p-Phenylenediamine reacts with Sanger’s reagent in hot ethanol to give the expected mono- and di-substitution products, but in ethanol at room temperature, it gave exclusively 2-nitro-5-fluorophenyl-p-phenylenediamine, where a hydrogen atom is displaced by attack at an activated, unsubstituted position. The reactions of p-phenylenediamine and aniline with Sanger’s reagent were compared in the cheap, ‘green’ solvent ethanol.
An unexpected product forms from reacting Sanger’s reagent with p-phenylenediamine
Sanger’s reagent, 2,4-Dinitrofluorobenzene (DNFB) 1, is used for the chromatographic detection and quantification of amino acids, peptides and proteins (Figure 1).1 Its effectiveness is based on the reaction of the reagent with free alpha and epsilon amino groups to form stable, yellow dinitrophenyl derivatives.1 It allows the terminal amino acid of a peptide chain to be determined.1
The formation of a terminal amino acid and peptide chains labelled with Sanger’s reagent 1.1 Compound 5 forms when a peptide bond along the chain hydrolyses.
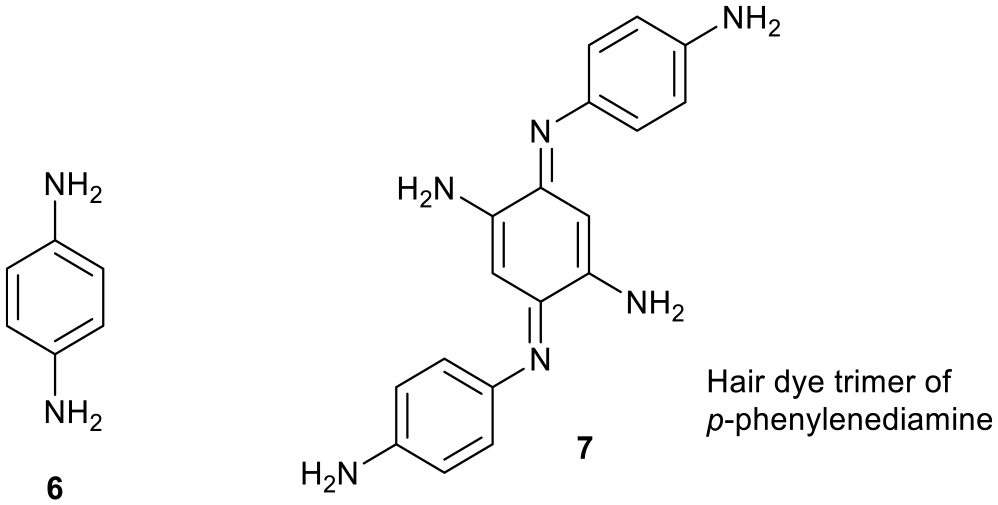
Selective cleavage of the labelled peptide chain allowed the amino sequence of insulin to be determined by examining the overlap of peptide sequences.1 DNFB 1 was a better reagent than 2,4-dinitrochlorobenzene because it reacted at room temperature and did not need heating.1 The other reagent studied in this paper, p-phenylenediamine 6, is commonly used as a hair dye precursor to structure 7 by oxidation with H2O2/aqNH3.2–12 (Figure 2). 2,4-Dinitrofluorobenzene 1 and p-phenylenediamine 6 were interesting reagents to react together with a view to making isomers of trimer 7, that might expand the range of hair dyes, and to explore the reactivity of DNFB 1.
p-Phenylenediamine 6 and its hair dye trimer 7.
Discussion
p-Phenylenediamine 6 was reacted with DNFB 1 in a 1:2 ratio at 50 °C in EtOH for 24 h (Figure 3).
The products 8-10 from reacting p-phenylenediamine 6 with DNFB 1.
The main product 8, from a bis adduct formation reaction, was difficult to purify by chromatography owing to its poor solubility and pigmentary properties. This accounts for the low yield. A quantity of the half-coupled product 9 was also formed. A front-running compound 1013 was also formed in variable but significant quantities and was characterised by its proton and carbon NMR spectra. This known compound forms here by the displacement of fluorine with ethanol in the hot solution. In hot isopropanol, the isopropyl analogue of compound 10 did not form, so the product was mainly compound 8 (89%) contaminated with a small amount of compound 9. Isopropanol is more hindered than ethanol so the formation of a substitution product from it and DNFB 1 is suppressed.
Compound 8 crystallises in the monoclinic space group P21/n with half a molecule in the asymmetric unit with the complete molecule generated by crystallographic inversion symmetry (Figure 4). The dihedral angle between the central and pendant aromatic rings is 39.94 (7)° and the N2/O1/O2 and N3/O3/O4 nitro groups are twisted from the latter ring by 4.84 (10) and 17.48 (7)°, respectively. An intramolecular N – H···O hydrogen bond occurs [H···O = 1.934 (19) Å, N – H···O = 138.0 (16)°] and the same group also participates in a much weaker intermolecular N – H···O interaction.
The molecular structure of compound 8 showing 50% displacement ellipsoids and hydrogen bonds as double dashed lines. Symmetry code: (i) 1–x, –y, 1–z.
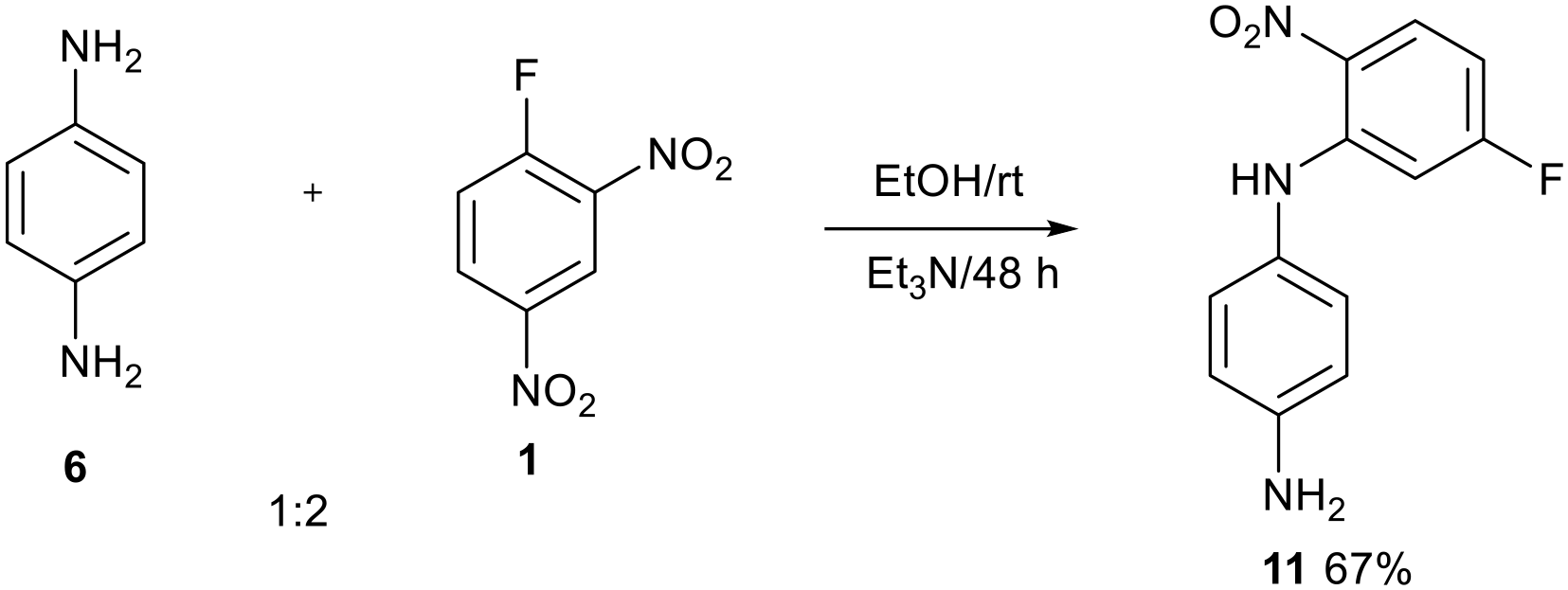
An experiment with p-phenylenediamine 6 and two equivalents of DNFB 1 at room temperature in EtOH in the presence of two equivalents of Et3N was conducted to minimise or avoid the reaction of EtOH with DNFB 1. An unexpected product 11 was formed in high yield (Figure 5). Compound 11 was not isolated previously from a column along with compounds 8-10 (Figure 4) but the column was complex in yellow colour and the failure to isolate compound 11 does not prove that none of it formed as a minor product.
Formation of unexpected compound 11.
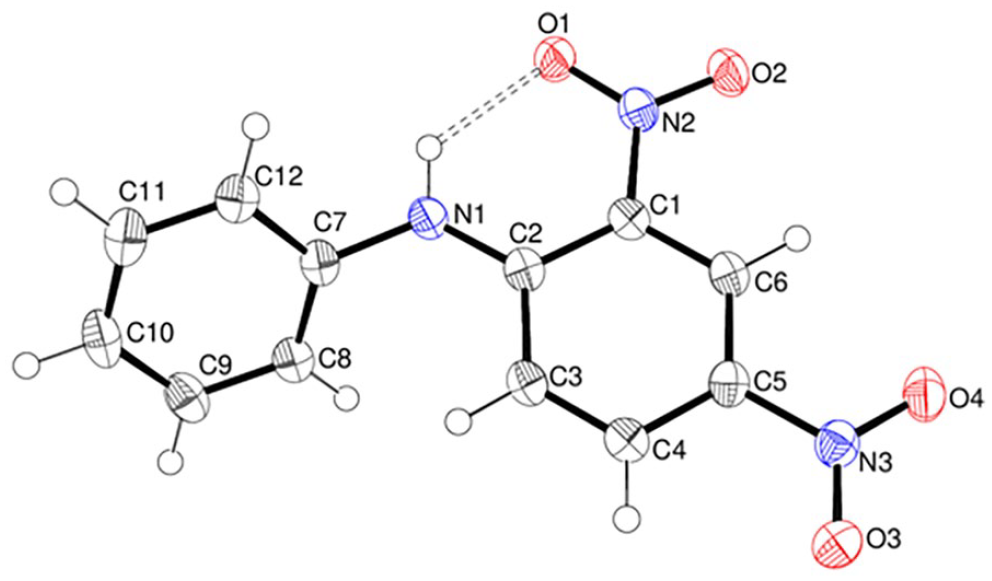
Compound 11 crystallises in the monoclinic space group P21/c with two molecules in the asymmetric unit (Figure 6). In the N1 molecule the dihedral angle between the aromatic rings is 54.29 (16)° and the equivalent angle in the N4 molecule is 51.22 (15)°. The nitro group is close to coplanar with its attached ring in both molecules [dihedral angles of 5.23 (7) and 8.46 (8) for the N1 and N4 molecules, respectively]. Each molecule features an intramolecular N – H···O hydrogen bond [H···O = 1.94 Å, N – H···O = 133° for the N1 molecule and H···O = 1.93 Å, N – H···O = 133° for the N4 molecule]. In the crystal, hydrogen-bonded (010) layers arise from N – H···O hydrogen bonds formed by the N2 and N5 terminal amine groups as well as weak N – H···F links arising from the N1 and N4 secondary amine groups.
The molecular structure of 11 showing 50% displacement ellipsoids. Hydrogen bonds are indicated by double-dashed lines.
Only a mono coupled product formed so the second primary amine is less reactive. A fluorine atom was clearly present from the NMR spectra as it couples with hydrogen and carbon. The primary amine of p-phenylenediamine 6 has reacted at an unsubstituted position of DFNB 1 and eliminated a nitro group as nitrous acid with Et3N. An addition–elimination mechanism is drawn in Figure 7.
A mechanism for the formation of unexpected product 11.
This is an unusual way for the reaction proceed. Et3N is present when the reaction was done at a low and high temperature (Figure 7) so is probably not changing the reaction pathway. p-Phenylenediamine behaves like a stronger nucleophile, owing to repulsion between the nitrogen lone pairs, a higher energy HOMO, and can drive the alternative reaction pathway. The para nitro group is eliminated rather than the ortho nitro group which is more sterically crowded. Both the fluorine substituted site and the unsubstituted site are activated by two nitro groups. The product is still bright yellow so it would be detectable if any of it formed with an amino acid or peptide chain.
One possible explanation for the experimental outcome might be the strength of the carbon–fluorine bond which is very strong. Related addition–elimination reactions are also known for 1,3-dinitrobenzene14,15 under oxidative conditions and the nitration of furan in acetic anhydride16,17 which is the least aromatic of the π-excessive heterocycles. A nitro group is not eliminated though. The displacement of hydrogen, rather than a halogen, has previously been called vicarious nucleophilic substitution where the leaving group is attached to the nucleophile.18,19
DNFB 1 was reacted with aniline 14 to compare with the products formed from p-phenylenediamine 6 (Figure 8). The expected and known substitution product 15 was formed20 in which the fluorine atom has been displaced. No other products were formed in significant amounts. The extra equivalent of aniline binds to the HF rather than using Et3N to do this.
The reaction of aniline 14 with DNFB 1 to form 15.
The asymmetric unit of 15 (space group P21/n) consists of one molecule (Figure 9) in which the dihedral angle between the aromatic rings is 50.32 (5)°, in agreement with the crystal structure of a sample of 15 which has been reported (Cambridge Structural Database reference code TAMHEX).21 It was prepared here under controlled, comparable conditions for a comparison with structure 11.
The molecular structure of compound 15 showing 50% displacement ellipsoids.
The amino acids glycine 16 and (R)-phenylglycine 17 (Figure 10) were reacted with Sanger’s reagent 1 under comparable conditions to see if any unexpected addition product formed (Figures 5 and 7).22,23 Only the expected displacement product was formed with these two amino acids (thin-layer chromatography (TLC) 50% DCM:50% MeOH and 400 MHz NMR) (Figures 11 and 12).
Glycine and (R)-phenylglycine.
A protocol for reacting amino acids with Sanger’s reagent 1 under controlled comparable conditions, used in this work, followed by chromatography.1
Compound formed from reacting Sanger’s reagent with phenylglycine.
Conclusion
p-Phenylenediamine 6 reacts with DNFB 1 in hot ethanol to give the expected products 8 and 9, but in ethanol at room temperature, an unexpected product 11 forms. The expected products form by displacement of fluorine and the unexpected product forms by the displacement of hydrogen from an unsubstituted position. Both sites of attack are activated by two nitro groups. The fluorine is strongly electron withdrawing but the carbon–fluorine bond is very strong, which might influence the outcome. A mechanism is drawn which rationalises how the reaction can proceed. It is an addition–elimination mechanism in which the negatively charged complex must protonate first before the nitro group is eliminated. Either nitro group could eliminate depending upon where the intermediate anion protonates. The energy barrier leading to the more stable product is higher as it requires a higher temperature to form. The reactions of p-phenylenediamine 6 and aniline 14 with DNFB 1 were compared and showed that aniline 14 gave the expected product. The product is still bright yellow so would act as a label if this reaction had occurred in the Sanger’s method. However, preliminary studies have shown that amino acids react with DNFB 1 entirely as expected. Compound 11 is unknown by a Reaxy’s search.
Experimental
Infrared spectra were recorded on a diamond-attenuated total reflection (ATR) Fourier transform (FTIR) spectrometer. Ultraviolet (UV) spectra were recorded using a Perkin Elmer Lambda 25 UV-Vis spectrometer with EtOH as the solvent. The term sh means shoulder. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at 400 and 100.5 MHz, respectively, using a Varian 400 spectrometer. Chemical shifts, δ, are given in ppm and using a solvent peak as an internal reference. Coupling constants, J, are given in Hz. High-resolution mass spectra were obtained at the University of Wales, Swansea, using an Atmospheric Solids Analysis Probe (ASAP) (positive mode) instrument: Xevo G2-S ASAP. Melting points were determined on a Kofler hot-stage microscope.
p-Phenylenediamine (500 mg, 4.6 mmol) and Et3N (0.94 g, 9.3 mmol) in EtOH (50 mL) were mixed with 2,4-dinitrofluorobenzene 1 (1.72 g, 9.2 mmol) and heated at 50 °C for 24 h.24 After cooling the mixture was diluted with water (200 mL), extracted with DCM (100 mL), dried over MgSO4 and concentrated in vacuo. TLC analysis with DCM showed that two yellow products were present and a spot running just ahead of the starting material 2,4-dinitrofluorobenzene 1 which was the first title compound10 (146 mg, 15%) m.p. 86–87 °C δH (400 MHz; CDCl3) 1.57 (3H, t, J = 8.0), 4.34 (2H, q, J = 8.0), 7.22 (1H, d, J = 8.0, B part of AB system), 8.44 (1H, d, J = 8.0, A part of AB system) and 8.74 (1H, s); δC (100.1 MHz; CDCl3) 14.2, 66.8, 114.3, 121.6, 129.1, 138.9, 140.0 and 156.6; Elution with DCM gave the second title compound8 (163 mg, 8%) as bright red crystals, m.p. > 225 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 220 (log ε 3.5) and 340(3.2); νmax (diamond)(cm−1) 3282w, 3088w, 1613m, 1584m, 1501s, 1421m, 1318m, 1275m, 1216m, 937m, 858m, 823s, 739s, 631s and 495s; δH (400 MHz; D7DMF) 7.45 (2H, d, J = 8.0, B part of AB system), 7.69 (4H, s), 8.35 (2H, d, J = 8.0, A part of AB system), 9.03 (2H, s) and 10.34 (2H, s, br); δC (100.1 MHz; CDCl3) 117.4, 123.5, 126.9, 129.9, 131.9, 136.5, 137.2 and 146.9; m/z (Orbitrap ASAP) 441.0793 (M+ + H, 100%) C18H12N6O8H requires 441.0795. Elution with DCM gave the third title compound9 (178 mg, 14 %) as crystals, m.p. 184–185 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 237 (log ε 3.7) and 356 (3.5); νmax (diamond)(cm−1) 3423w, 3349w, 3266w, 1614s, 1577s, 1511s, 1486s, 1413s, 1329s, 1300s, 1250vs, 1220vs, 1142s, 1122s, 1059s, 917s, 837s, 746s, 699s, 653s, 515s and 495s; δH (400 MHz; CDCl3) 3.88 (2H, s, br), 6.78 (2H, d, J = 8.0, B part of AB system), 7.04 (1H, d, J = 8.0, B part of AB system), 7.08 (2H, d, J = 8.0, A part of AB system), 8.15 (1H, d, J = 8.0, A part of AB system), 9.19 (1H, s) and 9.84 (1H, s, br); δC (100.1 MHz; CDCl3) 116.0, 116.1, 124.2, 126.9, 127.4, 129.7, 130.5, 136.8, 146.3 and 148.3; m/z (Orbitrap ASAP) 275.0786 (M+ + H, 100%) C12H10N4O4H requires 275.078
The reaction was repeated with p-phenylenediamine 6 (100 mg, 0.93 mmol) and Et3N (187 mg, 1.85 mmol) in iPrOH (30 mL) with 2,4-dinitrofluorobenzene 1 (345 mg, 1.85 mmol) and heated at 70 oC for 20 h. After cooling the mixture was diluted with water (180 mL), left standing for 1 h, filtered with a sinter No 4 and washed with water (10 mL). This gave the second title compound8 as a crude product which was air dried (363 mg, 89%). This product is contaminated with a small amount of 2,4-dinitro-p-phenylenediamine 9, which is hard to remove because of the poor solubility of the product. This was identified by TLC and comparison to a standard.
2-Nitro-5-fluorophenyl-p-phenylenediamine 11
p-Phenylenediamine 6 (200 mg, 1.85 mmol) and Et3N (0.374 g, 3.7 mmol) in EtOH (50 mL) were mixed with 2,4-dinitrofluorobenzene 1 (0.689 g, 3.7 mmol) and stirred in EtOH (50 mL) at room temperature for 48 h. The mixture was diluted with water (200 mL) and filtered. The precipitate was dissolved in DCM (200 mL), dried over MgSO4, filtered and evaporated to give a crude product (380 mg, 83%). This was purified by chromatography on silica. Ether eluted the title compound (308 mg, 67 %) as red/brown crystals, m.p. 167–168 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 220 (log ε 4.0), 277(sh) (3.7) and 412 (3.4); νmax (diamond)(cm−1) 3434w, 3358w, 3321w, 1621s, 1576s, 1496s, 1456s, 1399s, 1250s, 1189s, 1126s, 1078s, 991s, 851s, 827s, 749s, 588s and 570s; δH (400 MHz; CDCl3) 3.82 (2H, s), 6.58 (1H, dtd, J = 12.0 and 3.0), 6.60 (1H, dd, J = 12.0 and 3.0), 6.76 (2H, d, J = 8.0, B part of AB system), 7.06 (2H, d, J = 8.0, A part of AB system), 8.25 (1H, dd, J = 12.0 and 8.0) and 9.49 (1H, s, br); δC (100.1 MHz; CDCl3) 101.2 (d, F-C-C, J = 20.0), 105.2 (F-C-C, J = 20.0), 116.0, 127.5, 128.3, 128.4, 129.7 (d, C-C-C-F, J = 10.0), 145.3, 147.0 and 167.1 (d, C-F, J = 27.0); m/z (Orbitrap ASAP) 248.0839 (M+ + H, 100%) C12H10N3O2F+H requires 248.0835
2,4-Dinitrophenylaniline 15
2,4-Dinitrofluorobenzene 1 (200 mg, 1.1 mmol) in EtOH (15 mL) was mixed with aniline (200 mg, 2.2 mmol) for 3 h at room temperature.25 The precipitate was filtered and air dried to give the title compound (130 mg, 46 %) as orange/red crystals, m.p. 158–159 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 227 (log ε 4.0) and 351(4.1); νmax (diamond)(cm−1) 3315w, 1616s, 1595s, 1580s, 1516s, 1494s, 1419m, 1321s, 1251s, 1222s, 1144s, 1121s, 1058s, 921s, 741vs, 683s, 625s, 508s and 492s; δH (400 MHz; CDCl3) 7.19 (1H, d, J = 8.0), 7.34 (2H, d, J = 8.0), 7.41 (1H, t, J = 8.0 and 8.0), 7.53 (2H, t, J = 8.0 and 8.0), 8.19 (1H, dd, J = 8.0 and 2.0), 9.19 (1H, d, J = 2.0) and 10.0 (1H, s, br); δC (100.1 MHz; CDCl3) 116.1, 124.1, 125.6, 127.7, 129.9, 130.3, 131.1, 136.7, 137.5 and 147.1; m/z (Orbitrap ASAP) 260.0676 (M+ + H, 100%) C12H9N3O4H requires 260.0671
Amino acids with DNFB 1
General procedure
2,4-Dinitrofluorobenzene 1 (200 mg, 1.0 mmol), an amino acid ( 1 mmol) and Et3N (218 mg, 2.0 mmol) in EtOH (20 mL) were mixed and stirred at room temperature for 4 h.1,22,23 The glycine is a suspension which slowly dissolves. The solution turns yellow. The mixture was diluted with water (180 mL), treated with dilute aq. HCl (approx. 10 mL, 5.0 M) and extracted repeatedly with dichloromethane (50 mL, × 5). The combined extracts were dried over MgSO4, filtered or decanted and evaporate to dryness. The products were purified by chromatography on silica. Ether eluted unused DNFB 1 first. The amino acid adduct, 19 or 20, was eluted with Et2O/MeOH (50:50). They were identified and their purity confirmed by their carbon 13 spectrum. DNFB Glycine adduct 19 (115 mg, 48 %); δC (100.1 MHz; CDCl3) 47.5, (CH2)116.7, 123.6, 129.5, 130.4, 134.8, 147.5 and 170.6 (CO2H) DNFB (R)-Phenylglycine adduct 20 (111 mg, 35%); δC (100.1 MHz; CDCl3) 60.6 (CH2), 116.5, 123.8, 127.2, 128.2, 129.1, 130.3, 130.6, 135.8, 138.1, 146.6 and 171.5 (CO2H).
Crystal structure determinations
The crystal structure of compound 8 (red prism, 0.38 × 0.10 × 0.05 mm, recrystallised from dichloromethane:light petroleum ether), compound 11 (red plate, 0.18 × 0.13 × 0.05 mm, recrystallised from dichloromethane:light petroleum ether) and compound 15 (orange rod 0.32 × 0.05 × 0.03 mm, recrystallized from dichloromethane:light petroleum ether) were established using intensity data collected on a Rigaku CCD diffractometer (Cu Kα radiation, λ = 1.54178 Å) at 100 K. The structures were routinely solved by dual-space methods using SHELXT26 and the structural models were completed and optimised by refinement against |F|2 with SHELXL-2018.27 For compounds 8 and 15, the N-bound hydrogen atoms were found in difference maps and their positions were freely refined. The N-bound H atoms in compound 11 (N – H = 0.88 Å) and the C-bound H atoms in all structures were placed geometrically (C – H = 0.95 Å) and refined as riding atoms. The constraint Uiso(H) = 1.2Ueq(carrier) was applied in all cases. Full details of the structures and refinements are available in the deposited cifs. The crystal of compound 12 was found to be non-merohedrally twinned and data quality is poor, resulting in rather high R-factors, but the structure has been unambiguously determined.
Crystal data for compound 8 (C18H12N6O8): Mr = 440.34, monoclinic, space group P21/n (No. 14), a = 6.8428 (2) Å, b = 7.7513 (3) Å, c = 16.1753 (5) Å, β = 92.032 (3)°, V = 857.41 (5) Å3, Z = 2, T = 100 K, μ = 1.189 mm–1, ρcalc = 1.706 g cm–3, 8260 reflections measured (10.94 ⩽ 2θ ⩽ 148.7°), 1708 unique (Rint = 0.025), R(F) = 0.033 [1616 reflections with I > 2σ(I)], wR(F2) = 0.087 (all data), Δρmin, max (e Å–3) = −0.25, +0.30, CCDC deposition number 2268861.
Crystal data for compound 11 (C12H10FN3O2): Mr = 247.23, monoclinic, space group P21/c (No. 14), a = 21.8046 (9) Å, b = 12.6354 (6) Å, c = 7.9040 (4) Å, β = 90.469 (4)°, V = 2177.56 (18) Å3, Z = 8, T = 100 K, μ = 0.996 mm–1, ρcalc = 1.508 g cm–3, 4405 reflections measured (4.05 ⩽ 2θ ⩽ 135.4°), data not merged due to twinning [refined twin components 0.829 (5):0.171 (5)], R(F) = 0.104 [4006 reflections with I > 2σ(I)], wR(F2) = 0.308 (all data), Δρmin, max (e Å–3) = −0.56, +1.09, CCDC deposition number 2268862.
Crystal data for compound 15 (C12H9N3O4): Mr = 259.22, monoclinic, space group P21/n (No. 14), a = 3.77300 (10) Å, b = 11.0278 (4) Å, c = 26.8121 (11) Å, β = 90.420 (4)°, V = 1115.56 (7) Å3, Z = 4, T = 100 K, μ = 1.011 mm–1, ρcalc = 1.543 g cm–3, 38625 reflections measured (6.59 ⩽ 2θ ⩽ 150.3°), 2275 unique (Rint = 0.091), R(F) = 0.057 [2002 reflections with I > 2σ(I)], wR(F2) = 0.175 (all data), Δρmin, max (e Å–3) = −0.29, +0.44, CCDC deposition number 2268863.
Supplemental Material
sj-docx-1-chl-10.1177_17475198231212673 – Supplemental material for Expected and unexpected products from reacting Sanger’s reagent with p-phenylenediamine
Supplemental material, sj-docx-1-chl-10.1177_17475198231212673 for Expected and unexpected products from reacting Sanger’s reagent with p-phenylenediamine by Michael John Plater and William TA Harrison in Journal of Chemical Research
Footnotes
Acknowledgements
We thank the UK EPSRC National Mass Spectrometry Service Centre for mass spectrometric data and the UK National Crystallography Centre (University of Southampton) for the X-ray data collections. M.J.P. performed all synthesis and obtained the characterisation data and WTA Harrison solved the crystallographic data sets. Data sets were obtained free of charge from the National Crystallography Centre, University of Southampton.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
ORCID iD
Michael John Plater
Supplemental material
Supplemental material for this article is available online.
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.