
Abstract
Chemical modifications through substitution are observed to be effective in controlling the optoelectronic properties of various polymers for different applications. In this study, density functional theory–based calculations are employed to investigate the optoelectronic properties of several oligothiophenes based on poly(3-hexylthiophene-2,5-diyl) by varying the number of fluoro and cyano substituents attached. The resulting structures of the polymer derivatives are affected by the electrostatic interactions between the cyano or fluoro groups and the adjacent thiophene unit. Of the two, cyano substitution results in much lower frontier orbital energies for the same number of substituents. It was observed that a decrease in the highest occupied molecule orbital and lowest unoccupied molecular orbital energies correlates very strongly with the number of cyano and fluoro substituents. The effect of the cyano and fluoro groups on the frontier orbitals is also demonstrated and observed to correlate strongly with a lowering of the highest occupied molecule orbital and lowest unoccupied molecular orbital energies as the number of substituents is varied. The predicted solar cell characteristics reveal that most cyano and fluoro derivatives will have improved characteristics compared to unsubstituted poly(3-hexylthiophene-2,5-diyl). This theoretical study shows that by varying the number of electron-withdrawing substituents, the optoelectronic properties may be tuned for solar cell applications.
Keywords

Introduction
In the past decade, we have seen great interest in the study of various materials for solar cell applications, such as perovskite,1–3 small molecules, 4 silicene, 5 graphene,6–8 and conducting organic polymers9–11 as alternatives for Si-based solar cells. Conducting organic polymers can be very attractive commercially due to their various potential advantages, such as low weight, ease of fabrication, low-cost, and high absorption coefficients.12–16 However, conducting organic polymers also face several issues, such as low exciton diffusion lengths,17,18 and poor stability.19,20 These issues can be addressed by looking for new materials with superior properties.
The active layer in organic solar cells (OSCs) is typically a bulk heterojunction (BHJ)21,22 composed of blends consisting of conjugated polymers as the donor material and a fullerene-based material as the acceptor material. The most common donor material used in OSCs is poly(3-hexylthiophene-2,5-diyl), abbreviated as P3HT23,24 typically mixed with [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM)25,26 as the acceptor material. P3HT has a crystalline structure and good charge-transport properties compared to most donor materials used in OSCs; 15 however, P3HT has a relatively high band gap energy (EGap), low photochemical stability/ionization potential (IP), and a low open-circuit voltage (VOC) compared to recent donor materials,27–30 thus conjugated polymers with better characteristics: EGap, IP, and VOC are desirable.
The electronic and optical properties of conjugated organic polymers may be easily adjusted by chemical modifications.31–35 Electron-withdrawing substituents such as fluorine (F) and cyano (CN) have been proven to increase the performance of OSCs by decreasing the highest occupied molecule orbital (HOMO) energy of the polymer resulting in higher open-circuit voltages.36–45 In the study by Rudenko et al., 44 the incorporation of 15% CN groups on the P3HT backbone resulted in a very low HOMO of −5.78 eV and an open-circuit voltage of 0.83 V. Roth et al. 45 reported a significant increase in photochemical stability by introducing 10% of CN groups on the P3HT backbone compared to pristine P3HT and also observed an increase in the photochemical stability of P3HT-CN:PC61BM blends. For F substitution, improvement in charge mobilities of fluorinated poly(3-alkyl)thiophenes was observed which can be attributed to an increase in the planarity of the substituted polymers. 46 In the study by Homyak et al., 47 a strong correlation was observed between increasing %F and electronic properties, where the results suggested a decrease in HOMO energies from −5.08 to −5.47 eV. Blaskovits et al. 48 also observed the same trends with increasing fluorination (0%–50%) and based on their quantum mechanical calculations, favorable sulfur–F interactions resulting in reduction of the dihedral angles between monomer units. Increases in solar cell performances were also observed in fluorinated donor–acceptor-type acceptor materials. In the study by Shim et al., 37 an increase of 25% efficiency was observed using end-group fluorination. In another study carried out by Stuart et al., 40 fluorination of the benzodithiophene (BDT)-dithienylbenzothiadiazole (DTBT) backbone increased the efficiency by 75%. However, Kelly et al. 49 observed the same efficiencies for different fluorination strategies with the polymer poly(benzodithieno-dithienyltriazole) (PBnDT-XTAZ). These results suggest that electron-withdrawing groups such as F and CN are effective in improving the optical and electronic properties of the donor polymers in OSCs. Thus, a greater understanding of the effects that electron-withdrawing groups on the backbone of conjugated polymers have on optical and electronic properties is desired.
The optical and electronic properties of conjugated polymers have been successfully studied by density functional theory (DFT).34,50,51 Even though results from these calculations have been observed to deviate from experimental values, the trends have been observed to be consistent.52–54 Thus, observing changes in the optical and electronic properties by this method can be satisfactorily addressed. In a computational study by Oliveira et al., with n = 2–10 thiophene units, they showed that substitution of all H attached to the thiophene ring with F atoms in the P3HT backbone may result in increased solar cell characteristics compared to pristine P3HT. However, substitution with CN groups greatly reduced both the HOMO and lowest unoccupied molecular orbital (LUMO) energies which may negatively affect the dissociation of excitons due to unfavorable LUMO energy alignment between the donor and acceptor molecules. 55 In our previous study, we reported on the effects of chemical modifications on octamers of P3HT by substitution of various electron-withdrawing groups (Cl, CN, F, NO2, and COCH3) on its bithiophene monomer unit. 56 It was observed that although both F and CN decrease the HOMO and LUMO energies of P3HT, substitution with CN significantly reduces both the HOMO and LUMO energies which may result in better solar cell characteristics than F substitution. The difference between these two studies is the number of substitutions, that is, all thiophene units were substituted with F and CN in the study by Oliveira and Lavarda, 55 while F and CN were substituted for every bithiophene unit in the study by Franco and Padama. 56 From the experimental and computational studies mentioned above, it is possible that the variation of the number of F and CN substituents on the polymer backbone may be the key in predicting their optoelectronic properties and which derivatives will have better solar cell characteristics. Thus, the systematic variation of the substitutions is carried out in this study.
In this work, we further studied the effect of these groups on P3HT by varying the number of F and CN substitutions and determining the effects on the optical and electronic properties of the polymers and on its solar cell characteristics. The hydrogens attached to the thiophene rings were replaced with F and CN groups, and the frontier orbitals energies (EHOMO and ELUMO) and fundamental energy gap (EGap) were determined using DFT. The effect of substitution on relevant solar cell characteristics was then predicted.
Materials and methods
Figure 1 shows the structures of the chemically modified oligothiophenes (P3HT-xCN and P3HT-xF) based on P3HT used in this study. Previously, 56 it was observed that the electronic properties of oligothiophenes converge at around n = 8–12, and that the energy values at n = 8 are closest to the experimental values; thus, n = 8 was used in this study to represent P3HT. This is consistent with previous reports that the energy gap decreases linearly with 1/n only until n = 12 and follows the second-order polynomial for n > 12.57,58 The hydrogens attached to the thiophene rings were replaced with F and CN groups and the number of substituents and their positions are shown in Figure 1. The substituents were varied symmetrically from the central thiophene units toward the outer units, since the effect of the central units on the frontier orbitals is greater than on the outer units. 56 The hexyl group at the 3-position was replaced with methyl to reduce computational costs. Previously, it was observed that the alkyl chain length does not significantly affect the optoelectronic properties of conjugated polymers.57–60 The termini of the oligomers were all terminated with hydrogen. All quantum-chemical calculations were performed using the PC-GAMESS/Firefly QC package, 61 which is partially based on the GAMESS (US) source code. 62 The gas phase equilibrium geometries (opttol = 0.0001 Ha/Bohr) of the oligomers were determined using DFT 63 using B3LYP, a hybrid functional with 20% Hartree-Fock (HF) exchange and 6-31G(d) as the basis set; all atoms were relaxed. Due to the limitations of van der Waals free methods such as pure and hybrid DFT in treating organic materials, Grimme’s D3 dispersion correction with the Becke–Johnson damping, DFT-D3(BJ), was employed.64,65 The Cartesian coordinates of the optimized structures are given in the Supporting Information (Table S1). The DFT/B3LYP method has been reported to satisfactorily predict trends in conjugated systems without significant computational costs.50–54 Total energy calculations were also carried out using DFT-D3(BJ)/B3LYP/6-31G(d). The frontier orbital energies were fitted to the linear correlation between the experimental values 56 and calculated values for P3HT and PC61BM shown in the Supporting Information (Figure S1). Excited states were determined using time-dependent functional theory (TD-DFT)66,67 also at the B3LYP/6-31G(d) level. The fundamental energy gap (EGap) is the difference between ELUMO and EHOMO, while the optical gap (EOpt) is the first singlet excitation energy. 31 The difference between EGap and EOpt is called the exciton binding energy: EB = EGap − EOpt. The open-circuit voltage is calculated using equation (1)

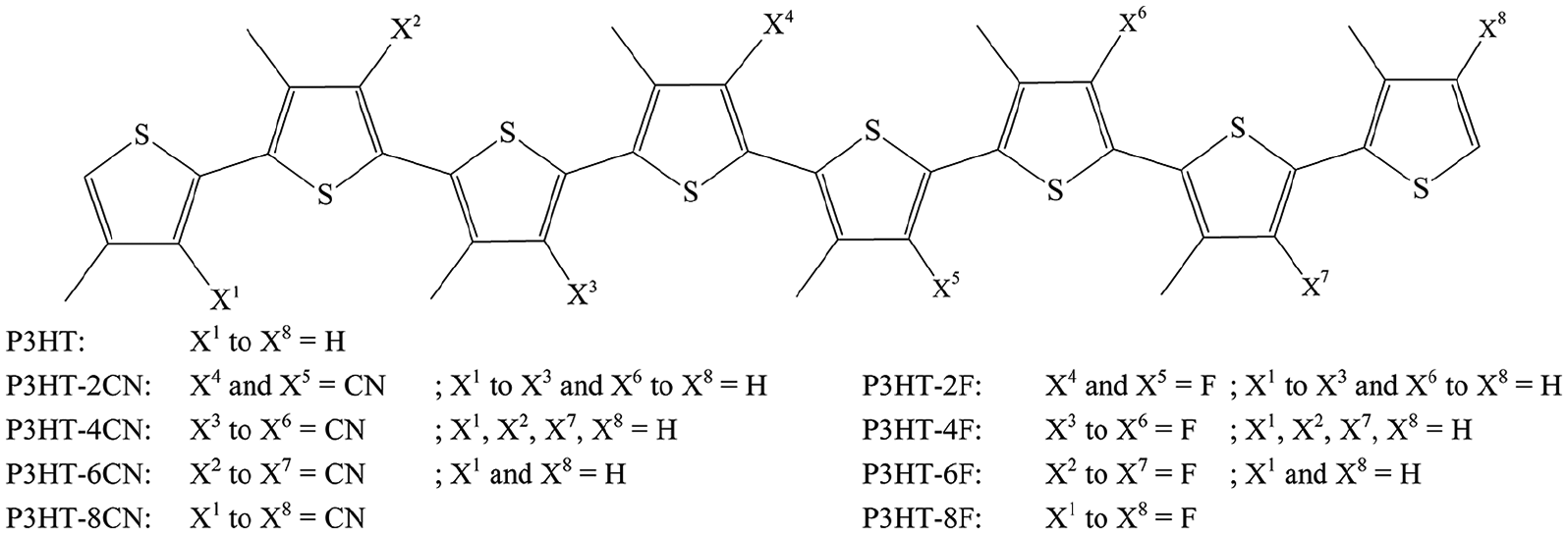
Chemical structures of the oligothiophenes P3HT-xCN and P3HT-xF used in this study.
where e is the elementary charge and 0.3 eV is an empirical factor. 31 The IP was estimated from the negative of EHOMO based on Koopmans’ theorem. 68 Population analysis was performed by employing the Hirshfeld distribution analysis69,70 using the Multiwfn 3.3.8 program. 71 Although the values for the Hirshfeld method are typically lower than other methods, the atomic charges calculated by the Hirshfeld method have been shown to be independent of the basis set used and correlate well with chemical substitution trends.69,72,73 The density of states was evaluated using the GaussSum package. 74 Molecular orbital (MO) surfaces and electrostatic potential surfaces were calculated using wxMacmolPlt. 75
Results and discussion
Electronic properties
The resulting structures for both F and CN derivatives were observed to be planar trans-conformation similar to previous theoretical46,48 and experiment results.43,44 Substitution with CN and F results in planar conformations due to the increased electrostatic interactions between the S atoms of the thiophene ring and the substituted CN and F. Hirshfeld charge analysis reveals that the average Hirshfeld charges (QHirsh) of the S atoms for P3HT-2CN, P3HT-4CN, P3HT-6CN, and P3HT-8CN are +0.114 e−, +0.124 e−, +0.128 e−, and +0.134 e−, respectively; and the average charges of the S atoms for P3HT-2F, P3HT-4F, P3HT-6F, and P3HT-8F are +0.089 e−, +0.091 e−, +0.090 e−, and +0.092 e−, respectively. The QHirsh values for the substituted CN groups in P3HT-2CN, P3HT-4CN, P3HT-6CN, and P3HT-8CN are −0.154 e−, −0.148 e−, −0.143 e−, and −0.141 e−, respectively, and the QHirsh values for the substituted F groups in P3HT-2F, P3HT-4F, P3HT-6F, and P3HT-8F are −0.090 e−, −0.089 e−, −0.089 e−, and −0.090 e−, respectively. It can also be observed that the magnitude of the QHirsh values for the CN-substituted derivatives are greater than those of the F-substituted derivatives, regardless of the number of substituents. The electrostatic potential surfaces of the derivatives also reveal very low potential for CN compared to F, as shown in the Supporting Information (Figure S3). Thus, the electron-withdrawing capability of CN is greater than F, which results in a greater effect on the frontier orbitals as will be discussed in the next section.
Figure 2 shows the fitted energy values for the F and CN derivatives. It can be observed that all the derivatives resulted in lower HOMO and LUMO energies compared to P3HT due to the electron-withdrawing effect of both CN and F groups on the polymer backbone. As the number of substituents increases for both CN and F, the HOMO and LUMO energies also decrease. Of the two substituents, it can be observed that HOMO and LUMO energies of P3HT-xCN derivatives decrease much more than those of P3HT-xF derivatives. These results show that the CN group has a stronger electron-withdrawing effect on the polymer backbone compared to an F group, as mentioned earlier.

Predicted energy values for the P3HT-xCN and P3HT-xF derivatives: EHOMO (bottom of the rectangular boxes), ELUMO (top of the rectangular boxes), and EGap (length of the rectangular boxes) for n = 8 oligomers using DFT at the B3LYP/6-31G(d) level. The experimental EHOMO (−6.12 eV) and ELUMO (−3.76 eV) values for PC61BM are also included for reference (dashed and dotted lines, respectively).
The decrease in HOMO and LUMO energies for CN and F derivatives were observed to be linear with respect to the number of CN and F substituents. Excellent correlations (R2 = 1.0) were observed for the HOMO and LUMO energies with respect to the number of CN and F substituents according to the linear equations, as shown in Figure 3. The linear equations show that the electronic properties of oligothiophenes may easily be predicted and tuned by varying the number of CN and F substituents.

Correlations between the HOMO/LUMO energies of P3HT-xCN and P3HT-xF derivatives.
Density of states
All CN and F derivatives were observed to shift the total density of states (TDOS) of P3HT to lower energy values, as shown in Figure 4. The shifts in the TDOS of CN-substituted derivatives (Figure 4(a)–(d)) were observed to be greater than the shift in TDOS of F-substituted derivatives (Figure 4(e)–(h)). From the projected density of states (PDOS), the contribution of CN π-orbitals to TDOS is greater than the contribution of the p-orbitals in F, which confirms that the CN substituent is much more effective in controlling the frontier orbital energies compared to the F substituent. The frontier MOs as shown in the Supporting Information (Figure S4) reveal that the CN group makes larger contributions to the HOMO and LUMO orbitals compared to the F group. For CN derivatives, the CN substituents were observed to contribute 2.13%, 4.09%, 6.52%, and 7.51% for P3HT-2CN, P3HT-4CN, P3HT-6CN, and P3HT-8CN, respectively, to the HOMO while contributing 4.79%, 5.94%, 6.30%, and 6.66% for P3HT-2CN, P3HT-4CN, P3HT-6CN, and P3HT-8CN, respectively, to the LUMO. However, for F derivatives, the F substituents were observed to contribute 1.36%, 2.43%, 3.04%, and 3.20% for P3HT-2F, P3HT-4F, P3HT-6F, and P3HT-8F, respectively, to the HOMO while contributing 0.51%, 0.97%, 1.51%, and 1.97%, for P3HT-2F, P3HT-4F, P3HT-6F, and P3HT-8F, respectively, to the LUMO. The effect of these contributions was used to explain the decrease in the HOMO and LUMO energies of the CN and F derivatives, respectively.

Total density of states (TDOS) and projected density of states (PDOS) for (a) P3HT-2CN, (b) P3HT-4CN, (c) P3HT-6CN, (d) P3HT-8CN, (e) P3HT-2F, (f) P3HT-4F, (g) P3HT-6F, and (h) P3HT-8F.
Optical properties
Table 1 shows the calculated first singlet excited states (EOpt) and oscillator strengths (fOsc) of the CN and F derivatives using the TD-DFT/B3LYP/6-31G(d) level of theory. The simulated absorption spectra are shown in the Supporting Information (Figure S2). The calculated EOpt is consistent with the experimental EOpt for P3HT at 2.23 eV. 76 The calculated EOpt values for the CN-substituted P3HT are observed to be lower than P3HT for 2CN and 4CN but higher in 6CN and 8CN. A possible explanation of the increase in EOpt and EGap is due to the decrease in the degree of conjugation in P3HT-xCN as the number of CN substituents increases which can be observed in the frontier orbitals in the Supporting Information (Figure S4). However, F-substituted P3HT derivatives are observed to be lower than unsubstituted P3HT. The EOpt values are observed to have the same trends as EGap and all the values are lower than P3HT. Thus, the CN and F derivatives in this study may have wider absorption of the solar spectrum than P3HT. However, the CN and F derivatives generally have lower oscillator strengths compared to P3HT, which may result in a lower absorption coefficient, a crucial parameter for OSCs. Lower absorption coefficients were also reported for CN-substituted43,45 and F-substituted46–48 polythiophenes. This problem could be overcome by a greater increase in other parameters such as VOC, lower EOpt, and increased charge mobilities of the material. It may also be possible to slightly increase the thickness of the material to overcome the reduction of light absorption. The EB values are also shown in the last column of Table 1. It is found that all the values are very close to the value for P3HT and those of the CN derivatives are slightly higher, while those of the F derivatives are slightly lower. In our previous study, we observed that large increases in the dihedral angles between thiophene units contribute to large increases in the EB values. Since the CN and F derivatives are all planar, very minimal effects are observed on EB values.
First singlet excitation energies (EOpt), oscillator strengths (ƒOsc) of n = 8 oligothiophenes calculated at the TD-DFT/B3LYP/6-31G(d) level and the exciton binding energies (EB) of the oligothiophenes calculated from the difference between the calculated EGap and EOpt values (the corresponding main electronic configurations are also shown).

HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital.
Solar cell characteristics
Table 2 summarizes the predicted solar cell characteristics: IP, VOC, EGap/Eopt and ED-R of the P3HT derivatives in this study. From the IP and VOC values, all CN and F derivatives will have higher oxidative stability and open-circuit voltages compared to P3HT, consistent with the reported increase improvement due to CN and F substituents.44,45,47,48 For EGap and EOpt, the increase or decrease in the gap depends on the number of CN and F substituents. The dependence on the increase and decrease of EGap and EOpt were observed in CN-substituted43,44 and F-substituted47,48 P3HT. ED-R is the ability of the polymer to dissociate excitons and prevent recombination and is calculated using ED-R = ELUMO(P3HT-xF/xCN) − ELUMO(PC61BM) − EB. A negative value for ED-R may undesirably affect the exciton dissociation since the LUMO energy values for the acceptor and donor molecules are very close and might not be enough to overcome the exciton binding energy. From Table 2, P3HT-6CN and P3HT-8CN have negative values and may be unfavorable as acceptor molecules for organic photovoltaics (OPVs).
Summary of the predicted solar cell characteristics of the P3HT derivatives in this study.
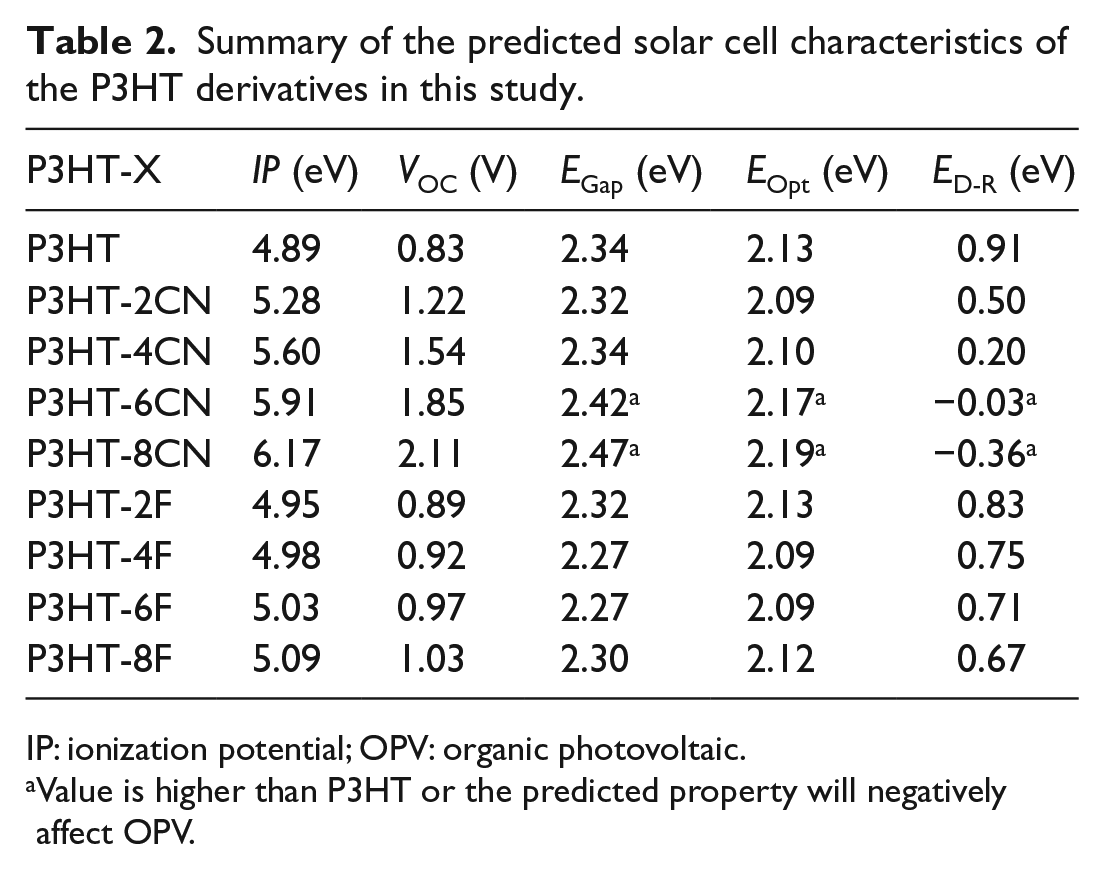
IP: ionization potential; OPV: organic photovoltaic.
Value is higher than P3HT or the predicted property will negatively affect OPV.
Conclusion
In this study, the electronic and optical properties of various oligothiophenes with varying numbers of fluoro or CN substituents were studied via DFT and TD-DFT and the corresponding P3HT-xF or P3HT-xCN solar cell characteristics were predicted. The results show that the CN and F substituents possess negative Hirshfeld charges (QHirsh) while the S atoms of the adjacent thiophene ring possess positive charges which results in planar structures due to electrostatic interactions between the CN or F substituents and S. The CN groups have higher electron-withdrawing ability than the F groups which results in much lower frontier orbital energies. The number of CN and F substituents, and the frontier orbital energies were demonstrated to have a linear relationship, where a decrease in the CN-substituted derivatives is faster compared to F. This is due to the larger effect of the CN groups on the frontier orbitals of P3HT compared to F. The predicted solar cell characteristics reveal that most of the CN and F derivatives could potentially improve the characteristics of P3HT. The theoretical results in this study show how the optoelectronic properties of P3HT can be further improved by varying the number of electron-withdrawing substituents. Moreover, this study may aid in the design of polymers for solar cell applications by chemical substitutions of conjugated polymer materials.
Supplemental Material
Supporting_Information – Supplemental material for Tuning the optoelectronic properties of oligothiophenes for solar cell applications by varying the number of cyano and fluoro substituents for solar cell applications: A theoretical study
Supplemental material, Supporting_Information for Tuning the optoelectronic properties of oligothiophenes for solar cell applications by varying the number of cyano and fluoro substituents for solar cell applications: A theoretical study by Francisco C. Franco in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the University Research Coordination Office (URCO) of De La Salle University, Manila under Project No. 27 F U/S 2TAY15-3TAY16.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.