytokines, and the complement system. Monocytes are bone marrow–derived circulating phagocytic cells that have the capacity to differentiate into dendritic cells and macrophages in the tissues where they sample the environment and present foreign pathogens, along with human leucocyte antigen-D-related molecules (HLA-DR), a major histocompatibility complex class II receptor, on their cell surface, to T-cells. In this way they form a key component of the early immune response by orchestrating adaptive immunity as well as enabling the recognition of self from non-self.
Neutrophils are the major myeloid vascular population, and as terminally differentiated phagocytes, they control infection by rapidly migrating into tissues, engulfing pathogens, and releasing lysosomal enzymes and superoxide anions.
The adaptive immune response, in contrast, is normally quiescent and only comes into play when the defences of the innate response fail to control pathogen invasion. While its response is initially much slower, with effector cell activity only evident after one week, it is more specific to the invading pathogen, and results in a faster and more exuberant response on re-exposure. Pathogen-specific antibodies, secreted by plasma cells that have differentiated from activated B-cells, mediate the humoral element of adaptive immunity. These B-cells also differentiate into memory cells that remain quiescent, but have the capacity for clonal expansion and the production of antibodies to the original pathogen, if the antigen is encountered in future. The cell-mediated arm of adaptive immunity is orchestrated by CD4+ and CD8+ lymphocytes. CD4+ T-cells, also called ‘helper’ or ‘regulatory’ T (T-reg) cells, are activated by antigen-presenting cells (APCs) and play a central role in controlling adaptive immunity, while CD8+ lymphocytes or cytotoxic T-cells kill targeted cells like tumour or virally infected cells.
Mechanisms of sepsis-induced immune suppression with immune biomarkers, their measurement modalities and selected immune augmentation therapies.
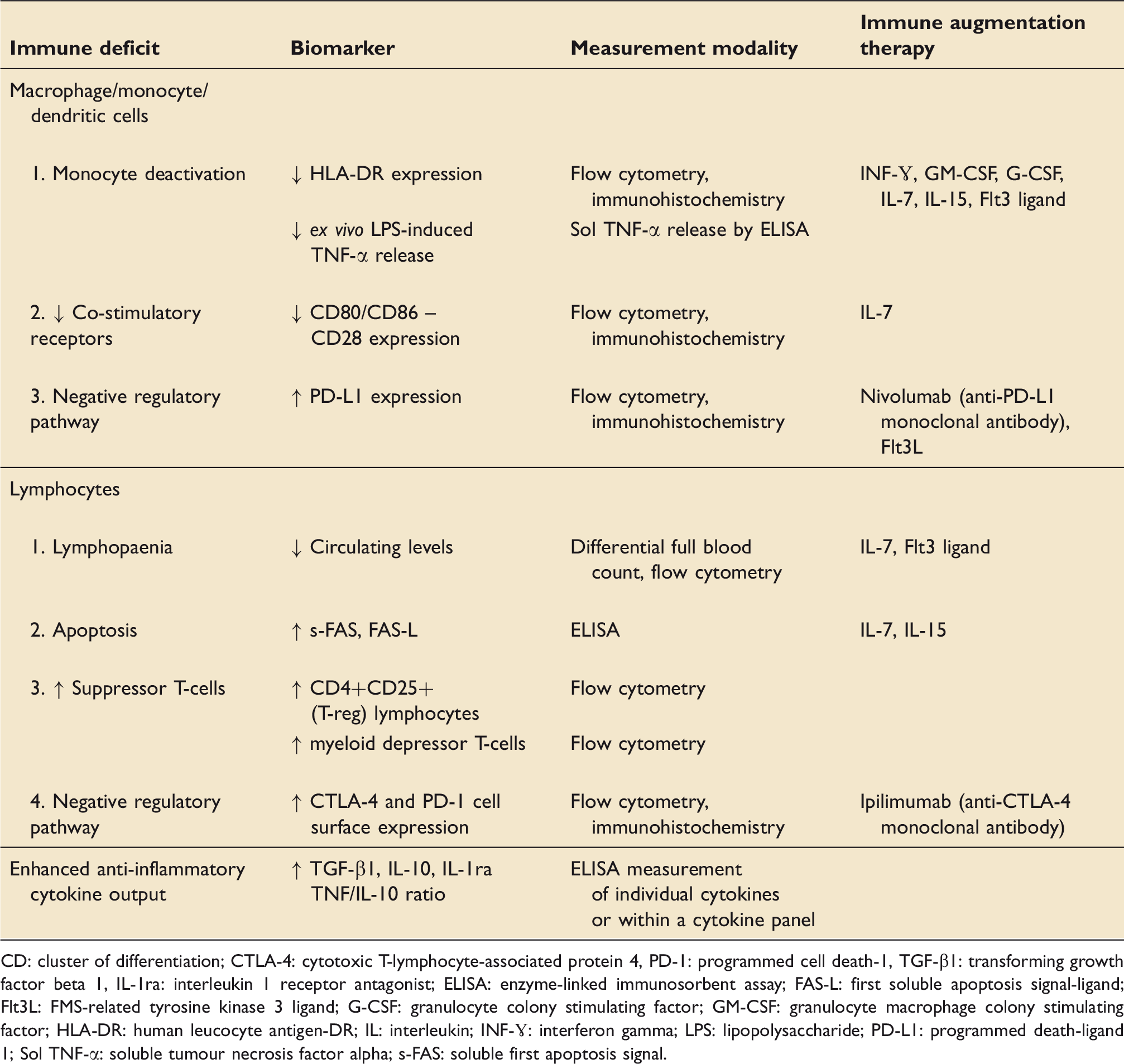
CD: cluster of differentiation; CTLA-4: cytotoxic T-lymphocyte-associated protein 4, PD-1: programmed cell death-1, TGF-β1: transforming growth factor beta 1, IL-1ra: interleukin 1 receptor antagonist; ELISA: enzyme-linked immunosorbent assay; FAS-L: first soluble apoptosis signal-ligand; Flt3L: FMS-related tyrosine kinase 3 ligand; G-CSF: granulocyte colony stimulating factor; GM-CSF: granulocyte macrophage colony stimulating factor; HLA-DR: human leucocyte antigen-DR; IL: interleukin; INF-ɣ: interferon gamma; LPS: lipopolysaccharide; PD-L1: programmed death-ligand 1; Sol TNF-α: soluble tumour necrosis factor alpha; s-FAS: soluble first apoptosis signal.
In addition to monocytes, depressed immune function is evident across multiple leucocyte subpopulations during sepsis. Immunosuppressive lymphocyte subpopulations, including T-regs and myeloid-derived suppressor cells, that normally prevent autoimmune disease and limit chronic inflammation, increase in number,31–33 and there is a simultaneous upregulation of negative co-stimulatory molecules, such as programmed death 1 (PD-1) and its ligand, PD-L1 (Figure 1). 34 The expression of membrane-bound receptors, such as CD86 and CD8021,35 on APCs, which form part of the normal co-stimulatory signal for T-cell activation, are reduced. These deficits result in an impaired capacity to activate T-cells and a subsequent attenuation of cytokine production (Figure 1). Widespread leucocyte apoptosis leads to a loss of T-cells, B-cells, macrophages, and dendritic cells, 21 and complement (C5a)-mediated neutrophil function becomes dysregulated,36,37 which further impairs leucocyte functional capacity. Lastly, there is a shift from an initial pro-inflammatory to a predominantly anti-inflammatory cytokine profile, thought to be driven by differentiation in the macrophage phenotype.
Superimposed on these sepsis-mediated alterations are demographic factors, in particular, our ICU patient population is aging and is at a disproportionate increased risk of sepsis. 38 This phenomenon is in part due to ‘immunosenescence,’ which has been shown to result from complex dysfunctions within both innate and adaptive immunity. One of the prominent deficits is an increase in the number of highly differentiated (mainly CD8+) memory T-cells that, while maintaining their cytotoxic capacity, exhibit a reduced receptor repertoire and lose their proliferative capacity, 39 thereby impairing their ability to mount effective responses. Along with other pre-existing immune deficiency states, such as cancer and HIV that may co-exist during critical illness, ‘immunosenescence’ may further exacerbate sepsis-induced immunosuppression.
The future of immune therapy in sepsis: A personalised approach
The concepts of sepsis immune-pathogenesis have been somewhat mirrored by the changing attitudes to immunomodulatory therapy and their success or failure in implementation. Thus, the litany of failures in targeting pro-inflammatory or pro-coagulant mediators based on the assumption that their overproduction was a primary cause of death 40 encouraged fundamental re-appraisal of the sepsis disease process and, consequently, are now viewed with a greater appreciation of the dynamics and complexity of the immune response and patient population. 41 From the perspective of clinical trial design, one potential reason for the failures was the lack of a stratified approach in delivering the immunomodulatory therapy. It is therefore unlikely that modulating the levels of only one of these targets in a non-patient specific manner, as was the case with anti-TNF therapy in the 1990s, 42 would have a significant effect on outcome. 43 Furthermore, sepsis is a heterogeneous syndrome where a variety of different pathogens and sites of infection affect a diverse patient population, making investigations in this field even more challenging.
Immune biomarkers in sepsis
To enable a personalised and directed prescription of immune therapy, a stratified immune biomarker approach would need to be employed. Such biomarkers would need to accurately measure the individual patient’s immune balance (varying levels of hyperinflammatory or immune deficiency) within the circulation and tissues and should ideally be available at the point of care. Their measurement would need to be reproducible over time, in order to monitor disease progression and guide the intensity and duration of therapy, and they should be able to predict patients at high risk of adverse outcomes such as secondary infection, progression to septic shock, and death.
Although around 180 sepsis biomarkers have been reported in the literature, 44 currently monocyte HLA-DR and immune cell ex vivo cytokine release are the only immune biomarkers that have been used to guide immune adjuvant therapy in clinical trials (see below). An overview is provided of some of the biomarkers that have been investigated in sepsis management, which in future could be used as part of an integrated approach in guiding immune adjuvant therapy.
C-reactive protein and procalcitonin
C-reactive protein (CRP) and procalcitonin (PCT) are presently the only biomarkers in widespread clinical use in detecting infection during sepsis management. CRP is a non-specific acute-phase reactant produced in the liver, whilst PCT is a precursor of the redundant human hormone calcitonin that is normally produced by thyroid C-cells, under the control of the CALC-1 gene. During sepsis, CALC-1 gene expression is substantially upregulated and results in the release of PCT from all differentiated cell types in the body. Though data is conflicting on whether PCT outperforms CRP as a sepsis biomarker, PCT could be used to more accurately gauge the onset and magnitude of the hyper-inflammatory response since levels rise (within 2–4 h vs 12–24 h for CRP) and fall quicker than CRP (half-life of 24 h vs 3–7 days for CRP). 45 At levels of >10 ng/mL, PCT has also been shown to correlate with progression to septic shock, 46 and PCT clearance is correlated with survival.47,48 Whilst further work is needed, these data suggest that PCT could be used as a marker to guide immune therapy targeting excess inflammation.
Cytokines
TNFα, IL-6, and IL-1β are the principle pro-inflammatory cytokines that regulate the initial hyper-inflammatory response in sepsis, and elevated levels of these mediators have been associated with worse outcomes.49,50 In contrast, IL-10 is a major anti-inflammatory cytokine, synonymous with the CARS response and implicated in the down-regulation of monocyte HLA-DR and co-stimulatory receptors, such as CD80 and CD86. Elevated levels of IL-10 have been shown to correlate both with severity and outcome in sepsis. 51 However, the utility of these cytokines as immune biomarkers is limited by their non-specific release in other inflammatory conditions such as trauma, surgery, and autoimmune disease.
To address this, many investigators have explored the use of multi-array panels of cytokines in predicting outcomes in sepsis.52–54 Bentzer et al. 55 recently reported that a cytokine triplet of IL-3 (a pro-inflammatory cytokine), IL-6, and CCL4 (a T-cell chemoattractant) identified a subset of patients treated with corticosteroids that had an increased survival at 28 days (OR 19, 95 % CI 3.5–140, p = 0.02).
One of the hallmarks of sepsis is the diminished capacity of monocytes to release cytokines, such as TNF and IL-6, in response to an ex vivo lipopolysaccharide (LPS) challenge, also known as endotoxin tolerance. Rather than being completely refractory, evidence suggests that these cells remain responsive but that their intracellular signaling shifts toward the production of anti-inflammatory mediators like IL-10. 56 This state, which has been termed monocyte ‘deactivation’ or ‘reprogramming,’ goes hand-in-hand with a reduced expression of HLA-DR, and is associated with worse outcomes and an increased risk of nosocomial infection in sepsis. 29 Clinical studies using granulocyte-macrophage colony-stimulating factor (GM-CSF) and interferon-gamma (IFN-γ) to reverse sepsis immunosuppression have used ex vivo monocyte responsiveness to guide and monitor therapy.18,57
Currently, these ex vivo assays are confined to the research setting; however, further validation, standardisation, and ultimately automation could lead to their greater uptake in clinical practice. 58
Immune cell surface receptors
TREM-1
Triggering receptor expressed on myeloid cells-1 (TREM-1) is a cell surface marker that is upregulated and released into the circulation by activated neutrophils, monocytes, and macrophages. TREM-1 plasma levels of >60 ng/mL have been shown to accurately differentiate infection from non-infectious inflammation, 59 and in a cohort of patients who received early goal directed therapy, it was able to identify a subset with a poor prognosis despite complete initial resuscitation for sepsis. 60 However, a recent meta-analysis and systematic review of nine clinical trials by Su et al. 61 concluded that on its own sTREM-1 exhibited only moderate prognostic ability and may be best used as part of a panel of biomarkers in predicting prognosis in infection.
HLA-DR
Arguably, the best marker of sepsis-induced immune suppression to date is monocyte HLA-DR expression, which unlike the measurement of individual cytokines, is a good indicator of the sum effect of the complex humoral and cellular interactions that occur in sepsis. It has long been used to guide immune therapy in small early-phase clinical trials62,63 where, until recently, inter-test/laboratory variability had limited its widespread use. However, a system that allows the standardised quantitative measurement of cell surface antigens (QuantiBRITE system, Beckton Dickinson) is now available, which enables determination of HLA-DR antibodies per cell (AB/C) and has demonstrated excellent inter-laboratory correlation. 64 This approach has already been used in a multi-center clinical trial investigating GM-CSF therapy in septic patients, where investigators used a monocyte HLA-DR cut-off level of less than 8000 AB/C on two consecutive days to guide the appropriateness and timing of therapy. 65 Also of note in a recent prospective observational study in patients with sepsis, monocyte HLA-DR outperformed a whole blood LPS-stimulation assay at predicting mortality and the incidence of nosocomial infection at 28 days, 66 which further highlights its potential use as a single, clinically relevant biomarker of global immune cell function.
Lymphopenia
One of the characteristic features of sepsis-induced immune suppression is a marked reduction in circulating lymphocytes, including CD4+ and CD8+ T-cells and B-cells. Lymphocyte counts fall at the onset of sepsis through sequestration into the tissues and via apoptotic loss, and can remain low for up to 28 days. 67 Drewry et al. 68 recently showed that persistently low lymphocyte counts on day 4 after sepsis onset independently predicts both short-term and 28-day mortality. Additionally, they found that the presence of severe persistent lymphopenia (<0.6 × 103 cells/µL) was significantly associated with the development of secondary infections (OR 2.11, 95% CI 1.02–4.39; p = 0.04). These data suggest that persistent lymphopenia may serve as both a marker and a target in trials investigating immune augmentation therapy.
Immunomodulatory pathway markers
Immune ‘checkpoints’ refer to a series of inhibitory pathways within the immune system that are crucial in modulating the duration and amplitude of the normal immune response (Figure 1). They are initiated by a plethora of ligand-receptor interactions that are upregulated in sepsis and inhibit the co-stimulatory signals needed to augment normal T-cell activation by APCs. These immune checkpoints have already been targeted successfully by blocking antibodies in cancer immune therapy.69,70
Mechanism of action of immune checkpoint inhibitors in sepsis. (a) Normal T-cell activation through antigen presentation by antigen-presenting cells (APCs) in conjunction with membrane bound HLA-DR to the T-cell receptor. To ensure optimal T-cell priming and activation, a co-stimulatory signal is provided through the binding of CD80 and CD86 to their ligand CD28. (b) Sepsis results in the upregulation of negative co-stimulatory receptors on APCs, such as PD-1, which interacts with its ligand PD-L1 and blocks the normal T-cell activation pathway, resulting in T-cell exhaustion. (c) Immune checkpoint inhibitors such as Nivolumab are monoclonal antibodies that block the binding of PD-1 to PD-L1 and result in the restoration of normal stimulatory T-cell pathways.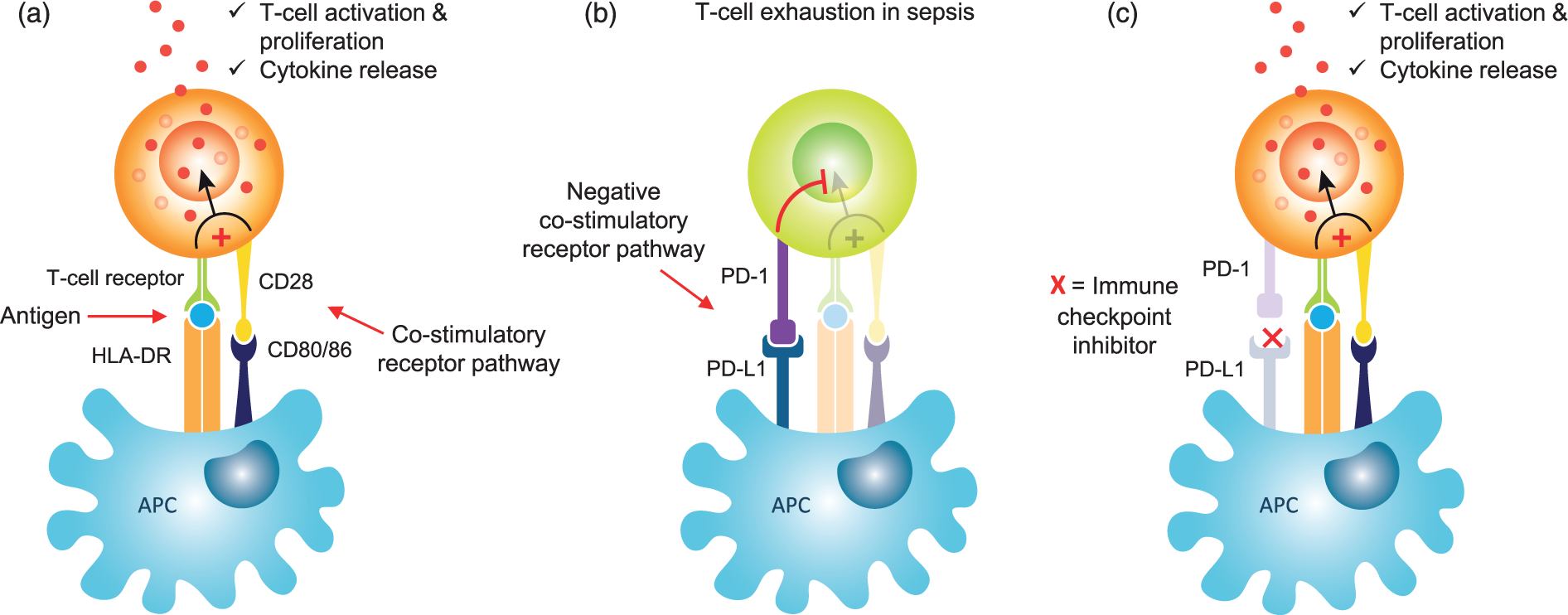
Two such interactions, the programmed death receptor-1 (PD-1) with its ligands PD-L1 and PD-L2, and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) with its ligands CD80 and CD86, have recently been investigated in sepsis pathophysiology.
In septic shock increased expression of PD-L1 and PD-1 have been found on circulating monocytes and CD4+ lymphocytes, respectively, which correlated with increased levels of IL-10 and was significantly associated with an increased mortality and the occurrence of secondary infections.71,72 Boomer et al. 73 reported that the expression levels of CTLA-4 on CD4+ and CD8+ lymphocytes increased with sepsis progression and this phenomenon has been implicated in the pathophysiological down-regulation of helper T-cell activity and the enhancement of T-reg cell immunosuppressive activity. 70 These data not only provide a mechanistic basis for the use of ‘immune checkpoint inhibitors’ in sepsis but also suggest that PD-1 and CTLA-4 could be used as biomarkers to diagnose sepsis-induced immunosuppression.
The ‘omic’ approach
There is an emerging body of evidence linking immune transcriptomic biomarkers to patient outcome and the risk of nosocomial infection in the ICU.74–77 Peronnet et al. 75 recently showed that the whole blood mRNA expression levels of CD74, the monocyte HLA-DR antigen-associated invariant chain, and IL-10 were able to predict and risk-stratify patients at risk of ICU-acquired infections. They found that independent of shock status, patients with low levels of CD74 on day 3 relative to day 1, and those with low levels of IL-10 on day 3 had a higher risk of developing ICU-acquired infections and may benefit from immune augmentation therapy.
Davenport et al. 15 recently demonstrated that global genomic profiling of circulating leukocytes was able to identify amongst patients with community-acquired pneumonia a subphenotype that exhibited relative immune suppression, endotoxin tolerance, and T-cell exhaustion, and had a worse prognosis than patients without immune suppression. Similar findings were also seen in patients with faecal peritonitis. 78 These data highlight the importance of identifying patients with discrete immune signatures, within the heterogeneous sepsis population who might benefit from specific immune-augmentation therapies.
An integrated approach – An immune ‘bioscore’
No one immune marker is likely to adequately identify and monitor the complex immune deficits that occur during sepsis. Instead, multi-parameter ‘bios-coring’ systems incorporating clinical risk scores 71 and innate and adaptive immune biomarkers are likely to provide a more accurate indication of the individual patient’s immune status.
The Immune Failure in Critical Therapy (INFECT) study (NCT02186522) is currently investigating the validity of combining markers of innate and adaptive immune dysfunction (monocyte HLA-DR, neutrophil CD88 and percentage T-regs) to risk-stratify patients who may develop nosocomial infection. 79
Moving toward point-of-care testing
To support a personalised immune-based therapeutic approach, laboratory tests that can provide rapid reliable and accurate results, most likely at the point of care, would need to be developed.
This is highlighted by the fact that if monocyte HLA-DR is not measured within 2–4 h of sampling, results become inaccurate. Currently, this means that a flow cytometer and a skilled technician would need to be available 24/7.80,81 Such limitations have hampered its widespread utility in the clinical setting.
However, advances in point of care technology have meant that a rapid, automated cartridge-based flow cytometry device that is able to measure monocyte HLA-DR expression (Accellix-HLA-DR, LeukoDx, Jerusalem, Israel) within 25 min has now been developed. Sprung et al. 82 have used similar technology to show that measurement of CD64 (Accellix-CD64), the neutrophil activation marker, correlates well with standard flow cytometry 82 and that it is able to differentiate infection from non-infectious inflammation in a timely manner. 83
Further development of this type of automated technology is likely to lead to improved immune biomarker assays, which could allow a stratified medicine approach to immunomodulation in sepsis.
Immune therapy in sepsis – Current and future insights
Immune augmentation therapies
Interest in the field of adjuvant immune therapy in sepsis has been revitalised in recent years, partly driven by recent developments in the treatment of cancer, and mostly targeting secondary immune suppression. To highlight this we conducted a search of the United States National Library of Medicine ClinicalTrials.gov website and the UK clinical trials gateway using the search terms sepsis and immune therapy, immune modulation therapy, immune augmentation/stimulation therapy, immune suppression, immune paralysis, interleukin, interferon, GM-CSF, granulocyte colony stimulating factor (G-CSF), immune checkpoint inhibitor, Ipilimumab, Nivolumab, immunoglobulin, Polymyxin B Hemoperfusion, Cytosorb® (Table 2). The search was then narrowed using the following filters: date range of 1 January 2013 to 1 January 2018, adults over 18 years, interventional (clinical) trial, registered as active (either recruiting or not recruiting), not yet recruiting or completed but not yet published on-line.
Summary of the clinical trials investigating adjuvant immune therapy in sepsis within the last 5 years.
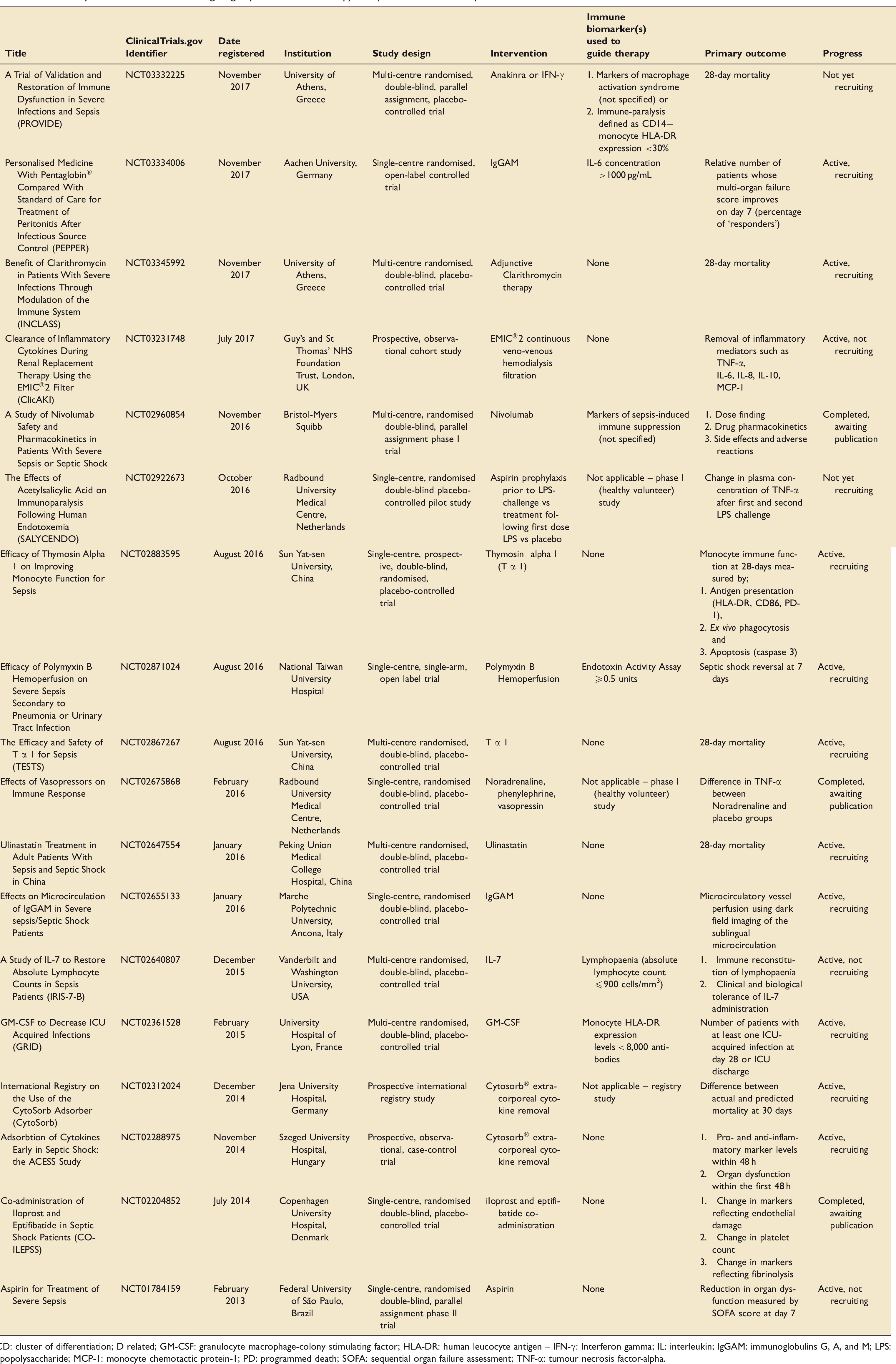
CD: cluster of differentiation; D related; GM-CSF: granulocyte macrophage-colony stimulating factor; HLA-DR: human leucocyte antigen – IFN-γ: Interferon gamma; IL: interleukin; IgGAM: immunoglobulins G, A, and M; LPS: lipopolysaccharide; MCP-1: monocyte chemotactic protein-1; PD: programmed death; SOFA: sequential organ failure assessment; TNF-α: tumour necrosis factor-alpha.
Most recently, Grimaldi et al. 86 reported success in treating a young patient involved in the Brussels bomb attacks in 2016 who had developed intractable invasive mucormycosis, a rare fungal infection with a poor prognosis that typically affects immunocompromised patients. Immunological examination revealed a picture in keeping with post-traumatic immune-paralysis; a low absolute lymphocyte count, depressed monocyte HLA-DR expression, and increased PD-1 receptor expression on T-cells. Combination immune therapy with Nivolumab, which reverses PD-1-mediated T-cell and cytokine inhibition, 88 and IFN-γ, which restores monocyte immune function, 18 were initiated on compassionate grounds and resulted in successful resolution of this life-threatening infection.
Nivolumab is a blocking monoclonal antibody that targets the PD1-PD-L1 ‘immune-checkpoint’ (Figure 1), an effector T-cell inhibitory pathway that is co-opted by cancer cells and viruses to confer immune resistance. Nivolumab has been shown to improve survival in animal models of fungal sepsis, 89 reverse T-cell dysfunction, and augment viral clearance in the treatment of chronic viral infections like hepatitis C.90–92 A phase-I clinical trial investigating the use of Nivolumab in the treatment of sepsis and septic shock is currently underway (NCT02960854).
Another potential target within the ‘immune checkpoint’ is the negative co-stimulatory molecule CTLA-4. Anti-CTLA-4 monoclonal antibodies like Ipilimumab which, alone or in combination with anti-PD-1 antibodies, have dramatically influenced the outcome of patients with advanced metastatic melanoma, as well as showing promise in other forms of cancer.69,93 Anti-CTLA-4 antibodies have been shown to improve outcome in animal models of polymicrobial 94 and fungal sepsis, 89 and levels of CTLA-4 on T-cells are elevated in human sepsis,31,73 making this an attractive therapeutic target. These potent immunomodulatory agents have, however, been associated with serious adverse effects (up to 24% in cancer chemotherapy), which may limit their use in critical illness and highlights the importance of finding the right therapy, to deliver to the right patient, at the right time.
IFN-γ and GM-CSF are both potent stimulators of neutrophils, monocytes, and macrophages, and are probably the most investigated immune-stimulating therapies in the treatment of sepsis-induced immune suppression to date.9,57,62,65,95–101 IFN-γ is predominantly secreted by Th-1 T-cells and has pleiotropic effects on innate immunity. It has been shown to reverse monocyte deactivation and endotoxin tolerance in vitro and in vivo, in both animal and healthy volunteer models of sepsis.57,100 A small pilot study investigating the use of subcutaneous IFN-γ in nine patients with septic shock, guided by levels of monocyte HLA-DR expression, showed that monocyte immune function was restored, with eight out of the nine patients recovering from their sepsis without any adverse effects. 62 Furthermore, IFN-γ has been shown to be effective adjuvant therapy for Cryptococcal meningitis in HIV infection, 102 as well as in the treatment of fungal 103 and bacterial infections 87 in other immunocompromised patient groups, such as following renal transplantation and in those on long-term corticosteroids. IFN-γ is approved for the treatment of fungal infection in patients with chronic granulomatous disease, 104 a hereditary immune deficiency disorder, and is currently being investigated as adjuvant therapy in septic shock in a pilot placebo-controlled clinical trial (NCT01649921).
GM-CSF is a cytokine with growth factor properties, secreted by Th-1 and B-cells, that stimulates neutrophil, monocyte/macrophage, and dendritic cell proliferation and differentiation,101,105 as well as priming for cytokine production, phagocytosis, and bactericidal ability. 106 Like IFN-γ, it increases monocyte HLA-DR expression and promotes inflammatory dendritic cell maturation and function, thereby promoting antigen presentation capacity and indirectly boosting adaptive immunity.
GM-CSF and its related cytokine, G-CSF, have recently been the subject of both a meta-analysis 9 and a systematic review 101 that included 12 randomised controlled trials (RCTs), three of which investigated GM-CSF as adjuvant therapy in sepsis. Though these trials showed an improvement in infection clearance (RR 1.34, 95% CI 1.11 to 1.62, p = 0.002), mostly attributed to G-CSF therapy, there was no effect on 14-day or 28-day mortality, although it should be noted that none of the individual studies were powered for this outcome measure. 9
Only one of the trials included in these reviews was biomarker-guided, and used monocyte HLA-DR expression to determine the intensity of GM-CSF administration. Patients with monocyte HLA-DR < 8000 AB/C where randomised to either receive 4 µg/kg/day GM-CSF or placebo for five consecutive days. GM-CSF was then continued for an additional 3 days at a dose of 8 mcg/kg/day if monocyte HLA-DR was ≤15,000 AB/C or 4 mcg/kg/day if monocyte HLA-DR was >15,000 AB/C. The investigators found that monocyte HLA-DR expression was normalised in all GM-CSF-treated patients (n = 19) compared to only 3/19 placebo-treated patients (p < 0.001), and that this was accompanied by restoration of ex vivo monocyte immune function (increased pro-inflammatory cytokine release). 65 Notably, no adverse effects were reported.
In contrast to its immune-stimulating effects, GM-CSF has also been shown to exhibit significant anti-inflammatory properties, such as a reduction in the progression of atherosclerosis, 107 attenuation of lung remodeling in pulmonary fibrosis, 108 and a dose-dependent expansion of immunosuppressive T-reg cells in tumour vaccine therapy. 109 There is therefore a theoretical risk of exacerbating sepsis-induced immune suppression, if GM-CSF is delivered at the wrong moment during sepsis progression. This uncertainty further emphasises the importance of delivering immune therapy guided by immune biomarkers in order to optimise their therapeutic effect and reduce the risk of harm. The GRID trial (GM-CSF to decrease ICU-acquired infections) is a large multicenter double-blind, placebo-controlled RCT that is currently investigating the efficacy of GM-CSF in preventing secondary infections in patients with sepsis-induced immune suppression, guided by monocyte HLA-DR expression levels (<8000 AB/C at day 3). Investigators will also look at secondary outcomes such as organ failure scores and 28-day mortality (NCT02361528).
IL-7 is a cytokine mainly produced by thymic and bone marrow stromal cells that plays a crucial role in B- and T-lymphocyte development, maturation, expansion, and homeostasis.110,111 In fact, it has diverse effects on both innate and adaptive immunity, which is why it has been termed the ‘maestro of the immune system.’ 112 Recombinant IL-7 (rIL-7) has been shown to effectively restore immunity in patients with HIV and cancer.113–117 It has also been shown to improve survival in clinically relevant animal models of bacterial and fungal sepsis.118–121 These and other pre-clinical studies have elicited a number of important mechanisms by which IL-7 might be beneficial in sepsis-induced immune suppression. Through its potent anti-apoptotic and lymphopoietic effects it results in the proliferation of both naïve and memory T-cells, 122 which could reverse the lymphocyte depletion associated with sepsis. This increase in T-cell numbers results in an expansion of the T-cell repertoire, 117 which along with enhanced expression of adhesion molecules like LFA-1 and VLA-4, 120 would improve T-cell trafficking to sites of infection, thereby broadening the immune response. Furthermore, IL-7 enhances IFN-γ secretion,120,123 which has been shown to reverse monocyte deactivation 62 and boost innate immunity in sepsis.
rIL-7 is well tolerated and has an excellent clinical safety profile.123–125 In contrast to IL-2, a cytokine with similar effects on T-cell function, IL-7 does not result in an increase in the numbers of suppressive T-reg cells 126 nor does it result in T-cell activation where there is a potential for a subsequent pro-inflammatory ‘cytokine storm’, 115 both of which might be detrimental in sepsis. The IRIS-7B (NCT02640807) trial is a multi-centre placebo-controlled RCT currently investigating IL-7’s ability to restore lymphocyte counts in sepsis.
Thymosin α-1 (T α 1) is a naturally occurring, thymus-derived peptide with widespread in vitro immunomodulatory effects.127–130 It is currently licenced for the treatment of Hepatitis B and C131–133 and has been shown to improve recurrence-free survival and ameliorate chemotherapy-induced myelosuppression in malignant melanoma and hepatocellular carcinoma. Importantly, T α 1 is well tolerated with few side effects; in one study the only reported adverse event was discomfort at the injection site. 134
A recent systematic review of 19 RCTs investigating T α 1 in sepsis by Liu et al. 135 included 1354 patients, and reported a significant reduction in mortality (RR 0.59, 95 % CI 0.45 to 0.77, p = 0.0001) and APACHE II scores (Standard mean difference: −0.80, 95 % CI −1.14 to −0.47, p < 0.0001) with a once-daily dosing regimen. Additionally, they showed a significant increase in HLA-DR expression levels, CD3+, CD4+ T-cell numbers as well as the CD4+/CD8+ ratio. There were however no differences in ICU length of stay, organ failure scores, or time on the ventilator. Despite including the largest study to date, the Efficacy of Thymosin Alpha 1 for Severe Sepsis (ETASS) trial (n = 381), 136 the review had significant limitations, including variable heterogeneity among the different outcome measures and the inclusion of predominantly underpowered, single-centre studies.
T α 1 has also been investigated in combination with Ulinastatin (UTI), a protease inhibitor found in human urine and blood that has anti-inflammatory properties.137,138 Analysis of pooled data from eight RCTs (n = 1112) in a recent meta-analysis by Liu et al. 139 showed that UTI + T α 1 therapy reduced 28-day (RR, 0.64; 95% CI, 0.54–0.75; p < 0.01) and 90-day mortality (RR, 0.69; 95% CI, 0.59–0.80, p < 0.01) by 36% and 31%, respectively. Interestingly combination therapy also reduced the time on mechanical ventilation and APACHE II scores, and suppressed pro-inflammatory cytokine release, suggesting that there may be a synergistic effect. However, this data included trials with significant heterogeneity and small sample sizes (only two studies had >200 patients), therefore further high-quality multi-centre trials are needed to provide conclusive evidence for the use of T α 1 and UTI in sepsis.
Two trials are currently underway; The Efficacy and Safety of T α 1 for Sepsis (TESTS, NCT02867267) and the Ulinastatin Treatment in Adult Patients With Sepsis and Septic Shock (NCT02647554).
Strategies targeting excess inflammation
Despite more than 100 clinical interventions over the last 40 years investigating the sepsis hypothesis, that the endogenous inflammatory response of the host determines the outcome of life-threatening infection, an anti-inflammatory therapeutic strategy in sepsis has thus far failed to improve outcomes.
Since the 1980s Intravenous Immunoglobulin (IVIg) has been proposed for the treatment of sepsis. It has multiple modes of action, including attenuation of inflammatory gene transcription, promotion of pathogen clearance, and anti-apoptotic innate and adaptive immune cell effects. 140 Polyclonal IVIg is a sterile mixture of purified antibodies manufactured from pooled human plasma that typically contains more than 95% unmodified immunoglobulin G (IgG), with intact Fc-dependent effector functions and only trace amounts of immunoglobulin A (IgA) or immunoglobulin M (IgM). The use of monoclonal, as well as polyclonal IVIg, in sepsis has been the subject of over 42 RCTs and multiple meta-analyses over the years. These have yielded conflicting results and have failed to demonstrate an overall benefit, therefore IVIg is not currently recommended in the 2016 surviving sepsis guidelines. 141 However, these studies were mostly underpowered and used different formulations, dosing regimens, and durations of therapy. Notably, a recent meta-analysis by Busani et al. 142 showed that studies using monoclonal IgM have shown a more consistent reduction in mortality than those using standard polyclonal IVIg. The results of a retrospective cohort study by Cavazzuti et al. 143 investigating IgM-enriched polyclonal IVIg in septic shock make a compelling argument for the use of early, targeted IVIg. The authors included 168 patients in their analysis, 92 (54.8%) of whom received 250 mg/kg/day of IgM over 3 days, starting within 24 h of sepsis onset. They found that when compared to the control group, and after adjusting for confounders, the 30-day mortality rate was reduced by 21.1% (p < 0.05) in those who received IgM. While a larger prospective multicenter RCT is urgently needed to clarify the use of IVIg in sepsis, some authors have proposed that additional mechanistic and dose-finding studies need to be undertaken to avoid further fruitless investigations in this field. 144 It is worth noting that despite conflicting data on efficacy, IVIg (2 g/kg/day for 3 days) is currently the only immune therapy advocated in the treatment of infection. It is recommended as adjuvant therapy, along with surgical debridement and IV antibiotics, in the treatment of streptococcal toxic shock syndrome due to necrotising fasciitis in the UK (http://bestpractice.bmj.com/best-practice/monograph/821/treatment/details.html).
Extracorporeal blood removal of endogenous pro-inflammatory mediators from the circulation was first proposed as a means of reducing systemic inflammation in experimental models of sepsis in the 1990s. 145 This concept was supported by findings that high-volume haemofiltration, in the higher dose range of 35–100 mL/kg/h than is traditionally used to support renal failure, stabilised haemodynamics, decreased vasopressor requirements, reduced serum lactate, and could improve survival in septic shock.146–148 Though this approach has not been adopted into clinical practice, a recent advance in the field of haemofiltration is the addition of haemoadsorption technology to conventional extra-corporal circuits, which has not only been shown to effectively remove bacterial products and inflammatory mediators like TNF-α and IL-10 but also improve outcome in animal models of sepsis.149–152 In 2009, the EUPHAS trial used polymyxin B haemoperfusion (PMX-HP) to show an improvement in organ failure, haemodynamics, and 28-day mortality in abdominal septic shock. 153 Polymyxin B is an antibiotic with a high affinity for endotoxin on the gram-negative bacterial cell wall which, when bound to the polystyrene fibers of a haemofilter effectively binds endotoxin in vitro and in vivo and could interrupt the noxious inflammatory cytokine cascade. Unfortunately, the results of this study were not replicated in the subsequent French ABDO-MIX RCT, 154 which failed to show clinical benefit in the setting of emergency abdominal surgery for peritonitis-related septic shock.
Despite these conflicting results, this technology has been widely adopted in the treatment of proven or suspected gram-negative sepsis throughout Japan and parts of Western Europe. The results of the EUPHAS2 study, a European retrospective registry of sepsis patients receiving PMX-HP therapy, was published in 2016 and, though prone to selection and recall bias, does provide some support for the clinical benefit of endotoxin removal, without significant adverse events. 155 The EUPHIRATES trial, 156 a 466-patient multi-centre RCT investigating the use of PMX-HP therapy in patients with septic shock and endotoxaemia, guided by a bedside endotoxin activity assay (EAA), is yet to be published, but a press release in May 2017 reported a non-significant 5% difference in mortality in the per protocol group. 157 Despite an overall lack of efficacy, the study did show significant improvements in cardiovascular function and ventilator-free days (median difference of 14 days; p = 0.0043) compared to the control group. In addition, at post hoc analysis, in the 194 patients with abdominal infection, a multiple-organ dysfunction score of >9, and an EAA 0.6–0.9 (highly endotoxaemic), there was a significant 10% (p = 0.0474) absolute reduction in 28-day mortality compared to control, which warrants further investigation into the use of PMX-HP therapy in this subgroup of patients.
Furthermore, an extracorporeal cytokine absorber (Cytosorb®, Cytosorbents corp.) is currently being investigated in 17 trials in a range of settings such as intra-operative cardiopulmonary bypass and pancreatitis, many of which are in the analysis phase. A case series by Kogelman et al. 158 recently showed that when Cytosorb® was added to conventional continuous hemodialysis for the treatment of severely ill (APACHE II > 25) septic shock patients, there was subsequent rapid stabilisation of haemodynamic parameters and a reduction in serum lactate. Though there was no control arm, the mortality rates were lower than would have been predicted by APACHE II scores, and the effect seemed to be more pronounced in those where therapy was started within 24 h of sepsis onset. Cytosorb® extracorporeal cytokine removal therapy is currently being investigated in the ACESS (Adsorbtion of Cytokines Early in Septic Shock) Study (NCT02288975).
Advances in nanotechnology have led to the development of a haemoadsorption device coated with a genetically engineered form of the human opsonin, mannose binding lectin (MBL), which is linked to an antibody Fc domain.159,160 Similar to the cleansing effect of the spleen, MBL has the capacity to bind the cell walls of gram-positive and gram-negative bacteria, viruses, fungi, and parasites, as well as LPS-endotoxin, without affecting mammalian cells. When tested in in vitro human and in vivo animal models of sepsis, this ‘biospleen’ produced a highly efficient (90–99%) removal of pathogens, and resulted in a reduction of organ pathogen loads, the suppression of inflammation, and improvements in vital signs compared to controls treated with antibiotics alone. 160 This technology is currently undergoing further refinement in pre-clinical studies.
As well as these specific immune modulating therapies it has also become apparent that our current routine ICU therapies, such as vasopressors used for hemodynamic support in septic shock, may have important immune-modulating effects. Noradrenaline has been shown to inhibit macrophage migration and influence cytokine release in vitro,161–164 while vasopressin receptors have been identified on B-cells and macrophages and it has been shown to suppress pro-inflammatory cytokine release in in vitro human and animal models.165–168 Noradrenaline’s immune-modulating effects (compared to vasopressin and phenylephrine) are currently being investigated in vivo in a human experimental model of LPS-induced endotoxaemia (NCT02675868). In septic shock, Russel et al. 169 showed that vasopressin suppressed 24-hour plasma cytokine levels more than noradrenaline. While vasopressin has not been shown to improve clinical outcomes, a recent study by Gordon et al. 170 showed that it is a safe and effective alternative to noradrenaline in the treatment of septic shock. These data raise the possibility that using vasopressin early in the course of septic shock, when excess inflammation predominates, rather than later as a ‘catecholamine-sparing’ agent, may have beneficial effects on immune function. Greater understanding of the immune effects of our current ICU medications may help guide future management decisions in sepsis.
Conclusions
The initial response to sepsis consists of both pro- and anti-inflammatory responses, the intensity of which is dependent on multiple factors specific to the pathogen and host. Immunosuppression occurs early on in its course, and if sepsis progresses many patients may enter a period of protracted immune suppression, resulting in increased mortality. Adjuvant immune therapy to manipulate the hyper-inflammatory and/or immune-suppressive phase of sepsis is an attractive therapeutic option, which may improve outcome and ease the burden of antimicrobial resistance. However, before this can become a clinical reality, we must recognise that sepsis is a clinical syndrome, where significant heterogeneity exists. The challenge in developing effective adjuvant immune-modulating therapies is to better characterise this heterogeneity by not only defining disease-specific cohorts but also identifying subphenotypes who might benefit from specific interventions. If we can identify these treatable traits, we may be able to deliver targeted, personalised immune therapy, guided by the bedside measurement of immune biomarkers, and not rely solely on non-specific clinical parameters.
Footnotes
Acknowledgement
The authors thank Nicola Davies for help with the manuscripts artwork.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Gordon reports that outside of this work he has received speaker fees from Orion Corporation Orion Pharma and Amomed Pharma. He has consulted for Ferring Pharmaceuticals, Tenax Therapeutics, Baxter Healthcare, and GSK, and received grant support from Orion Corporation Orion Pharma, Tenax Therapeutics, and HCA International with funds paid to his institution.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Davies is supported by the New Investigators Award from the Intensive Care Society and a grant from The Wellington Hospital in London. Gordon is an NIHR Research Professor and his department receives support from the NIHR Imperial Biomedical Research Centre.