
Abstract
Chemical warfare agents, such as nerve agents (GD and VX) and blister agents (HD), have strong toxicities to mankind. In recent years, zirconium-based metal-organic frameworks have been found to be attractive materials for chemical warfare agent degradation. Among them, metal-organic framework-808 (MOF-808) and porous coordination network-222 (PCN-222) were the best. However, few papers pay attention to their practical application. In this work, we prepared MOF-808 and PCN-222 using water phase and organic solvothermal methods, respectively. Their performance for the catalytic degradation of chemical warfare agents under practical decontamination conditions was studied. The results showed that MOF-808 displayed a high potency for catalytic hydrolysis of VX (10,000 mg L−1) in unbuffered solution. PCN-222 exhibited weaker reactivity with a half-life (t1/2) of 28.8 min. Their different performances might stem from the different connectivity of the Zr6 nodes and framework structures. The results illustrated that the hydrolysis of high-concentration GD required a strong alkaline buffer to neutralize the hydrolysis product of hydrofluoric acid (HF) to avoid catalyst poisoning. When H2O2 was used as the oxidant instead of O2, both zirconium-based metal-organic frameworks performed with effective catalytic potency for HD degradation without any special lighting and so was suitable for practical application, whereas the products obtained from HD, such as HDO2 and V-HDO2, still possessed vesicant toxicity. Overall, MOF-808 prepared via a water-phase synthesis performed with effective catalysis for the degradation of high-concentration VX, GD, and HD with t1/2 of < 0.5, 3.1 and 2.2 min, respectively, exhibiting its potential for practical applications.

Introduction
Chemical warfare agents (CWAs), as weapons of mass destruction, have caused a large number of casualties since World War I due to their acute toxicities. 1 Among these agents, nerve agents, such as Soman (pinacolyl methylphosphonofluoridate, GD) and VX (O-ethyl-S-[2-(diisopropylamino)ethyl] methylphosphonothioate), and blister agents, such as sulfur mustard (bis-(2-chloroethyl)sulfide, HD), are the most toxic.2–4 Their structures are displayed in Figure 1. Although the Chemical Weapons Convention (CWC) was signed in 1993, CWAs have been used against civilian populations by terrorist organizations in recent decades. 5
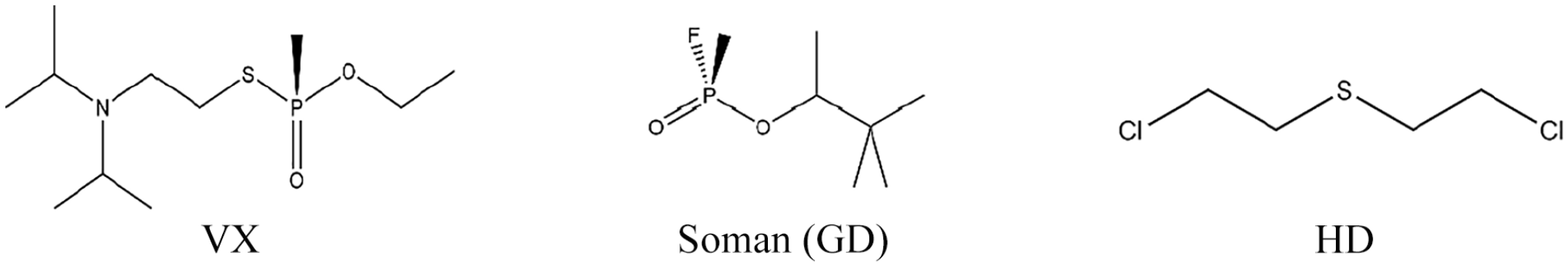
Chemical structure of VX, GD, and HD.
For this reason, various decontaminants were developed to destroy these agents rapidly.6–8 Typical decontaminants are broadly classified into two categories based on their reactivities. Some are oxidants, such as hypochlorite, 6 chloramines,6,9 BX24,6,10 and DF-200. 11 Others are nucleophilic reagents, for example, alkalis, 7 DS2,7,12 GD6, 6 and GDS2000. 7 Both types of decontaminants have high degradation efficiencies for CWAs. However, they are corrosive and unfriendly to the environment and so a few metal oxides, such as MgO, 13 CaO, 14 and Al2O3, 15 and metal hydroxides, including Zr(OH)416,17 and Ce(OH)4, 18 have been developed to degrade CWAs. In recent years, metal-organic frameworks (MOFs) with permanent porosities, exceptional surface areas, and functional tunability have caught the attention of researchers for CWA decontamination.8,19,20 In particular, zirconium-based metal-organic frameworks (Zr-MOFs) have shown useful catalytic activity toward the degradation of CWAs.3,21–25
Zr-MOFs comprised Zr6 clusters [Zr6(μ3-O)4(μ3-OH)4], secondary building units (SBUs), and carboxylate organic linkers held together via strong coordination bonds.3,23 The Lewis acidic Zr6(μ3-O)4(μ3-OH)4 and the SBUs provide catalytic sites for the catalytic hydrolysis of nerve agents. 23 By altering the organic linkers, Zr-MOFs with various pore sizes, node connectivities, and linker functionalities were obtained which influenced the reaction activities of the MOFs.3,23,25 The 12-connected UiO-66 was the first Zr-MOF used for the hydrolysis of nerve agents and their simulants. 26 It relied on structural defects as the catalytic sites with a t1/2 of 45 min for the hydrolysis of dimethyl (4-nitrophenyl) phosphate (DMNP, a simulant for nerve agents) in basic N-ethylmorpholine (NEM) solution. 23 The 12-connected UiO-67 with a larger pore size that facilitated nerve agent diffusion into the framework to provide access of to the Zr6 nodes delivered higher catalytic activity for DMNP hydrolysis with a t1/2 of 4.5 min.27,28 Zr-MOFs functionalized with basic amino-linker groups gave even better performance (e.g. UiO-66-NH2 and UiO-67-NH2) compared with unmodified ones.28,29 This was attributed to the fact that the amino groups affected subtle changes in the microsolvation environment around the active sites on the Zr6 nodes, ultimately resulting in increased nucleophilicity of the water molecules that attacked the organophosphorus substrate coordinated to the Zr6 node. 23 Halide-containing linkers also contributed to better performance of Zr-MOFs, due to the strong halogen-bonding interaction between an iodine atom and the methoxy group on organophosphorus nerve agents. 30 This higher catalytic potency was also achieved using 8-connected and 6-connected Zr-MOFs. NU-1000 with 8-connected Zr6 nodes and 1,3,6,8-tetrakis-p-benzoic acid-pyrene (H4TBAPy) linkers displayed a shorter t1/2 of 15 min for DMNP catalytic hydrolysis than UiO-66.23,31 Of note, MOF-808 built with lower connectivity Zr6 nodes (6-connected) and 1,3,5-benzenetricarboxylic acid possessed more available Zr6 nodes and larger pores for the catalytic hydrolysis of nerve agents and their simulants with t1/2 in NEM solution of less than 0.5 min.23,32–34 Overall, MOF-808 was the best catalyst for hydrolysis of nerve agents among Zr-MOFs.
However, the outstanding performance of the above Zr-MOFs was demonstrated with relatively low concentrations (1000–4000 mg L−1) of the nerve agents in basic NEM-buffered solutions that were used to avoid catalyst poisoning and low catalytic turnovers. 35 In practical implementation of decontamination, high concentrations of nerve agents are unavoidable. Thus, it is necessary to investigate the performance of Zr-MOFs for the degradation of nerve agents with high concentrations. In addition, MOF-808 was usually synthesized by solvothermal reactions that employed organic solvents and high temperatures. These solvents are toxic, flammable, and can decompose during the solvothermal progress resulting in increased cost compared with syntheses under aqueous conditions.
The degradation of HD via hydrolysis is challenging due to its low water solubility.27,34 Oxidative degradation is preferable. The Farha team developed a Zr-MOF comprising 8-connected Zr6 nodes and porphyrinic photosensitizer organic linkers, namely porous coordination network-222 (PCN-222).36,37 It not only catalyzed nerve agent hydrolysis but also could photo-oxidize the HD simulant, 2-chloroethyl ethyl sulfide (CEES), with a t1/2 of about 13 min under blue light. 38 However, the use of such specific lighting conditions would be difficult to realize in practical applications.
In this work, MOF-808, synthesized using aqueous conditions, and PCN-222, prepared by an organic solvothermal method, were synthesized and characterized. Their catalytic performance for the hydrolysis of nerve agents (VX and GD) with high concentration was investigated. To avoid the photocatalytic PCN-222 oxidation of blister agents, 35 we substituted O2 by the more active H2O2 oxidant and investigated the catalytic efficiency of PCN-222 and MOF-808 for HD degradation without the need for specific lighting, hence developing a practical strategy for HD decontamination with MOFs.
Results and discussion
Characterization of MOF-808 and PCN-222
The structural properties of MOF-808 and PCN-222 synthesized in this work were studied by Powder X-ray diffraction (PXRD), scanning electron microscope (SEM) and Brunauer–Emmett–Teller (BET). The PXRD patterns of our synthetic MOF-808 and PCN-222 indicated high crystallinity. The diffraction patterns of MOF-808 at 4.3°, 8.3°, 8.8°, and 10.1° and PCN-222 at 2.4°, 4.8°, and 7.1° matched well with the reported simulated patterns, Figure 2, demonstrating the successful synthesis of MOF-80833,39 and PCN-222. 40 It can be seen from Figure 3 that the SEM images depict particles with the anticipated octahedral morphology for MOF-808 with a uniform crystallite size of about 1 μm. Its apparent morphology was consistent with the literature, and its particle size was larger than the size of MOF-808 (about 0.2 μm) synthesized by the solvothermal method. 23 The PCN-222 displayed a rod-like morphology with a crystallite length of about 1–3 μm, in agreement with the reported synthetic material. 40 Strikingly, visible agglomeration was observed in the SEM image of PCN-222.

PXRD patterns of MOF-808 (a), PCN-222 (b) and their simulated patterns.

SEM images of MOF-808 (a) and PCN-222 (b).
N2 adsorption–desorption was performed to investigate the BET areas of MOF-808 and PCN-222, see Figure 4. The BET analysis for MOF-808 illustrated a specific surface area (SBET) of 1344 m2 g−1 with a pore volume of 0.82 cm2 g−1, which was smaller than the SBET of dimethylformamide (DMF)-synthesized MOF-808 (1962 m2 g−1). 34 The lack of further activation using high temperature and vacuum conditions was probably responsible for this slightly lower SBET. The pore size distribution of MOF-808 was mainly about 18 Å, in conformity with the solvothermal synthesis. 39

N2 adsorption–desorption isotherms of (a) MOF-808 and (b) PCN-222 at 77 K.
The SBET of PCN-222 was about 1625 m2 g−1 with a pore volume of 1.22 cm2 g−1, and pore width of 11 and 31 Å, respectively. Its surface area was lower than the reported synthesis (2223 m2 g−1), 40 possibly due to less thorough cleaning in the post-synthetic procedure, leaving unreacted guest molecules in the channel of the framework of PCN-222 and the agglomeration of the crystallite structure.
VX degradation
The degradation of high-concentration VX (about 10 g L−1) with the two prepared Zr-MOFs was performed in the absence of alkaline buffers. Without the MOFs, the hydrolysis of VX in an acidic solution with a pH of about 5 was extremely slow, with a t1/2 of about 2400 h (Table 1). 6 In the presence of MOF-808 and PCN-222, the degradation of VX was sharply enhanced, as shown in Figure 5 and Table 1. We conjectured that in the acidic MOF solutions (Table 1), the alkaline N-group of VX could react with H+ forming a charged VX, which not only could improve the solubility of the VX but also, more importantly, would be favorable for its adsorption onto the surface of the MOF due to the effect of charge interaction leading to accelerated degradation.
t1/2 of VX hydrolysis by different catalysts.

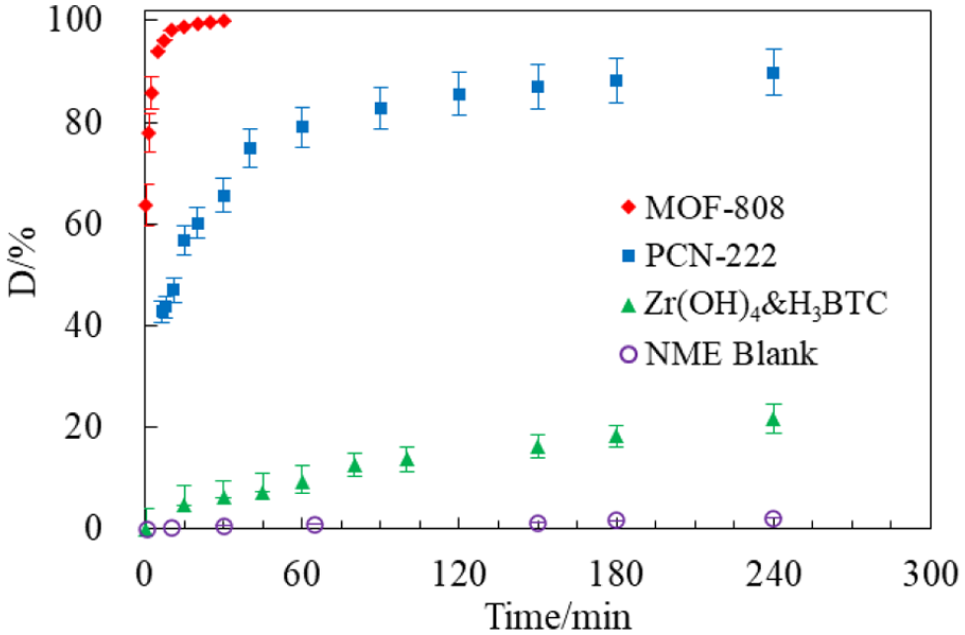
Kinetic profiles for degradation of VX with different materials (5 mL 1:1 ethanol water solution; 0.031 mmol catalyst; 0.187 mmol VX, 10,000 mg L−1; room temperature; vortex, 500 r min−1).
These results show that MOF-808 prepared via a water-based method had better reactivity toward VX hydrolysis with a t1/2 of less than 0.5 min leading to complete degradation within 15 min (Figure 5 and Table 1). Its potency was superior to that prepared by a solvothermal method which had a t1/2 of 8.7 min 32 ; this suggested that the MOF-808 prepared using our water-based method had more defects available to catalyze the VX degradation. The differences for VX hydrolysis between MOF-808 and PCN-222 are probably partly due to their different Zr6 node connectivity as demonstrated in the literature.3,23,25 MOF-808 was built with 6-connected Zr6 nodes and organic linkers (H3BTC) that form channels of about 18 Å in diameter in its three-dimensional framework. These might not only allow VX molecules with sizes about 11 Å to permeate the framework but also could improve the chance of VX interaction with catalytic sites for rapid hydrolysis. In contrast, PCN-222, with relatively small pores of 11 Å and larger cages of about 31 Å, might have less suitable spatial features.
To further seek confirmation of our hypothesis for the hydrolysis of VX with MOF-808, Zr(OH)4 and H3BTC were investigated for VX catalytic degradation. Zr(OH)4 possesses Lewis acidic/basic sites similar to the Zr6 nodes of MOF-808. When Zr(OH)4 was used in conjunction with H3BTC, although it had a similar pKa and Lewis acidic/basic sites as MOF-808, its t1/2 of VX degradation was 693.1 min, much longer than that of MOF-808 and PCN-222. It was concluded that the spatial framework of MOFs performs a vital role in VX catalytic hydrolysis and that the pore size of MOF-808 was suitable for increased nerve agent degradation, as speculated above.
In addition, without basic buffer solutions, the catalytic sites for VX hydrolysis can be inhibited by its hydrolysis product, as reported in the literature.23,35,41 This can result in incomplete degradation such as that found with PCN-222 (Figure 5). In contrast, MOF-808 maintained a fast rate until complete degradation of the nerve agent. This may be related to the many catalytic active sites in MOF-808.
GD degradation
Generally, the more labile P–F in GD agents is hydrolyzed more rapidly than the P–S bond in VX, 6 see Figure 6.3,25 However, it can be seen from Figure 7 that the degradation of GD was much slower than VX with MOF-808 in the absence of basic buffer solutions; this is consistent with the literature. 32 The acidic product of GD hydrolysis, HF, can damage the framework of MOFs and lead to the loss of the hydroxyl groups causing the catalysts to be poisoned. 23 Thus, basic buffer solutions, such as borax and NEM, were introduced for GD catalytic hydrolysis with MOFs leading to higher hydrolysis rates for GD as illustrated in Figure 7.
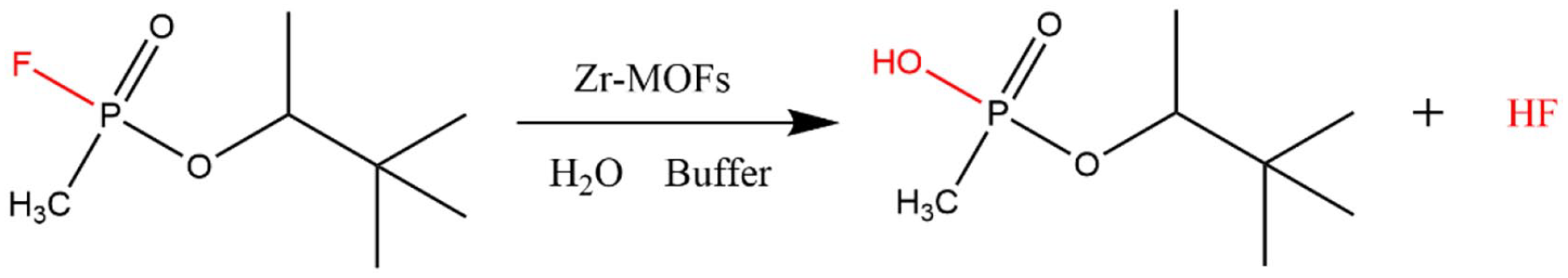
Mechanism of GD catalytic hydrolysis with MOFs.
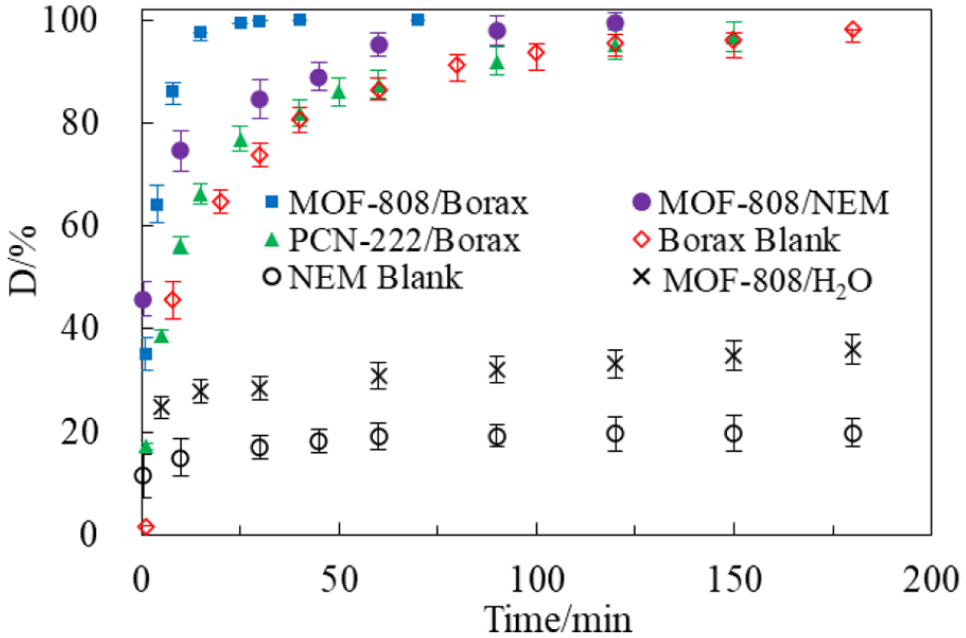
Kinetic profiles for catalytic hydrolysis of GD in different solutions with MOF-808 and PCN-222 (5 mL various solutions; 0.031 mmol catalyst; 0.274 mmol GD, (10,000 mg L−1); room temperature; vortex, 500 r min−1).
The initial degradation of GD in NEM solutions was fast, 50% being degraded within 0.5 min. However, it took a long time, 120 min, to degrade completely (Table 2). In contrast, MOF-808 in borax solution exhibited outstanding performance with a t1/2 of about 3.1 min and complete degradation within 30 min (Figure 7 and Table 2). This interesting result could stem from the buffer capacities of borax and NME solutions for the hydrolysis of high concentrations of GD. We observed that the pH in an aqueous solution of NEM dropped dramatically from 10.98 to 7.56 during the hydrolysis of GD (Table 2) because of the generation of large amounts of HF from the high-concentration GD. Thus, the rate slowed down significantly, requiring longer time for complete degradation. In contrast, the borax solution with a relatively large buffer capacity could keep the basic pH steady enabling the MOFs to catalyze the GD hydrolysis rapidly and constantly. These results illustrate the necessity of using alkaline buffers for the hydrolysis of high concentrations of GD by MOFs.
pH changes of different buffers and rates of GD degradation with MOF-808.

Read from Figure 7.
Calculated according to equations (1) and (2).
The catalysis of GD degradation by PCN-222 was investigated in an aqueous borax solution. Unfortunately, it had almost the same GD hydrolysis rate as in a borax solution without PCN-222. This indicated that PCN-222 had no noticeable catalytic effect on GD hydrolysis. We speculate that the hydrolysis products of high concentrations of GD might be inhibiting the catalytic sites of PCN-222.
HD degradation
In these studies, H2O2 was used as an oxidant instead of O2 to investigate the catalytic performance of Zr-based materials for HD degradation under standard room lighting. The results are shown in Figure 8. All Zr-based materials, including two Zr-MOFs and Zr(OH)4, enhanced the degradation of HD in contrast to the blank with H2O2 alone. This was similar to the reports in the literature where MOFs and Zr oxides (hydroxides) could activate H2O2 oxidation.42–45

Kinetic profiles for catalytic oxidation of HD in H2O2 ethanol solution using different materials as catalysts (4.5 mL ethanol; 0.5 mL 30 wt% H2O2, (3 wt%, 4.4 mmol), 0.390 mmol HD, room temperature, vortex, 500 r min−1; room light; blank, with H2O2.).
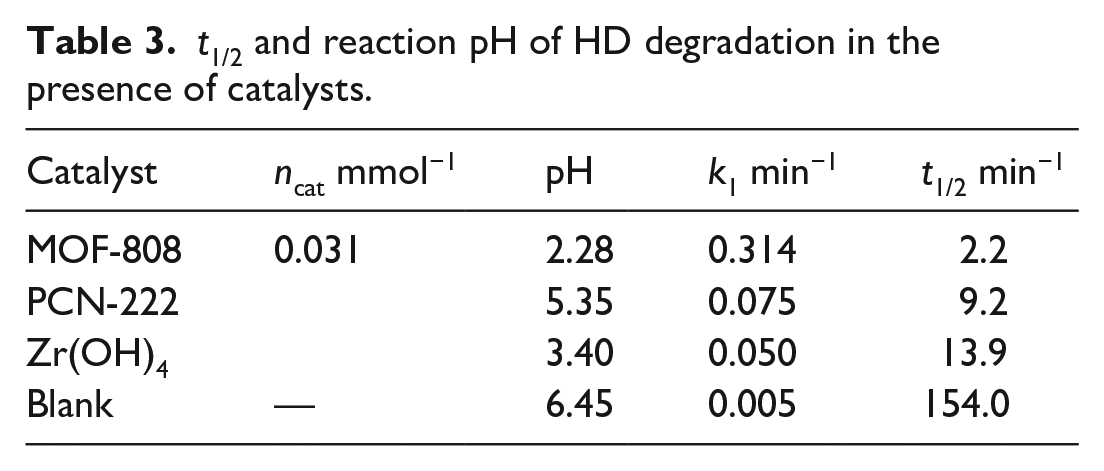
Figure 8 also shows that the degradation rates of HD varied with different Zr-based materials. MOF-808 showed the highest rate with a t1/2 of 2.2 min and complete degradation within 15 min; next was PCN-222 with a t1/2 of 9.2 min (see Table 3). Zr(OH)4 was the slowest. Generally, low pH solutions enhanced the oxidation capability and leading to faster degradation of HD. However, it appears that although the pH of the solution of PCN-222 was higher than that of Zr(OH)4, it exhibited faster oxidation of HD. It has been reported that the porphyrin ligands of PCN-222 can enhance the oxidation of HD by H2O2 due to a photocatalytic process.36,37 A control experiment was then conducted using H2TCPP (the porphyrin ligand of PCN-222) for HD degradation under standard room lighting. The result shows that its rate was similar to the blank, with no evident acceleration (Figure 8). The active catalytic sites of Zr-materials are probably their Lewis acidic/basic centers. The higher catalytic activity of MOFs is mainly due to its Zr6 nodes and three-dimensional framework. PCN-222 also with Zr6 nodes was more efficient for H2O2 activation than Zr(OH)4. The larger surface area of PCN-222 could enhance the adsorption of HD through its hydrophobic pore walls enabling diffusion into its pores. This would facilitate the interaction of H2O2 coordinated to the Zr6 nodes with HD adsorbed into the MOFs, resulting in rapid oxidation rates. MOF-808, with the same framework, has pores that are more suitable (18 Å), more open Zr6 nodes (lower connectivity), and a lower pKa, enabling it to display higher catalytic performance for HD degradation than PCN-222.
t1/2 and reaction pH of HD degradation in the presence of catalysts.
To determine the mechanism of HD degradation by H2O2 catalyzed by Zr-MOFs, we used gas chromatography-mass spectrometry (GC–MS) to analyze and monitor the products during HD degradation. From Figure 9 and Table 4, three main products were observed in the oxidation of HD. They were 2-chloroethyl vinyl sulfone (V-HDO2), bis(2-chloroethyl) sulfone (HDO2), and bis(2-chloroethyl) sulfoxide (HDO). It can be seen from Figure 10 that the degradation of HD by H2O2 alone, without any catalyst, tends to generate more sulfoxide (HDO) and less sulfone (HDO2 and V-HDO2). When Zr-MOFs, such as PCN-222, were used as the catalyst, the amount of sulfone significantly increased while the amount of HDO showed a trend of first increasing and then decreasing. This means that the HDO was being further oxidized to the sulfone consistent with the literature.6,45 The generation of V-HDO2 was mainly due to the elimination of HDO2, which has been studied in the literature. 46 The mechanism is shown in Figure 11.

GC–MS total ion chromatograms of HD oxidation.
Possible degradation products of HD oxidation detected by GC–MS.


The distribution of the product of HD degradation monitored by GC–MS. Blank reaction without any catalysts (a) and catalysis with PCN-222 (b).

Mechanism of HD oxidation by H2O2.
Using H2O2 instead of O2 as the oxidant and Zr-MOFs as the catalysts, HD could be degraded rapidly under room lighting without any special lighting (such as blue light). Such a procedure would be more suitable for the practical application of HD degradation. However, the introduction of H2O2 increased the formation of HDO2 and V-HDO2 that possess certain vesicant toxicities, which is not entirely satisfactory for the objective of the degradation of HD to nontoxic products. Further studies, such as the adjustment of the pH of the solution, need to be done to reduce the formation of these vesicant products.
Conclusion
PXRD, BET, and SEM characterization demonstrated that MOF-808 had been successfully prepared via a water-phase synthesis and PCN-222 by a solvothermal method. Their catalytic performance for the degradation of high concentrations of CWAs was investigated. Without basic buffer solutions, although the solutions with MOF-808 and PCN-222 were acidic, VX could be degraded rapidly. Comparatively, MOF-808 performed as a more effective catalyst for the degradation of high concentrations of VX with a t1/2 of < 0.5 min. Hydrolysis of high concentrations of GD required strong alkaline buffers to maintain the catalytic activity of the MOFs. The stronger buffer capacity of borax solutions maintained GD degradation with a t1/2 of 3.1 min. It was observed that Zr-MOFs could catalyze the oxidation of HD by H2O2. The products of HD oxidation including HDO2 and V-HDO2 had vesicant toxicities. This process may be improved by adjusting the pH of the reaction solution.
To summarize, MOF-808 prepared via a water-phase synthesis was a useful catalyst for the degradation of high concentrations of VX, GD, and HD and may be suitable for practical application.
Experimental
Reagents and instrumentation
The toxic agents of VX, GD, and HD had a purity of > 95%.
Because of their high toxicity, they were handled only by well-trained personnel using appropriate safety procedures
ZrOCl2.8H2O (99%), ZrCl4 (99%), Zr(OH)4 (97%), 1,3,5-benzenetricarboxylic acid (H3BTC, 98%), tetrakis(4-carboxyphenyl) porphyrin (H2TCPP, 99%), NEM, dichloromethane (DCM, HPLC), borax (Na2B4O7·10H2O, 99%), and Na2HPO4 (99%) were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd. Absolute ethyl alcohol (⩾ 99.5%) and H2O2 (30 wt%) were purchased from Jindong Tianzheng Fine Chemical Reagent Factory.
PXRD analysis was performed on a Rigaku SmartLab SE X-ray diffractometer (Rigaku Corp., Tokyo, Japan) in the range of 3°–40° using Cu Kα radiation (λ 0.15405 nm) as an X-ray source and at a scanning rate of 5° min−1. SEM was performed on a ZEISS Gemini 300 (Carl Zeiss SMT AG, Oberkochen, Germany). N2 adsorption–desorption isotherms were run on an Autosorb-1 Surface Area and Pore Size Analyzer (Quantachrome Instruments, Boynton Beach, FL, USA) at a temperature of 77 K. Gas chromatography-mass spectrometry (GC–MS, 7890B/5977A) equipped with an HP-5 capillary column (30 m × 0.32 mm × 0.25 µm), employing a temperature program (60 °C–280 °C at 15 °C min−1) and N2 as the carrier gas, was for the analysis of VX, GD, and HD.
Preparation of MOF-808 and PCN-222
H3BTC (1.05 g, 8 mmol) was dissolved in deionized H2O/acetic acid (40 mL/40 mL) with ultrasound for 10 min. ZrOCl2.8H2O (4.8 g, 32 mmol) was then added, and the mixture was heated in an oven at 85 °C for 24 h. The resulting white MOF-808 powder was collected by centrifugation. It was then washed with H2O (40 mL × 2) and ethanol (40 mL × 2) under ultrasound for 10 min to remove the unreacted reagents. Finally, the sample was dried at 85 °C for 12 h in a vacuum oven without further activation procedures.
The PCN-222 was synthesized as reported.38,40 It was activated under dynamic vacuum conditions at 120 °C for 24 h before its characterization and detoxification evaluation.
Degradation reactions
Overall, 0.031 mmol of MOF-808 (40 mg), PCN-222 (75 mg), or Zr(OH)4&H3BTC was added to a tube containing 5 mL ethanol-H2O (1:1) solution. Using a vortex agitator, 50 μL VX (0.187 mmol) was then added to form an agent concentration of 10 g L−1 for degradation reactions under room temperature. After a certain time, 30 μL of the sample was diluted with 3 mL DCM for VX determination with GC–MS. The residual ratio of VX (R, %) and its degradation ratio (D, %) were calculated as described previously. 47
The degradation of GD was conducted the same as that using VX, except the solvent was replaced by 0.5 M NEM or 0.1 M aqueous borax.
As for HD degradation, 0.5 mL of 30 wt% H2O2 was introduced in 4.5 mL ethanol under room lighting. Samples were analyzed using the T-135 method. 48 The residual ratio of HD (R, %) and degradation efficiency (D, %) were obtained as reported.27,34 Degradation products were detected by GC–MS. Since the molar content of H2O2 was larger than HD, the degradation of HD could be regarded kinetically as a pseudo-first-order reaction. The first-order constant (k1) and half-life (t1/2) could be calculated according to equations (1) and (2)


Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.