
Abstract
The vascular endothelial growth factor receptor-2 signaling pathway promotes the formation of new blood vessels, and vascular endothelial growth factor receptor-2 tyrosine kinase exists in both active and inactive conformations. Novel indole–benzimidazole and indole–benzothiazole derivatives joined by different linkers are designed and synthesized as inhibitors of vascular endothelial growth factor receptor-2 tyrosine kinase. All the synthesized compounds were evaluated for their cytotoxicity against four human cancer cell lines (HeLa, HT29, A549, and MDA-MB-435) and human umbilical vein endothelial cell. Meanwhile, the inhibitory activities against vascular endothelial growth factor receptor-2 are estimated in vitro and the binding interactions with dual conformations of vascular endothelial growth factor receptor-2 tyrosine kinase are evaluated by molecular docking. Compounds
Keywords

Introduction
Cancer, the uncontrolled, rapid, and pathological proliferation of abnormal cells, is one of the most formidable afflictions in the world, 1 which causes about 550,000 deaths per year and represents the second leading cause of death in the world. 2 Current cancer chemotherapy is severely limited by side effects and/or drug resistance, and thus it remains essential to develop new and safer anticancer drugs. At present, the development of tumor angiogenesis inhibitors has become an important field in anticancer (drug) research due to the low toxicity to the human body, the broad-spectrum of activity against tumors, and tolerance to the development of resistance.
Vascular endothelial growth factor (VEGF) shows high specificity and plays an important regulatory role in angiogenesis and vascular migration. It is frequently overexpressed in a variety of malignant tumors and is closely related to the growth, metastasis, and prognosis of tumors. Vascular endothelial growth factor receptor-2 (VEGFR-2), belonging to the protein tyrosine kinase family, 3 is a transmembrane receptor and is mainly expressed in vascular endothelial cells and hematopoietic stem cells. As a key signal sensor in the processes of physiological and pathological angiogenesis, it mainly regulates the physiological response of VEGF, including permeability, proliferation, and migration. As well as VEGF, VEGFR-2 is overexpressed in malignant tumors, including ovarian and thyroid cancer, melanoma, and medulloblastoma. Research has shown that VEGFR-2 tyrosine kinase inhibitors can interrupt VEGF/VEGFR signal pathways and control tumor cell growth. Such inhibition is a good way to develop new anticancer drugs by targeting VEGFR-2 tyrosine kinase inhibitors.
In the intracellular domain, tyrosine kinase contains ATP-binding and downstream signaling protein binding sites. One kind of VEGFR-2 kinase inhibitor occupies the ATP-binding site keeping ATP from combining with VEGFR-2 and preventing VEGFR-2 autophosphorylation. In fact, VEGFR-2 tyrosine kinase exists in both active and inactive conformations; the two conformations can be transformed by aspartate, phenylalanine, and glutamic acid (DFG) modules. 4 Tyrosine kinase is active when the phenyl ring on phenylalanine is directed to the N-terminal alpha helix, that is, the DFG-in conformation; however, when the benzene ring flips to the ATP binding site, it prevents the kinase from binding to ATP and tyrosine kinase is in the inactive state, that is, the DFG-out conformation.5,6 In general, class I inhibitors bind to the ATP binding site, class II inhibitors bind to the allosteric sites of the DFG-out conformation and indirectly compete with ATP. In addition, DFG ring movement is a slow process, being a balance between the active and inactive conformational transition. Therefore, it is significant to develop inhibitors that bind to the active conformation of tyrosine kinase and/or the inactive conformation for better efficacy.
Considering that ATP binding sites are near the allosteric sites of the DFG-out conformation (DFG-out sites), dual inhibitors of the DFG-in conformation and the DFG-out conformation with double linking groups can be designed, which can bind two sites selectively. According to the research on inactive kinase inhibitors (imatinib, nilotinib, and ponatinib), a diarylamide may be one active group which can bind DFG-out sites.7,8 Moreover, several studies also report that benzoheterocyclic compounds, such as indoles,9-11 benzimidazoles,12,13 benzothiazoles, 14 acridines, quinolines, and quinazolines, show tyrosine kinase inhibitory activities on different cancer cell types. Therefore, it can be speculated that the compounds will also work if diaryl moieties can be replaced by benzoheterocyclic moieties (Figure 1). Furthermore, for the DFG-in conformation, the two benzoheterocyclic moieties can bind at ATP binding sites.

The structures of the designed target compounds.
In our previous study, and based on speculation, a series of novel indole–benzimidazole or indole–benzothiazole VEGFR-2 inhibitors were designed by de novo drug design based on a linked-fragment approach.15,16 The compounds were synthesized and showed certain VEGFR-2 inhibitory activity. 17 However, the activities were not high enough to be applicable to inhibit cancer cell proliferation. Herein, we have optimized the inhibitor structures for high tumor cell cytotoxicity and broad-spectrum properties.
All the target compounds were synthesized and their antitumor activity against the human lung adenocarcinoma cell line (A549), the human colon cancer cell line (HT-29), the human breast cancer high metastatic cell line (MDA-MB-435), the human cervical carcinoma cell line (HeLa), and the human umbilical vein endothelial cells (HUVECs) were evaluated in vitro, comparing with sunitinib malate as a positive control drug. Molecular docking studies were performed to understand the binding mode of the synthesized compounds with VEGFR-2 tyrosine kinase and the interactions with the kinase active site were analyzed with the aim of explaining their VEGFR-2 kinase inhibitory activity.
Results and discussion
Anti-tumor activity
The indole derivatives
The IC50 values of the synthesized compounds for five types of tumor cell lines in vitro.

HeLa: human cervical carcinoma cell line; HT-29: human colon cancer cell line; HUVEC: human umbilical vein endothelial cell; A549: human lung adenocarcinoma cell line; MDA-MB-435: human breast cancer high metastatic cell line; NA: no inhibitory activity.
It can be observed that three compounds
It is obvious that compound
For the MDA-MB-435 cell line, compounds
As shown in Table 1, most of the compounds can inhibit the growth of the A549 cancer cell effectively, and the relative IC50 values were in the micromolar range. The positive drug and compounds
Although HUVEC belongs to the normal cell category, it has the potential of stem cells and can be subcultured 50–60 times in theory. It is usually selected as a cell model for vascular endothelial cell experiments. The positive drug sunitinib malate demonstrated significant angiostatic effects toward HUVEC. Compounds
Enzyme inhibitory activity
According to results of enzyme inhibitory assays (Table 2), the new compounds (
Inhibition rates of the newly synthesized compounds against VEGFR-2 tyrosine kinases at 1 μM.

Molecular docking studies
To understand the possible mechanism of action, molecular docking studies of the six compounds (
The docking-free-energy score of six compounds bound to the binding site of VEGFR-2 tyrosine kinase with active or inactive conformations.
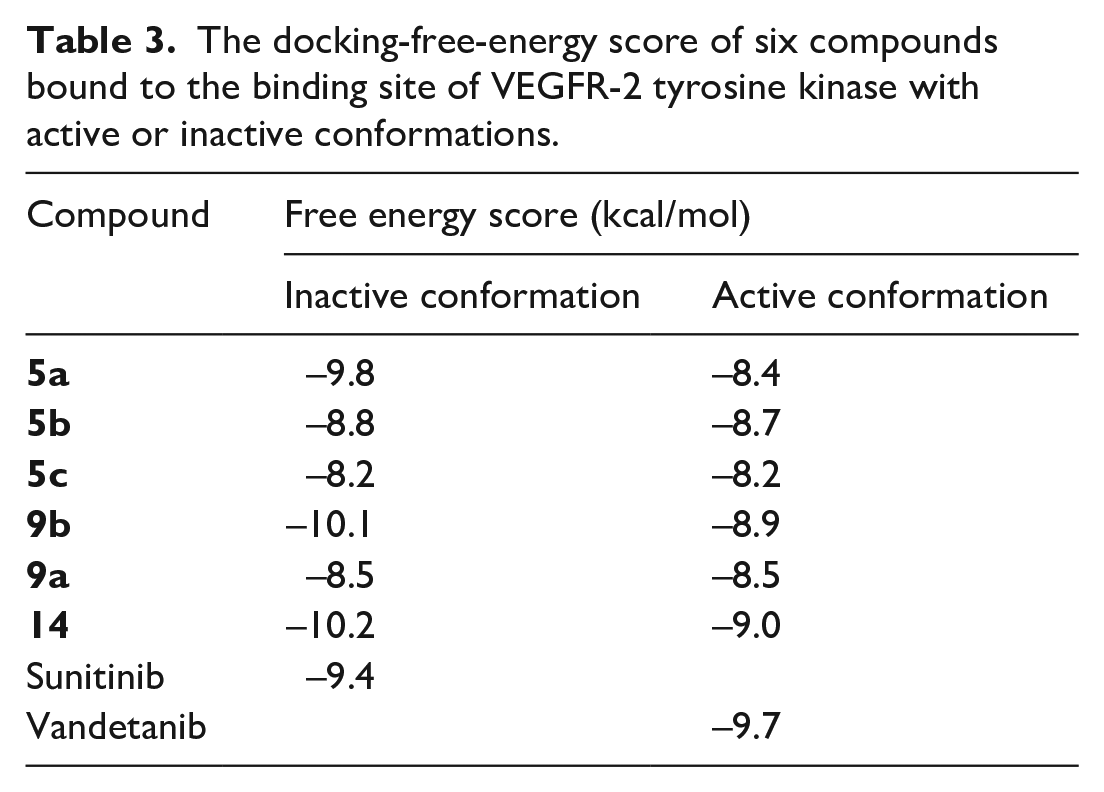
Inactive DFG-out conformation
First of all, the confirmation of the molecular docking protocol was obtained by docking of the crystallized sunitinib ligand in the VEGFR-2 inactive site. The docking confirmation step produced the experimental binding of the crystallized ligand between the docked pose and the crystallized ligand (free energy score −9.4 kcal/mol). Meanwhile, all the key interactions between the crystallized ligand with the key amino acids in the active site (Val80, Glu149, Phe150, Cys151, Phe153; Figure 2) were produced by capability of the docking pose.

2D diagram and 3D representation of compound
From the docking results (Figure 2), it can be observed that compound
In the inactive conformation, the NH on the indole and imidazole rings of compound

2D diagram and 3D representation of compound
Active DFG-in conformation
According to the above results, compounds
As can be seen from the docking results of compound
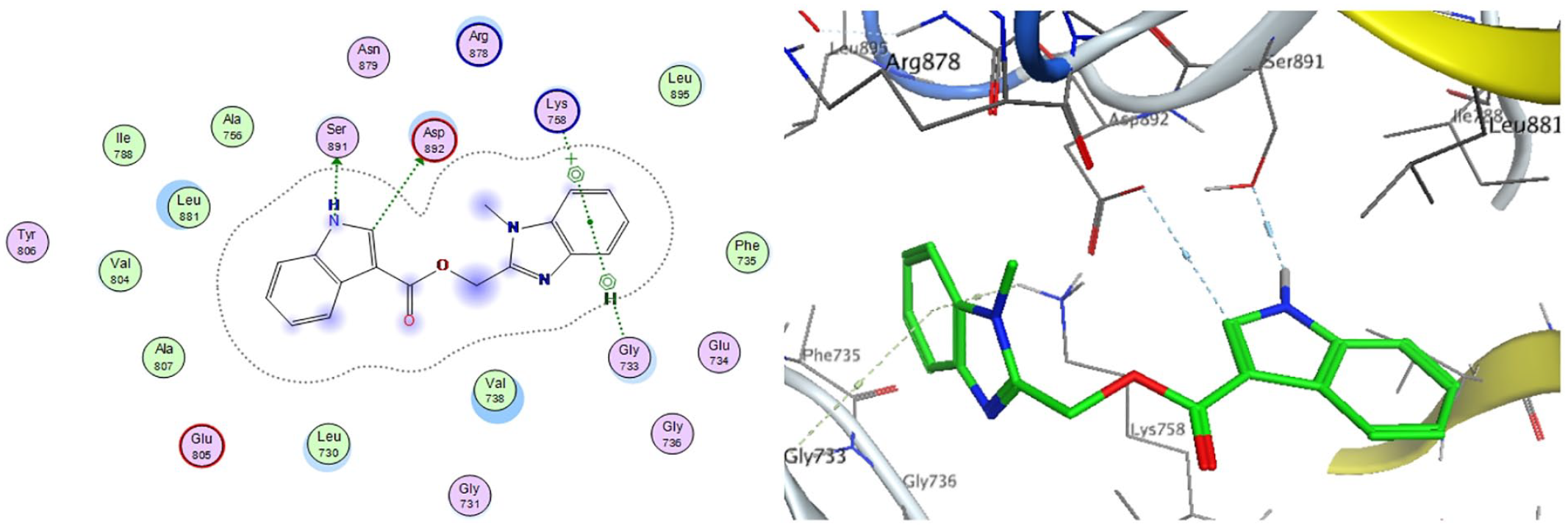
2D diagram and 3D representation of compound
As shown in Figure 5, the indole ring of

2D diagram and 3D representation of compound
Compound

2D diagram and 3D representation of compound
Discussion and conclusion
The target products are synthesized by condensation dehydration reaction (ester and ether) or deacidification reaction (amide). The reaction conditions are mild and easy to control. The difference is that there are many by-products in the synthesis of ether, therefore, the yields of
Though the benzoheterocyclic compounds show good antitumor activity, the mechanism of action is complicated. In this study, the VEGFR-2 tyrosine kinase inhibitory activity was consistent with antitumor activity. For example, compounds
Undoubtedly, the structure of the compound affects the enzyme activity, which in turn influences its antitumor activity. The data illustrated that (1H-benzimidazole-2-base-1H-3-)methyl indole acid ester compounds
The different linking moieties between the benzimidazole groups and indole groups also show different activities. Comparing compounds
Conversely, the activity of N-(benzothiazole-2-base)-1H-indole-3-carboxamide
According to molecular docking results, the compounds bind to the different conformations of VEGFR-2 tyrosine kinase. In the DFG-out conformation, one benzoheterocyclic moiety of
Overall, the indole–benzoheterocycle compounds
Experiment
Synthesis of the target compounds
Materials and methods
All solvents and reagents were obtained from commercial sources and were used without further purification. Thin-layer chromatography (TLC) and silica gel column chromatography used with silica gel GF254 and 200–300 mesh, respectively (Qingdao Haiyang Chemical Co. Ltd, Shandong, China). Melting points were determined on a digital melting point apparatus (WRS-1B; Shanghai Jingke Scientific Instrument Co. Ltd, Shanghai, China) and were uncorrected. 1H NMR and 13C NMR were recorded on Bruker Avance DMX 500 or 300 MHz instruments, using tetramethylsilane (TMS) as the internal standard and DMSO-d6 as the solvent. FTIR spectra were measured on a Mattson 2020 Galaxy Series FTIR spectrometer. High-resolution mass spectrometry (HRMS) data were acquired on an Agilent Technologies LC/MSD TOF mass spectrometer, using electron ionization (EI) in either positive or negative ion mode.
Synthesis of (1H-benzoimidazol-2-yl)methyl 1H-indole-3-carboxylate derivatives (5a–c)
As shown in Scheme 1, to a mixture of

Synthesis of compounds
To produce indole-3-carboxylic acid derivatives
Finally, compounds
1-Methyl-1H-benzo(d) 1H-indole-3-carboxylate (5a )
White solid; 47% yield; m.p. 207–209°C. 1H NMR (500 MHz, DMSO-d6): δ 3.65 (s, 3H, CH3), 5.42 (s, 2H, CH2), 7.02 (t, J = 7.5 Hz, 1H, ArH), 7.11 (t, J = 7.5 Hz, 1H, ArH), 7.18 (t, J = 7.5 Hz, 1H, ArH), 7.24 (t, J = 7.5 Hz, 1H, ArH), 7.35 (s, 1H, CH–N), 7.51 (d, J = 8.0 Hz, 1H, ArH), 7.55 (d, J = 8.0 Hz, 1H, ArH), 7.60 (d, J = 8.0 Hz, 2H, ArH). 11.86 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6): δ 163.08, 148.96, 136.42, 134.21, 132.92, 131.51, 125.85, 125.69, 125.58, 122.64, 121.64, 120.18, 115.03, 112.64, 112.56, 104.46, 55.37, 31.38. FTIR: 3447, 3087, 2920, 1690, 1537, 1441, 1365, 1340, 1244, 1174, 778, and 748 cm–1. HRMS (EI): m/z [M]+calcd for C18H15N3O2: 305.1164; found: 305.1261.
1-Methyl-1H-benzo(d) 5-methoxy-1H-indole-3-carboxylate (5b )
White solid; 65% yield; m.p. 227–228°C. 1H NMR (500 MHz, DMSO-d6), δ 3.65 (s, 3H, CH3), 3.75 (s, 3H, OCH3), 5.42 (s, 2H, CH2), 7.11 (t, J = 7.5 Hz, 1H, ArH), 7.18 (t, J = 7.5 Hz, 1H, ArH), 7.24 (t, J = 7.5 Hz, 1H, ArH), 7.35 (s, 1H, CH–N), 7.51 (d, J = 8.0 Hz, 1H, ArH), 7.55 (d, J = 8.0 Hz, 1H, ArH), 7.60 (d, J = 8.0 Hz, 2H, ArH), 11.86 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6): δ 163.12, 155.22, 148.96, 134.25, 132.79, 131.27, 131.16, 126.62, 125.98, 125.66, 114.87, 113.36, 112.69, 112.52, 104.05, 101.94, 55.22, 31.42, 24.39. FTIR: 3090, 2934, 1714, 1648, 1508, 1490, 1438,1270, 1250, 1181, 1150, 1070, 977, and 735 cm–1. HRMS (EI): m/z [M]+calcd for C19H17N3O3: 335.1270; found: 335.1458.
1-Methyl-1H-benzo(d) 5-bromo-1H-indole-3-carboxylate (5c )
White solid; 60% yield; m.p. 213–215°C. 1H NMR (500 MHz, DMSO-d6): δ 3.72 (s, 3H, CH3), 5.39 (s, 2H, CH2), 7.14 (t, J = 7.5 Hz, 1H, ArH), 7.18 (t, J = 7.5 Hz, 1H, ArH), 7.26 (t, J = 7.5 Hz, 1H, ArH), 7.37 (s, 1H, CH–N), 7.51 (d, J = 8.0 Hz, 1H, ArH), 7.55 (d, J = 8.0 Hz, 1H, ArH), 7.63 (d, J = 8.0 Hz, 2H, ArH). 11.86 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6): δ 162.82, 148.91, 135.35, 135.21, 133.09, 132.14, 127.48, 125.60, 125.40, 125.30, 122.37, 115.27, 114.70, 114.46, 112.47, 104.27, 55.70, 31.28. FTIR: 2581, 2375, 1685, 1654, 1541, 1508, 1438, 1242, 1162, and 745 cm–1. HRMS (EI): m/z [M]+calcd for C18H14BrN3O2: 383.0269; found: 383.2781.
Synthesis of N-(benzothiazol-2-yl)-1H-indole-3-carboxamide derivative (9a )
As shown in Scheme 2, thionyl chloride (8 mL) was added dropwise to a solution of 1H-indole-3-carboxylic acid (
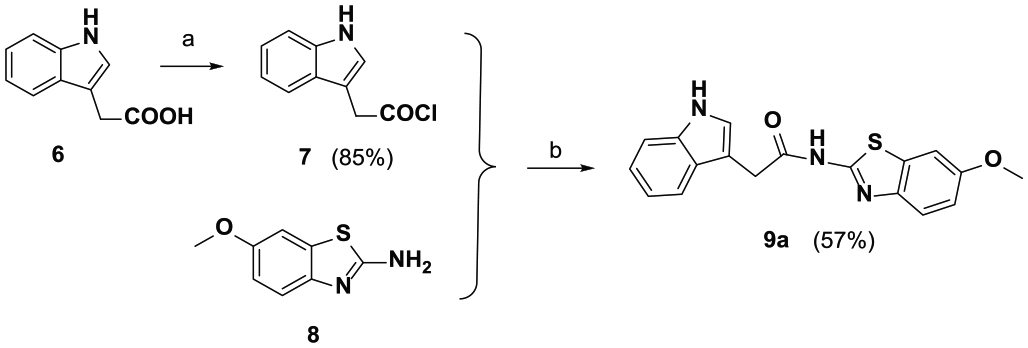
Synthesis of compound
2-(1H-indol-3-yl)-N-(6-metho[d]xybenzothiazol-2-yl)acetamide (9a)
Khaki solid; 57% yield; m.p. 254–257°C. 1H NMR (500 MHz, DMSO-d6): δ 2.83 (s, 3H, CH3), 4.53 (s, 2H, CH2), 7.06 (d, J = 6.5 Hz, 1H, ArH), 7.56 (t, J = 7.5 Hz, 2H, ArH), 7.61 (d, J = 5.5 Hz, 1H, ArH), 7.65 (d, J = 8.5 Hz, 1H, ArH), 7.69 (d, J = 8.5 Hz, 1H, ArH), 8.02 (d, 1H, J = 6.5 Hz, CH–N), 8.13 (s, 1H, ArH), 12.77 (s, 1H, NH). 13C NMR (126 MHz, DMSO-d6): δ 170.41, 156.04, 142.56, 136.05, 132.69, 127.08, 124.29, 121.06, 121.01, 118.56, 118.52, 114.82, 111.43, 107.27, 104.66, 55.55, 32.32. FTIR: 3395, 3174, 3065, 2598, 2832, 1602, 1567, 1438, 1365, 1302, 1267, 1224, 1150, 1122, 1058, 1027, 750, and 817 cm–1. HRMS (EI): m/z [M]+calcd for C18H15N3O2S: 337.0885; found: 337.0914.
Synthesis of 2-(((1H-indol-3-yl)methoxy)methyl)-1H-benzoimidazole derivatives (13a,b)
To a mixture of N-substituted o-phenylenediamine
In the second step, indole-3-carbaldehyde derivatives

Synthesis of compounds
2-(((1H-indol-3-yl)methoxy)methyl)-1-methyl-1H-benzoimidazole (13a )
Khaki solid; 35% yield; m.p. 139–140°C. 1 H NMR (500 MHz, DMSO-d6), δ 3.75 (s, 3H, CH3), 4.64 (s, 2H, CH2), 5.72 (s, 2H, CH2), 7.02 (t, J = 7.5 Hz, 1H, ArH), 7.11 (t, J = 7.5 Hz, 1H, ArH), 7.18 (t, J = 7.5 Hz, 1H, ArH), 7.24 (t, J = 7.5 Hz, 1H, ArH), 7.37 (s, 1H, CH–N), 7.51 (d, J = 8.0 Hz, 1H, ArH), 7.55 (d, J = 8.0 Hz, 1H, ArH), 7.60 (d, J = 8.0 Hz, 2H, ArH). 13C NMR (126 MHz, DMSO-d6): δ 149.56, 141.67, 136.38, 135.89, 127.17, 122.96, 122.71, 122.01, 120.69, 119.30, 118.61, 117.98, 114.17, 111.27, 110.38, 63.02, 59.73, 29.92. FTIR: 3020, 1495, 1464, 1415, 1397, 1182, 1162, 735, and 702 cm–1. HRMS (EI): m/z [M]+calcd for C18H17N3O: 291.1372; found: 291.1154.
2-(((1-benzyl-1H-indol-3-yl)methoxy)methyl)-1H-benzo[d]imidazole (13b )
Orange solid; 32% yield; m.p. 149–151°C. 1H NMR (500 MHz, DMSO-d6): δ 5.13 (s, 2H, CH2), 5.39 (s, 2H, CH2), 5.73 (s, 2H, CH2), 6.95 (t, J = 7.5 Hz, 1H, ArH), 7.08 (t, J = 7.5 Hz, 1H, ArH), 7.14 (d, J = 7.5 Hz, 2H, ArH), 7.22 (t, J = 7.5 Hz, 3H, ArH), 7.27 (d, J = 7.5 Hz, 2H, ArH), 7.40 (t, J = 8.0 Hz, 2H, ArH), 7.55 (s, 1H, CH–N), 7.58 (d, J = 6.5 Hz, 1H, ArH), 7.64 (d, J = 7.5 Hz, 1H, ArH). 13 C NMR (126 MHz, DMSO-d6): δ 149.43, 141.90, 138.07, 136.16, 135.38, 128.43, 128.34, 127.28, 126.84, 126.47, 122.94, 122.00, 121.69, 119.45, 119.28, 118.54, 111.20, 110.50, 109.38, 48.94, 37.21. FTIR: 3100–3500, 3055, 2925, 2850, 1612, 1465, 1240, 1287, 1170, 1127, 1007, and 741 cm–1. HRMS (EI): m/z [M]+calcd for C24H21N3O: 367.1685; found: 367.0964.
Synthesis of (1H-benzoimidazol-2-yl)methyl 2-(1H-indol-3-yl)acetate (14 )
1H-Indole-3-acetic acid ((

Synthesis of compound
(1H-benzoimidazol-2-yl)methyl 2-(1H-indol-3-yl)acetate (14 )
White solid; 79% yield; m.p. 133–135°C. 1 H NMR (500 MHz, DMSO-d6: δ 12.77 (d, J = 25.2 Hz, 1H), 10.97 (s, 1H), 7.69–7.60 (m, 1H), 7.55 (d, J = 15.0 Hz, 1H), 7.51 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.29 (s, 1H), 7.23 (dd, J = 21.0, 8.5 Hz,1H), 7.07 (t, J = 7.5 Hz, 1H), 6.96 (t, J = 7.4 Hz, 1H), 5.31 (d, J = 3.0 Hz, 2H), 3.8 (s, 2H). 13C NMR (126 MHz, DMSO-d6): δ 171.20,149.16, 141.35, 136.05, 127.04, 124.19, 121.07, 118.49, 111.38, 106.59, 59.54, 30.45. FTIR: 3310, 3055, 2930, 1715, 1622, 1544, 1441, 1251, 1128, 1108, 995, and 738 cm–1. HRMS (EI): m/z (M)+calcd for C18H15N3O2: 305.1164; found: 305.1257.
VEGFR-2 kinase assay in vitro
The inhibitory activities of seven compounds (
The inhibition rate against VEGFR-2 (IRE) was calculated by the following formula

Anti-tumor assay in vitro

In this study, the cytotoxicities of seven new compounds and sunitinib malate (a positive drug) against four cancer cells (A549, HT-29, MDA-MB-435, and HeLa) and HUVEC were estimated in vitro. To explain the structure–activity relationship, the activities of (1H-benzoimidazol-2-yl)methyl-1H-indole-3-carboxylate
Molecular docking
The molecular docking study was performed using AutoDock Vina 1.1.2. The crystal structure of VEGFR-2 tyrosine kinase (PDB ID: 4AGD, 2IVU) was obtained from the RCSB Protein Data Bank. The ligand and receptor were prepared and the binding model was analyzed using Molecular Operating Environment.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the National Natural Science Foundation of China (21272131), and Funded by the Key Lab of Marine Bioactive Substances and Modern Analytical Techniques, SOA (MBSMAT-2017-06).