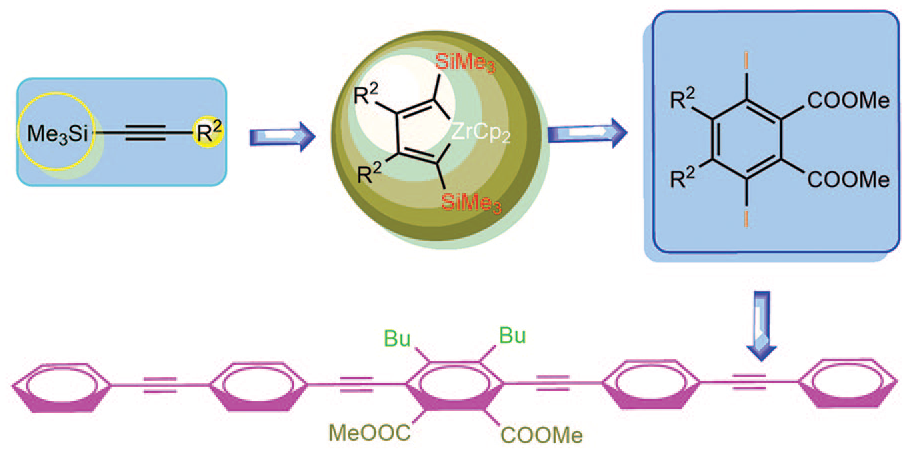
A series of novel 1,4-bis(alkynyl)benzene derivatives were synthesized from trimethylsilyl-substituted alkynes by the mediation of zirconocene with excellent regioselectivity in high yields. The 3,6-bis(trimethylsilyl)-4,5-dialkylphthalic acid dimethyl esters were prepared by cycloaddition of 2,5-bis(trimethylsilyl)zirconacyclopentadienes to dimethyl acetylenedicarboxylate. After iodination with iodine monochloride, 3,6-diiodo-4,5-dialkylphthalic acid dimethyl esters reacted with terminal alkynes to prepare the corresponding 1,4-bis(alkynyl)benzene derivatives by Sonogashira coupling reactions. After removal of trimethylsilyl, 4,5-dibutyl-3,6-bis(ethynyl)phthalic acid dimethyl ester (compound 3) reacted with 4-iodobenzoic acid ethyl ester and 2-iodothiophene, respectively, to obtain the corresponding products 4a and 4c. Compound 3 can be extended to higher oligomers, which reacted with 1-bromo-4-iodobenzene and phenylacetylene in a stepwise manner under Sonogashira conditions to give the phenylene-ethynylene oligomer 5 in an isolated yield of 85%. The structures of the products were confirmed by 1H NMR spectroscopy, 13C NMR spectroscopy, and MS. The optical properties of the 1,4-bis(alkynyl)benzene derivatives were studied by UV-Vis spectroscopy and fluorescence spectra. The results indicated that some can be developed into potential photovoltaic materials.
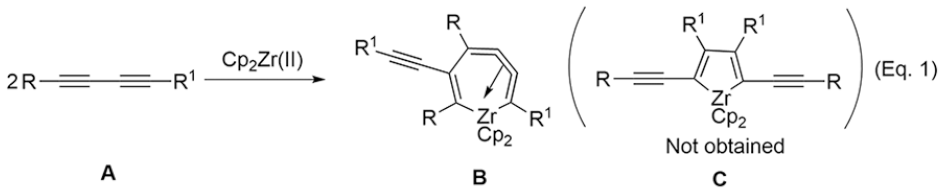
Molecular wires with linear conjugated structures are important components of electronic devices. Among these conjugated materials, oligo(phenylene-ethynylenes) (OPEs) with ideal conductivity and rigidity are widely used to prepare conductive and electroluminescent materials by the modification of functional groups.1,2 1,4-Bis(alkynyl)benzene is a crucial component as a monomer unit of OPEs, which can be developed into organic optoelectronic material by further coupling of the conjugated structure.3,4 The preparation of benzene derivatives has attracted much attention of chemists for several decades.5–7 Although the synthesis, properties, and applications of benzene derivatives have been studied for a long time, there are still only a few reports on the selective synthesis of 1,4-bis(alkynyl)benzenes.8–10 The synthesis of benzene derivatives by cycloaddition of zirconacyclopentadienes to a third molecular alkyne is well known,11 and chemists initially considered it as a potential way to prepare 1,4-bis(alkynyl)benzenes through the 2,5-bis(alkynyl)zirconacyclopentadiene C (Eq. 1).12 Unfortunately, although zirconacycle C seems to be a reasonable intermediate for the preparation of 1,4-bis(alkynyl)benzenes, the reaction of Cp2Zr(II) with 1,3-butadiyne A afforded seven-membered zirconacyclocumulenes B, and the zirconacycle C is not obtained (Eq. 1).13–15 Thus, it is necessary to develop an alternative method to prepare 1,4-bis(alkynyl)benzenes using the zirconocene complex and silylalkynes.
So far our group has developed zirconocene-mediated selective syntheses of benzene derivatives. On the basis of our previous works on the zirconocene-mediated cycloaddition of different alkynes, the fully substituted benzenes and 1,2,3,5-tetrasubstituted benzenes were prepared.16–18 The trimethylsilyl (TMS) groups can be easily substituted with other functional groups19 and the selective synthesis of 1,4-bis(alkynyl)benzenes from TMS-substituted alkynes by the mediation of Cp2Zr(II) species seemed to be an appropriate method. TMS-substituted alkynes were treated with Cp2Zr(II) species to afford the corresponding 2,5-bis(trimethylsilyl)zirconacyclopentadienes as single products in high yields with excellent regioselectivity. After the cycloaddition of the zirconacyclopentadienes with the third internal alkyne, 1,4-bis(trimethylsilyl)hexa-substituted benzenes were produced as important intermediates. The TMS groups were substituted by iodo groups to form the corresponding diiodo-substituted derivatives, which reacted with terminal alkynes to prepare the desired 1,4-bis(alkynyl)benzene derivatives through Sonogashira coupling reactions. After removal of the TMS groups from bis(trimethylsilylethynyl)benzenes, the corresponding bis(ethynyl)benzene derivatives were prepared. These can be extended to higher-order linear conjugated molecules through coupling with iodides. The basic unit of the current OPE is usually a para-disubstituted benzenes which can be extended only at the para-position. In this paper, some functional groups, such as –Pr, –Bu, and –CO2Me, can be introduced through the starting alkynes, and the ester groups can be further modified to obtain variable products. In this way, the 1,4-bis(alkynyl)benzene can be extended not only laterally but also longitudinally.
Herein, a method is proposed for the synthesis of 1,4-bis(alkynyl)benzenes that involves mediation of zirconocene followed by derivatization of the TMS groups. This method is applied with high selectivity and high yields using three alkynes as the starting materials.
Results and discussion
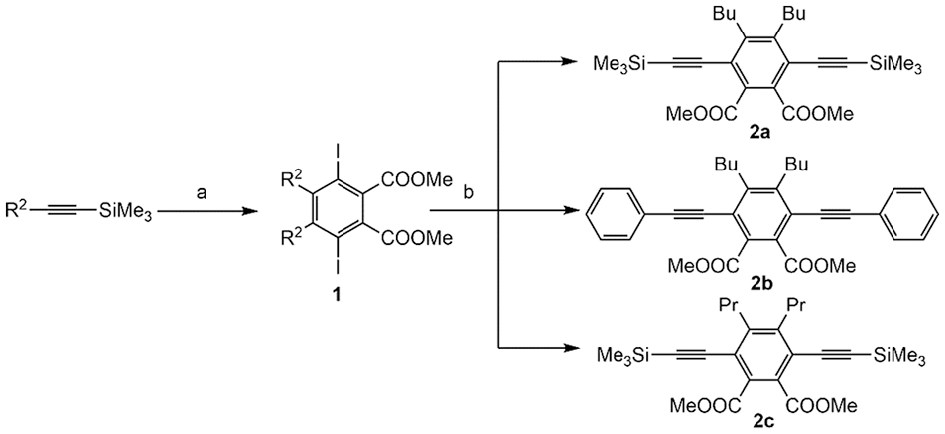
As shown in Scheme 1, 3,6-diiodo-4,5-dialkylphthalic acid dimethyl esters were prepared according to our previous work,16,18 wherein R2 was propyl or butyl. The iodine-substituted compounds 1 were reacted with a series of terminal alkynes by Sonogashira coupling to obtain the corresponding 1,4-bis(alkynyl)benzenes 2a–c. Using the TMS substituent, 2a was extended to higher oligomers. As shown in Scheme 2, the TMS group of 2a was removed to afford compound 3 that reacted with the corresponding aryl iodides to prepare dialkynes 4a–c through Sonogashira reactions. The dialkyne 4b was then reacted with phenylacetylene to obtain the phenylene-ethynylene oligomer 5 in high yield.
Synthetic procedures for compound 2a–c. Reagents and conditions: (a) i. n-BuLi, Cp2ZrCl2, THF, –78 °C, 3 h; ii. CuCl, DMAD, RT, 6 h; iii. ICl, 0 °C, 6 h. (b) (trimethylsilyl)acetylene or phenylacetylene, Pd(PPh3)4, CuI, 45 °C, 12 h, 2a: 93%, 2b: 82%, 2c: 85%.
UV-Vis absorption spectra of 2a–c, 3, 4a–c, and 5 are recorded in Figure 1. The absorbance peaks between 240 and 365 nm were attributed to the π–π* transitions of the conjugated molecules. The absorbance peaks of the 2a, 2c, and 3 were in the range of 240–300 nm, and the absorbance peak near 240 nm was the π–π* transition of the benzene ring. With the extension of the conjugated chain, the highest occupied molecular orbital (HOMO) level of the molecule increased and the lowest unoccupied molecular orbital (LUMO) level decreased and so the energy of π–π* transition decreased. The absorbance peaks of the alkynes 2b, 4a–c, and 5 were in the range of 249–363 nm. Since compounds 2b, 4a, 4c, and 5 were linear conjugated molecules containing three or more benzene rings, the spectroscopic absorption bands were red-shifted. The ultraviolet absorption of thiophene is close to that of benzene, resulting in the same red shift of the absorbance band of 4c containing the thiophene groups. The red-shifted distance of 5 was the largest because of its longest linear conjugated structure, and its maximum absorption wavelength (λmax) was 363 nm. The optical band gaps (∆Eopt) of the compounds were estimated from the onsets of their UV-Vis absorption spectra. The optical band gaps of 2a–c, 3, 4a–c, and 5 were determined to be 3.57, 3.26, 3.58, 3.71, 3.18, 3.24, 3.07, and 2.99 eV, respectively.
UV-Vis absorption spectra of 2a–c, 3, 4a–c, and 5 (measured at 10−5 M in CH2Cl2).
Figure 2 shows the photoluminescence (PL) spectra of dilute solutions of 2a–c, 3, 4a–c, and 5 in CH2Cl2 (10−5 mol/L). At a concentration of 10−5 M, the linear conjugated compounds 2b, 4a, 4b, and 5 containing three or more benzene rings exhibited good fluorescence response in CH2Cl2, and their emission peaks were in the range of 380–410 nm. The fluorescence intensity of 2a, 2c, 3, and 4c was very low, and the spectra showed that their emission peaks were in the range of 360–405 nm, corresponding to a purple color. By observing the spectra of 2b, 3, and 5, it was known that the degree of conjugation of π electrons increases with the extension of the linear conjugated structure, and the fluorescence spectra moved to longer wavelengths. The red-shifted distance of 5 was the longest and its fluorescence intensity was the strongest. The fluorescence spectra of 2b, 4a, and 4b showed that their fluorescence intensity was enhanced by the introduction of polar groups in the linear conjugate direction, which may be due to the fact that the electron-withdrawing group is beneficial to the movement of π electrons in the conjugated system so that the energy absorbed by the transition of the system is reduced, facilitating the generation of fluorescence. Their maximum absorption wavelengths remained substantially unchanged.
PL spectra of 2a–c, 3, 4a–c, and 5 (measured at 10−5 M in CH2Cl2).
Conclusion
In summary, we have developed an efficient method for the selective preparation of 1,4-bis(alkynyl)benzenes in high yields from internal alkynes. 3,6-Bis(trimethylsilyl)hexa-substituted benzenes are key intermediates which were prepared through cyclization of TMS-substituted alkynes and dimethyl acetylenedicarboxylate (DMAD) by the mediation of zirconocene. 3,6-Diiodohexa-substituted benzenes were reacted with terminal acetylenes by Sonogashira couplings to produce a series of 1,4-bis(alkynyl)benzene derivatives. After removal of the TMS groups, compound 3 was extended to obtain the products 4a–c in moderate to good yields. Finally, 4b was reacted with phenylacetylene through a Sonogashira coupling reaction to give the phenylene-ethynylene oligomer 5 in an isolated yield of 85%. The structures of the products were confirmed by 1H NMR spectroscopy, 13C NMR spectroscopy, and MS. The results of UV and fluorescence showed that 4a and 5 exhibited good optical properties. The maximum UV absorption wavelengths of 4a and 5 were 345 and 363 nm, respectively, and their corresponding UV absorption intensities were 0.794 and 0.587 in CH2Cl2 (10−5 mol/L), respectively. The fluorescence peaks of 4a and 5 were 382 and 405 nm, respectively, and their corresponding fluorescence absorption intensities were 6043 and 6707 in CH2Cl2 (10−5 mol/L), respectively. The fluorescence absorption peaks of 4a and 5 correspond to the purple color. They are promising candidates for use in electroluminescent materials.
Experimental
All organic solvents and materials for synthesis were of reagent grade and used without further purification. All reactions were carried out under a dry atmosphere of nitrogen using standard Schlenk technology. IR spectra were performed on a Nicolet 6700 FTIR spectrophotometer. Melting points were determined using a microscope apparatus and were uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III 400 MHz spectrometer in CDCl3 solution. High-resolution mass spectra (HRMS) were obtained with a Bruker Daltonics 1290 UPLC/micrOTOF-Q II spectrometer. UV-Vis absorption and fluorescent spectra were recorded on a Persee TU-1810APC and a Hitachi F-2500 FL spectrophotometer, respectively. Thin-layer chromatography (TLC) was performed on Energy GF254 plates. 3,6-Diiodo-4,5-dialkylphthalic acid dimethyl esters (1) were prepared according to our previous work.16,18
Synthesis of 4,5-dibutyl-3,6-bis(trimethylsilanylethynyl)phthalic acid dimethyl ester (2a)
Copper (I) iodide (76.2 mg, 0.40 mmol) and Pd(PPh3)4 (138.7 mg, 0.12 mmol) were added to a mixture of 3,6-diiodo-4,5-dibutylphthalic acid dimethyl ester (2.12 g, 4.00 mmol), (trimethylsilyl)acetylene (0.94 g, 9.60 mmol), and diisopropylamine (20 mL) in toluene (10 mL). The mixture was stirred at 45 °C for 12 h, and then it was quenched with saturated NH4Cl solution and extracted with ethyl acetate. The combined organic phase was washed with water and brine. The solution was dried over anhydrous Na2SO4. The solvent was evaporated, and the residue was purified by chromatography on silica gel (hexane/ethyl acetate = 5:1 as eluent) to afford 1.85 g compound 2a as pale brown solid.
Synthesis of 4,5-dibutyl-3,6-bis(ethynyl)phthalic acid dimethyl ester (3)
Compound 2a (0.99 g, 2.00 mmol) was dissolved in methanol (10 mL) and tetrahydrofuran (THF, 5 mL), and 0.85 g (8.00 mmol) potassium carbonate was added. The mixture was stirred at room temperature for 1 h before being poured into water. Then the organic layer was extracted with ethyl acetate and washed with brine. After drying over magnesium sulfate, the solvent was evaporated in vacuo, and the residue was purified by chromatography on silica gel (hexane: ethyl acetate = 5:1 as eluent) to afford 0.72 g product 3 as brown solid.
Synthesis of 3,6-bis(4-bromo-phenylethynyl)-4,5-dibutylphthalic acid dimethyl ester (4b)
Compound 3 (0.73 g, 1.94 mmol) obtained in the previous step was dissolved in toluene (10 mL) and triethylamine (15 mL), then 1-bromo-4-iodobenzene (1.13 g, 4.00 mmol), Pd(PPh3)4 (69.3 mg, 0.06 mmol), and copper (I) iodide (38.1 mg, 0.20 mmol) were added, and the mixture was stirred at room temperature for 3 h. Upon completion of the reaction, the mixture was quenched with water, and then the organic layer was extracted with ethyl acetate and washed with water and brine. The solution was dried over anhydrous Na2SO4 and the solvent removed in vacuo. The resulting crude product was purified by a column chromatography (silica gel, hexane/ethyl acetate = 1:1) to afford 1.00 g compound 4b as pale yellow solid.
Synthesis of 4,5-dibutyl-3,6-bis[4-(phenylethynyl)phenylethynyl]phthalic acid dimethyl ester (5)
Copper (I) iodide (76.2 mg, 0.40 mmol) and Pd(PPh3)4 (138.7 mg, 0.12 mmol) were added to a mixture of 3,6-bis(4-bromo-phenylethynyl)-4,5-dibutylphthalic acid dimethyl ester (4b, 2.68 g, 4.00 mmol), phenylacetylene (0.98 g, 9.60 mmol), and diisopropylamine (20 mL) in toluene (10 mL). The mixture was stirred at 80 °C for 6 h, and it was quenched with saturated NH4Cl solution and extracted with ethyl acetate. The combined organic phase was washed with water and brine. The solution was dried over anhydrous Na2SO4. The solvent was evaporated, and the residue was purified by chromatography on silica gel (hexane/ethyl acetate = 1:1 as eluent) to afford 1.20 g yellow solid.