2-Aminobenzylaniline was ditosylated and gave an unexpected product N,N-ditosylamino-2-benzylaminobenzene in which the primary amine had ditosylated, and the benzylamine was unreacted. The benzylamine, although more electron rich, is sterically crowded and less nucleophilic than the primary amine in this ditosylated system. Sterically crowded tosylamides were prepared by reacting o-phenylenediamine, p-phenylenediamine and 1,8-diaminonaphthalene with tosyl chloride.
o-Phenylenediamine 1 is readily monotosylated1–3 with 1 equiv. of tosyl chloride 2, to give compound 3, or ditosylated4,5 with 2 equiv. of tosyl chloride 2 in hot pyridine at 120 °C to give compound 4 (Scheme 1). A number of their uses are discussed in the following sections.
The tosylation of o-phenylenediamine 1 in hot pyridine at 120 °C/microwave 4 min.
Condensation with ethylglyoxalate in toluene gave compound 5, which was oxidatively cylised into the benzimidazole derivative 6 (Scheme 2).1
An oxidative cyclisation forming benzimidazole derivative 6.1
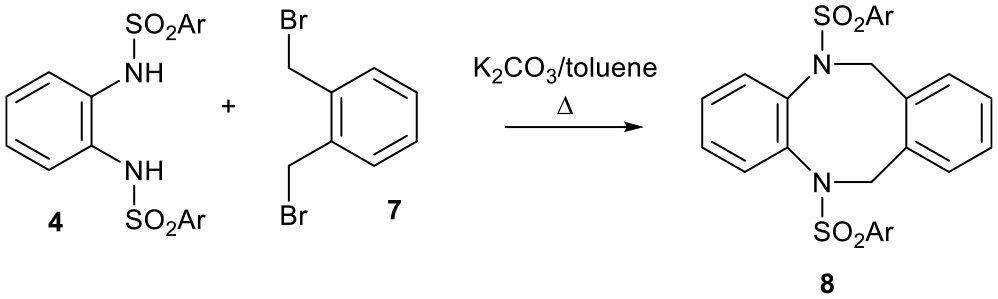
Ditosylate 4 condenses with α,α′-dibromo-o-xylene 7 and K2CO3 to give an unusual eight-membered ring heterocycle 8 and various derivatives, which were investigated for their biological properties (Scheme 3).6
Formation of an eight-membered ring heterocycle from ditosylate 4 and α,α′-dibromo-o-xylene 7.6
Ditosylate 4 condenses with 1,2-dibromopropanol 9 with sodium/EtOH to form the six-membered heterocycle 10 and various derivatives (Scheme 4).7
Ditosylate 4 condenses with SiCl4 to form a bis-spirocyclic heterocycle 11, which is bonded around the central silicon atom (Scheme 5).8 This utilises the subtending bite angle of the two N atoms and the large silicon size, and orthogonal bonding prevents steric crowding. Metal complex 12 is a typical and obvious application treating ditosylate 4 as a bidentate ligand.4
Formation of herocycle 118 and metal-ion complex 12.4
Discussion
Compound 4 was of interest for a new method of phenazine synthesis, and its preparation led to this programme of research.9,10 Initial studies focussed on repeating the syntheses of monotosylate 3 and ditosylate 4 by treating o-phenylenediamine 1 with either 1 or 2 equiv. of tosyl chloride 2 in hot pyridine at 100 °C, respectively. This works well, and only the ditosylate 4 forms using 3 equiv. of tosyl chloride 2 in pyridine. The Rf values of compounds 3 and 4 are identical with DCM as the eluent. The new compound o-phenylenediamine tritosylate 13 was formed in DCM with Et3N as the base. An X-ray single-crystal structure was obtained for compounds 3,1–344–5 and 13. The crystal structures of compounds 3 and 4 are in good agreement with previously published results11,12 (Cambridge Structural Database13 ref. codes UCUFOO and LAQDAJ, respectively).
There are two molecules in the asymmetric unit of compound 13, which crystallises in the triclinic space group P. In the C1 molecule (Figure 1), the dihedral angle between the central C1–C6 ring and the pendant C7–C12, C14–C19 and C21–C26 rings are 24.1° (2°), 31.9° (2°) and 70.03° (17°), respectively: The corresponding dihedral angles in the C28 molecule are 14.3° (3°), 28.9° (2°) and 84.29° (14°), respectively. The key torsion angles for the C1 molecule are C1–N1–S1–C7 [103.1° (4°)], C1–N1–S2–C14 [69.1° (4°)] and C2–N2–S3–C21 [52.7° (5°)], and the equivalent data for the C28 molecule are 123.3° (3°), 60.6° (4°) and 65.4° (4°), respectively. All this indicates that the C1 and C28 molecules have different conformations as shown in the overlay plot in Figure 2. In both molecules, the sulfonamide hydrogen atom is evidently too sterically crowded to form a hydrogen bond, and some weak intermolecular C–H···O links may help to consolidate the packing.
The C1 molecule in compound 13 (57% yield) showing 50% displacement ellipsoids.
Overlay plot of the C1 (red) and C28 (blue) molecules in the crystal of compound 13, with the C1–C6 and C28–C33 rings superimposed.
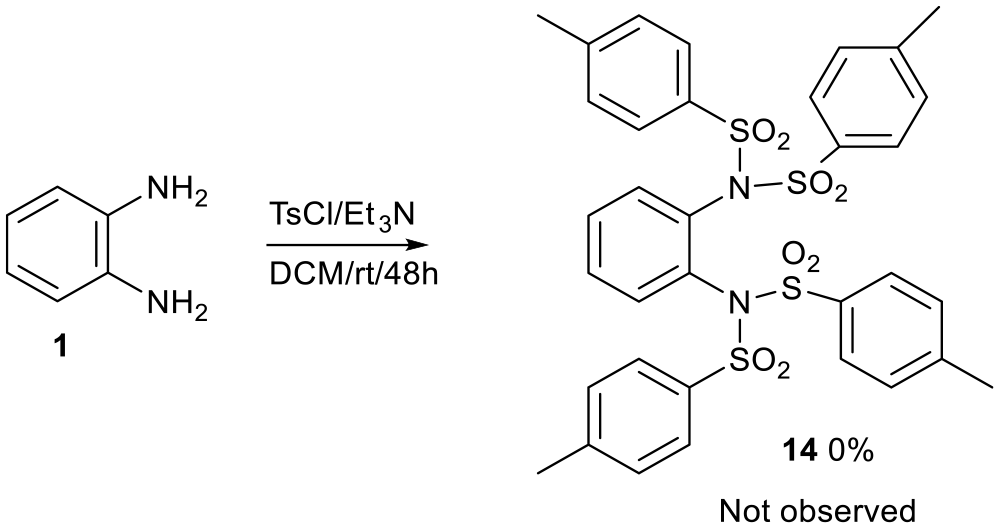
A more crowded tetratosyl-o-phenylenediamine derivative 14 did not form even when a reaction was performed with 4 equiv. of tosyl chloride 2 and o-phenylenediamine 1 (Scheme 6), presumably because compound 14 is too sterically crowded.
Tetratosyl-o-phenylenediamine derivative 14 which failed to form.
A successful synthesis of N,N-ditosylamino-2-benzylaminobenzene 16 was achieved by tosylating 2-aminobenzylaniline 1514 with 3 equiv. of tosylchloride (Scheme 7). This result was unexpected because the benzylamino group was expected to be more electron-rich and reactive towards tosyl chloride. The structure must be too crowded to form a tritosyl derivative. In the theoretical equilibrium shown in Scheme 8, compound 16 must be more stable than the unknown compound 17. Steric crowding by the benzyl group effectively makes the benzylamino group less nucleophilic in this system, and the primary amine is more nucleophilic. The nuclear magnetic resonance (NMR) data, both proton and carbon, easily distinguished compound 16 from compound 17.
The formation of N,N-ditosylamino-2-benzylaminobenzene 16.
Theoretical equilibrium between two ditosylated derivatives of 2-benzylaminoaniline 15.
The tosylation of 1,8-diaminonaphthalene 18 gave a mixture of two products, a tritosyl derivative 19 and a ditosyl derivative 20 (Scheme 9). An X-ray single-crystal structure determination was performed on compound 19.
The synthesis of tri- and di-tosylated-1,8-diaminonaphthalenes 19 and 20.
Compound 19 crystallises with one molecule in the asymmetric unit (Figure 3) in the orthorhombic space group Pbca. The dihedral angles between the C1–C10 naphthalene ring mean plane (root-mean-square deviation = 0.005 Å) and the C11–C16, C18–C23 and C25–C30 phenyl rings are 52.08° (3°), 14.40° (5°) and 84.41° (4°), respectively. The C1–N1–S1–C11 torsion angle is −56.40° (11°) with corresponding values of 94.13° (10°) for C1–N1–S2–C18 and 57.15° (13°) for C9–N2–S3–C25. An intramolecular N2–H1···N1 hydrogen bond with H···N = 2.177 (19) Å and N–H···N = 135.6° (16°) occurs [compare the C2–C1–N1–S1 and C2–C1–N1–S2 torsion angles of 78.86° (13°) and −84.24° (13°), respectively], and a weak C8–H8···O6 bond may help to establish the conformation.
The molecular structure of compound 19 showing 50% displacement ellipsoids with hydrogen bonds indicated by double-dashed lines.
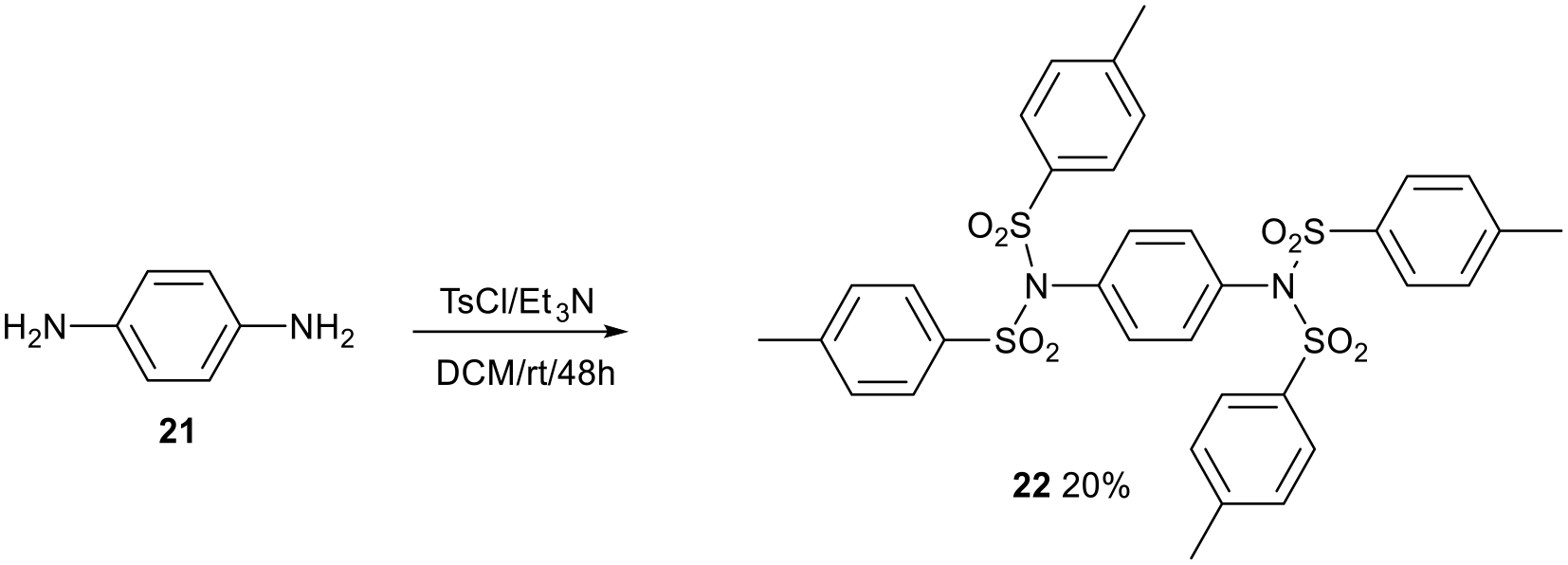
The synthesis of a tetratosylated derivative of p-phenylenediamine 21 is shown in Scheme 10. Less crowding occurs around each sulfonamide, so the synthesis is as expected. The compound is soluble in DCM. These molecules may find applications as rotamers as they are big enough to attach to a surface non-covalently and move across it.
The synthesis of N,N,N′,N′-tetratosyl-1,4-diaminobenzene 22.
Treatment of o-phenylenediamine 1 with 3 equiv. of (1S)-(+)-10-camphorsulfonyl chloride 23 gave the trifunctionalised derivative 24 (Scheme 11). This compound is waxy, and no X-ray single-crystal structure was obtained for it. The aliphatic proton NMR was too complicated to be interpreted with many overlapping signals, but the aromatic region showed two doublets and two triplets as expected. The carbon-13 spectrum was well resolved. The signals appeared in groups of three, which showed that three different camphor groups were present. There were six methyl groups, six aryl carbons and three carbonyl groups (Supplemental Material). This observation suggests that compound 24 is sterically crowded with the two different camphoryl groups attached to the same N atom. Restricted rotation around the Ar–N bond might explain this if it is slow on an NMR timescale.
The synthesis of N,N,N′-tricamphoryl-1,2-diaminobenzene tentatively assigned as structure 24.
Conclusion
o-Phenylenediamine 1 was mono-, di- and tri-tosylated with tosyl chloride in either hot pyridine at 100 °C or DCM to give mono-, di- or tri-tosyl-o-phenylenediamines 3, 4 or 13, respectively. 1,8-Diaminonaphthalene 18 was di- and tri-tosylated to give the corresponding di- and tri-tosylated-1,8-aminonaphthalenes, respectively. 2-Aminobenzylaniline 15 was ditosylated and gave an unexpected product N,N-ditosylamino-2-benzylaminobenzene 16 in which the primary amine had ditosylated, and the benzylamine was unreacted. The benzylamine, although more electron-rich, is sterically crowded and less nucleophilic than the primary amine in this ditosylated system. The two possible ditosylated products 16 and 17 were easily distinguished by proton and 13C NMR. X-ray single-crystal structure determinations were performed on compounds 3, 4, 13 and 19. In no case did we observe tetratosylated derivatives from o-phenylenediamine 1 which are too sterically crowded to form. Tritosylate 19 displayed an intramolecular hydrogen bond between the two nitrogen atoms similar to that seen in the protonation of proton sponge.15–17 Compound 24 has three different camphoryl groups in it (see Supplemental Material).
Experiment
Infrared (IR) spectra were recorded on a diamond attenuated total reflection Fourier-transform IR spectrometer. Ultraviolet (UV) spectra were recorded using a PerkinElmer Lambda 25 UV-Vis spectrometer with EtOH as the solvent. The term ‘sh’ means shoulder. 1H and 13C NMR spectra were recorded at 400 and 100.5 MHz, respectively, using a Varian 400 spectrometer. Chemical shifts, δ, are given in ppm and measured by comparison with the residual solvent. Coupling constants, J, are given in Hz. The term ‘br’ stands for broad. High-resolution mass spectra were obtained at the University of Wales, Swansea, using an Atmospheric Solids Analysis Probe (ASAP) (positive mode) Instrument: Xevo G2-S ASAP. Melting points were determined on a Kofler hot-stage microscope.
N-(2-Aminophenyl)-4-methylbenzenesulfonamide 3
o-Phenylenediamine (255 mg, 2.36 mmol) and tosyl chloride (450 mg, 2.36 mmol) were mixed in CH2Cl2 at room temperature with Et3N (477 mg, 4.72 mmol) for 24 h. The organic layer was extracted with water (100 ml), then dried with MgSO4 and filtered. The desired product was purified by chromatography on silica gel. Elution with Et2O/CH2Cl2 (20:80) then Et2O/CH2Cl2 (50:50) gave the title compound (394 mg, 64%) as a white solid, m.p. 140–141 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 219 (log ε 4.3) and 291 (3.5); νmax (diamond)(cm–1) 3467w, 3385w, 3208w, 1622w, 1597w, 1497w, 1464w, 1316s, 1148vs, 1091s, 813w, 751s, 672vs, 532vs and 473s; δH (400 MHz; CDCl3) 2.41 (3H, s), 6.54 (2H, d, J = 4.0), 6.76 (1H, d, J = 8.0), 7.03 (1H, m), 7.23 (2H, d, J = 8.0) and 7.64 (2H, d, J = 8.0); δC (100.1 MHz; CDCl3) 21.7, 117.3, 118.9, 121.5, 127.5, 128.5, 128.8, 129.7, 136.0, 143.8 and 143.9; m/z (Orbitrap ASAP) 263.0843 (M + H+, 100%) C13H14N2O2S + H+ requires 263.0849.
o-Phenylenediamine 1 (0.5 g, 4.6 mmol) and tosyl chloride 2 (2.65 g, 13.9 mmol) were mixed in CH2Cl2 (50 ml) at room temperature with Et3N (1.4 g, 13.9 mmol) for 24 h.18–20 The organic layer was extracted with water (100 ml) then dried with MgSO4 and filtered. The desired product was purified by chromatography on silica gel. Elution with Et2O/CH2Cl2 (20:80) then Et2O/CH2Cl2 (50:50) gave the title compound (1.5 g, 57%) as a white solid, m.p. 158–159 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 240 (log ε 3.8); νmax (diamond)(cm–1) 3375w, 1596w, 1492w, 1376s, 1158s, 1084w, 902s, 855w, 810w, 666vs, 546vs, 499w and 487w; δH (400 MHz; CDCl3) 2.41 (3H, s), 2.50 (6H, s), 6.65 (1H, dd, J = 8.0 and 4.0), 6.88 (1H, td, J = 8.0 and 4.0), 7.26-7.32 (3H, m), 7.38 (4H, d, J = 8.0), 7.44 (1H, dd, J = 8.0 and 4.0), 7.83 (4H, d, J = 8.0) and 7.85 (2H, d, J = 8.0); δC (100.1 MHz; CDCl3) 21.9, 117.8, 122.6, 123.0, 127.6, 129.0, 129.7, 129.9, 131.5, 132.7, 135.3, 136.5, 137.5, 144.1 and 146.0; m/z (Orbitrap ASAP) 588.1302 (M + NH4+, 100%) C27H26N2O6S3 + NH4+ requires 588.1297.
N,N-Ditosyl-1-amino-2-benzylaminobenzene 16
N-Benzyl-o-phenylenediamine 15 was prepared by a literature method.9 N-Benzyl-o-phenylenediamine 15 (200 mg, 1.0 mmol) in DCM (50 ml) was mixed with tosyl chloride (578 mg, 3.0 mmol) and Et3N (306 mg, 3.0 mmol) for 24 h. The organic layer was extracted with water (100 ml), then dried with MgSO4 and filtered. The desired product was purified by chromatography on silica gel. Elution with Et2O/CH2Cl2 (20:80) and then Et2O/CH2Cl2 (50:50) gave the title compound (290 mg, 57%) as an off-white solid, m.p. 151–152 °C (from dichloromethane:light petroleum ether). The product is an oil, which slowly crystallises over a few days with scratching. λmax (EtOH)/nm 316 (log ε 2.9) and 231sh (3.7); νmax (diamond)(cm–1) 1595w, 1507w, 1453w, 1350s, 1366s, 1161s, 1120s, 1161s, 1120w, 1080w, 890s, 839w, 817w, 725w, 694w, 662w, 564s and 536s; δH (400 MHz; CDCl3) 2.46 (6H, s), 4.27 (2H, d, J = 4.0), 4.64–4.69 (1H, br, NH), 6.63 (1H, t, J = 8.0 and 8.0), 6.72 (2H, m), 7.29 (5H, d, J = 8.0), 7.35–7.42 (5H, m) and 7.94 (4H, d, J = 8.0); δC (100.1 MHz; CDCl3) 21.8, 47.6, 112.5, 116.6, 119.5, 127.2, 127.3, 128.7, 128.9, 129.7, 131.8, 132.7, 136.4, 138.6, 145.4 and 147.6; m/z (Orbitrap ASAP) 507.1410 (M + H+, 100%) C27H27N2O4S2 requires 507.1412 (M + H+)
N-(8-{bis[(4-Methylphenyl)sulfonyl]amino}-1-naphthyl)-4-methylbenzenesulfonamide 19 and N,N′-naphthalene-1,8-diylbis(4-methylbenzenesulfonamide) 20
1,8-Diaminonaphthalene 18 (500 mg, 3.2 mmol) in DCM (50 ml) was mixed with tosyl chloride (2.42 g, 12.8 mmol) and Et3N (1.28 g, 12.8 mmol) for 48 h.21–23 The organic layer was extracted with dilute aqueous 2 M HCl (100 ml), then dried with MgSO4 and filtered. The desired product was purified by chromatography on silica gel. Elution with Et2O/CH2Cl2 (20:80) and then with Et2O/CH2Cl2 (50:50) gave the first title compound (931 mg, 47%) as an off-white solid, m.p. 210–211 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 320 (log ε 3.0) and 235 (3.8); λmax (diamond)(cm–1) 1595w, 1371s, 1349s, 1294s, 1187s, 1159s, 1084s, 1018s, 934s, 809s, 752w, 659s and 539s; δH (400 MHz; CDCl3) 2.21 (3H, s), 2.40 (6H, s), 6.75 (1H, d, J = 8.0), 7.09 (2H, d, J = 8.0), 7.14 (1H, t, J = 8.0 and 8.0), 7.21 (1H, t, J = 8.0 and 8.0), 7.30 (4H, d, J = 8.0), 7.39 (1H, d, J = 8.0), 7.47 (1H, d, J = 8.0), 7.76 (1H, d, J = 8.0), 7.80 (4H, d, J = 8.0), 7.81 (2H, d, J = 8.0) and 8.79 (1H, s); δC (100.1 MHz; CDCl3) 21.5, 22.0, 114.1, 122.1, 123.9, 124.5, 126.3, 128.2, 128.5, 129.4, 129.7, 130.3, 132.7, 132.8, 133.1, 133.9, 136.0, 136.5, 143.7 and 146.4; m/z (Orbitrap ASAP) 621.1187 (M + H+, 100%) C31H28N2O6S3H requires 621.1188 (M + H+), followed by the second title compound (122 mg, 8 %) as an off-white solid, m.p. 211–212 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 320 (log ε 3.5) and 225 (4.3); νmax (diamond)(cm–1) 3214s, 1597w, 1560w, 1454w, 1414w, 1341w, 1297s, 1150s, 1121w, 1092s, 1042s, 905w, 814s, 708s, 656s, 559s, 543 and 487s; δH (400 MHz; CDCl3) 2.38 (6H, s), 7.06 (2H, d, J = 8.0), 7.23 (4H, d, J = 8.0), 7.28 (2H, d, J = 8.0), 7.65 (2H, d, J = 8.0), 7.72 (4H, d, J = 8.0), 8.60 (2H, s); δC (100.1 MHz; CDCl3) 21.6, 123.4, 123.8, 125.6, 127.7, 128.2, 129.6, 131.3, 135.2, 136.2 and 144.3; m/z (Orbitrap ASAP) 467.1100 (M + H+, 100%) C24H22N2O4S2H requires 467.1099 (M + H+).
N,N,N′,N′-Tetratosyl-1,4-diaminobenzene 22
p-Phenylenediamine 21 (500 mg, 4.6 mmol) in DCM (50 ml) was mixed with tosyl chloride (3.53 g, 18.5 mmol) and Et3N (1.87 g, 18.5 mmol) for 48 h. The organic layer was extracted with dilute aqueous 2 M HCl (100 ml), then dried with MgSO4 and filtered. The desired product was purified by chromatography on silica gel. Elution with CH2Cl2/light petroleum ether (50:50) then CH2Cl2 gave the title compound (650 mg, 20%) as a white solid, m.p. 252–253 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 233 (log ε 3.4); νmax (diamond)(cm–1) 1596w, 1493w, 1377s, 1360s, 1158s, 1084s, 1017w, 928s, 811s, 702w, 677w, 657s, 606s and 546s; δH (400 MHz; CDCl3) 2.48 (12H, s), 7.03 (4H, s), 7.36 (8H, d, J = 8.0) and 7.82 (8H, d, J = 8.0); δC (100.1 MHz; CDCl3) 21.7, 128.6, 129.7, 132.3, 135.9, 136.2 and 145.2; m/z (Orbitrap ASAP) 725.1116 (M + H+, 100%) C34H32N2O8S4 requires 725.1120 (M + H+).
N,N,N′-Tricamphoryl-1,2-diaminobenzene 24
o-Phenylenediamine 1 (500 mg, 4.6 mmol) in DCM (50 ml) was mixed with (1S)-(+)-10-camphorsulfonyl chloride 23 (3.5 g, 14.0 mmol) and Et3N (1.4 g, 14.0 mmol) for 48 h. The organic layer was extracted with dilute aqueous 2 M HCl (100 ml) then dried with MgSO4 and filtered. The desired product was purified by chromatography on silica gel. Elution with Et2O/light petroleum ether (50:50) and then with Et2O gave the title compound (1.09 g, 31%) which solidified to a white, amorphous solid with scratching over 3 days. λmax (EtOH)/nm 222 (log ε 4.0) and 280 (3.3); νmax (diamond)(cm–1) 2958m, 1739s, 1494w, 1773s, 1351m, 1150s, 1051m, 886w, 850w, 756w, 720w, 599m, 564s, 525s and 498s; δH (400 MHz; CDCl3) (aromatic region) 7.19 (1H, t, J = 8.0), 7.43 (1H, d, J = 8.0), 7.46 (1H, t, J = 8.0), 7.83 (1H, d, J = 8.0) and 8.02 (1H, s, br, NH); δC (100.1 MHz; CDCl3) 19.6, 19.7, 19.71, 19.8, 19.9, 19.95 (6 methyl groups), 25.4, 26.1, 26.8, 26.9, 42.5, 42.7, 42.71, 42.8, 42.81, 42.9, 48.1, 48.2, 48.5, 51.8, 52.4, 53.7, 59.0, 59.1, 59.3, 122.4, 124.5, 124.8, 131.7, 132.0, 137.4 (6 aromatic carbons) 213.6, 214.3 and 215.6 (3 carbonyl groups) (two resonances are overlapping); m/z (Orbitrap ASAP) 751.2769 (M + H+, 5.0%) C36H50N2O9S3H requires 751.2757 (M + H+).
Crystal structures
The crystal structures of 3 (colourless needle, 0.61 × 0.03 × 0.03 mm, recrystallised dichloromethane:light petroleum ether), 4 (colourless needle 0.29 × 0.04 × 0.03 mm, recrystallised from dichloromethane:light petroleum ether), 13 (colourless lath, 0.30 × 0.04 × 0.01 mm, recrystallised from dichloromethane:light petroleum ether) and 19 (colourless prism, 0.22 × 0.20 × 0.06 mm, recrystallised from dichloromethane:light petroleum ether) were established using intensity data collected at 100 K on a Rigaku CCD diffractometer using Cu Kα radiation (λ = 1.54178 Å). The structures were routinely solved by dual-space methods using SHELXT,18 and the structural models were completed and optimised by refinement against |F|2 with SHELXL-2019.19 The N-bound hydrogen atoms were located in difference maps: For compound 3, they were refined as riding atoms in their as-found relative positions, and for compounds 4 and 13, their positions were freely refined. The carbon-bound hydrogen atoms were placed in idealised locations (C–H = 0.95–0.98 Å) and refined as riding atoms. The constraint Uiso(H) = 1.2Ueq (carrier) or 1.5Ueq (methyl carrier) was applied in all cases. The methyl groups were allowed to rotate, but not to tip, to best fit the electron density. Full details of the structures and refinements are available in the deposited crystallographic information files.
Crystal data for 3: C13H14N2O2S, Mr = 262.32, monoclinic, space group P21 (no. 4), a = 11.2901 (4) Å, b = 6.0529 (2) Å, c = 18.8537 (8) Å, β = 98.753 (3)°, V = 1273.42 (8) Å3, Z = 4, T = 100 K, μ = 2.231 mm–1, ρcalc = 1.368 g cm–3, 9542 reflections measured (8.6 ⩽ 2θ ⩽ 141.2°), R(F) = 0.058 [9086 reflections with I > 2σ(I)], wR(F2) = 0.173 (all data), Δρmin, max (e Å–3) = –0.49, +0.29, Flack absolute structure parameter –0.034 (16), refined as a non-merohedral twin, Cambridge Crystallographic Data Center (CCDC) deposition number 2277285.
Crystal data for 4: C20H20N2O4S2, Mr = 416.50, orthorhombic, space group Pccn (no. 56), a = 8.3251 (4) Å, b = 36.9411 (18) Å, c = 12.9160 (7) Å, V = 3972.2 (3) Å3, Z = 8, T = 100 K, μ = 2.681 mm–1, ρcalc = 1.393 g cm–3, 35539 reflections measured (4.8 ⩽ 2θ ⩽ 154.3°), 4140 unique (RInt = 0.060), R(F) = 0.055 [3695 reflections with I > 2σ(I)], wR(F2) = 0.136 (all data), Δρmin, max (e Å–3) = –0.67, +0.39, CCDC deposition number 2277286.
Crystal data for 13: C27H26N2O6S3, Mr = 570.68, triclinic, space group P (no. 2), a = 8.70715 (16) Å, b = 16.4892 (5) Å, c = 19.2569 (3) Å, α = 95.169 (2)°, β = 91.4000 (15)°, γ = 105.197 (2)°, V = 2653.93 (11) Å3, Z = 4, T = 100 K, μ = 2.943 mm–1, ρcalc = 1.428 g cm–3, 47498 reflections measured (4.6 ⩽ 2θ ⩽ 153.9°), 10495 unique (RInt = 0.089), R(F) = 0.086 [7617 reflections with I > 2σ(I)], wR(F2) = 0.243 (all data), Δρmin, max (e Å–3) = –0.89, +1.42, CCDC deposition number 2277287.
Crystal data for 19: C31H28N2O6S3, Mr = 620.73, orthorhombic space group Pbca (no. 61), a = 15.3534 (1) Å, b = 16.0086 (1) Å, c = 23.3294 (1) Å, V = 5734.05 (6) Å3, Z = 8, T = 100 K, μ = 2.774 mm–1, ρcalc = 1.438 g cm–3, 161236 reflections measured (7.6 ⩽ 2θ ⩽ 154.0°), 5874 unique (RInt = 0.053), R(F) = 0.031 [5710 reflections with I > 2σ(I)], wR(F2) = 0.083 (all data), Δρmin, max (e Å–3) = –0.39, +0.40, CCDC deposition number 2277288.
Supplemental Material
sj-docx-1-chl-10.1177_17475198231194866 – Supplemental material for A study of mono-, di- and tri-tosylated amines: An unexpected sulfonamide
Supplemental material, sj-docx-1-chl-10.1177_17475198231194866 for A study of mono-, di- and tri-tosylated amines: An unexpected sulfonamide by Michael John Plater and William T. A. Harrison in Journal of Chemical Research
Footnotes
Acknowledgements
We thank the UK EPSRC National Mass Spectrometry Service Centre for mass spectrometric data and the UK National Crystallography Centre (University of Southampton) for the X-ray data collections.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD
Michael John Plater
Supplemental material
Supplemental material for this article is available online.
References
1.
FuSJiangHDengY, et al. Adv Synth Catal2011; 353: 2795–2804.
2.
RivilloDGulyasHBenet-Buchholz Escudero-AdanEC, et al. Angew Chem Int Ed2007; 46: 7247–7250.
3.
BondarenkoNA.Russ J Gen Chem1996; 66: 584–589.
4.
OloyedeHOWoodsJAOGorlsH, et al. New J Chem2020; 44: 14849–14858.
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.