
Abstract
The structural, elastic, and electronic properties of multi-performance ternary phase MgCaSi have been investigated by density functional theory. The present results show that MgCaSi is thermodynamically and mechanically stable. The derived elastic constants indicate that the c axis is the easiest to compress, followed by the a and b axes. The bulk, shear, and Young’s moduli of MgCaSi are higher than these of the mother phase Ca2Si, demonstrating that the hardness of MgCaSi has been favorably improved. The higher Debye temperature of MgCaSi also indicates stronger interatomic interactions and better thermal conductivity. Although MgCaSi exhibits less brittleness based on Pugh’s empirical formula, Poisson’s ratio, and the Cauchy pressure, orthorhombic MgCaSi possesses lower anisotropy than Ca2Si based on several criteria. To reveal the bonding nature of MgCaSi, the electronic structures are further investigated. It is found that the strong Si−Si bond plays a significant role for structural stability and elastic properties.
Keywords
Introduction
Magnesium alloys have attracted significant attention due to their weight reduction when used in automobiles and aerospace.1–6 Furthermore, lightweight elements such as Si, Ca, and Al are also added to improve the strength and ductility of these Mg alloys,7–9 and hence lightweight elements are important additives for industrial magnesium alloys. It has been shown that the addition of calcium can improve the strength and creep resistance of Mg alloys, 10 and silicon is also found to improve the mechanical properties of Mg alloys; 11 hence, binary Mg-Ca and Mg-Si alloy systems have been developed and studied extensively.12–14 Moreover, several industrial Mg alloys containing both Ca and Si additives have been developed. 15 These new MgCaSi alloys not only are superior structural materials but also offer potential as new types of degradable biomaterials suitable for implant components with a minimized risk of rejection and toxicity.16,17 They can also serve as promising candidates for new and environmentally friendly thermoelectric materials due to the eight valence electrons and applicable electronic structures. 18 Thus, ternary MgCaSi alloys18,19 have attracted significant research interest.
With the technical development of addition of Ca and Si to Mg alloys, several ternary phases have been reported in MgCaSi alloys,9,11,16–19 such as MgCaSi, Ca7Mg7.5±δSi14, Ca2Mg3Si, and Ca2MgSi3. These ternary phases play an important role in the microstructures, properties, and applications of MgCaSi alloys, especially ternary MgCaSi which is a very typical and important phase. Experimentally, the observed precipitated MgCaSi phase increases the tensile strength of alloys.20–23 Moreover, this phase may also be a promising thermoelectric material18,24 and hydrogen storage material. 15 However, research on the ternary MgCaSi phase is very scarce because this ternary phase often occurs as adventitious byproducts. Recently, single crystals of MgCaSi were produced using the metal flux synthesis method 9 and can also be formed when a mixed powder of CaSi2 and Mg is heated; 24 it can even be formed at the Ca2Si/Mg2Si interface by heat treatment in Ca vapor. 23 Although these experiments have produced single-crystal MgCaSi, and some important physical and chemical properties such as hydrogen-storage behavior, heat capacity, and resistivity have also been studied, 9 crucial information on the mechanical, electronic, and other related properties is still scarce; thus, an understanding of its properties is far from complete. Specifically, to the best to our knowledge, theoretical investigations of this ternary phase are lacking.
The elastic properties describe the response of crystals to external stress, as characterized by the bulk, shear, Young’s moduli, and Poisson’s ratio, which directly relate to the hardness and strength of materials.25,26 The elasticity provides an essential criterion for the mechanical stability of crystal materials. Moreover, many microscopic processes of materials, such as dislocation interactions and crack propagation, can be studied based on the elasticity of materials. The elastic properties also concern the thermodynamic properties, such as phonon spectra, the melting points, and Debye temperatures. In particular, the elasticity of materials is quite helpful for studying structural phase transitions and searching for new materials. From an application standpoint, elasticity is very fundamental for structural materials because it provides the important parameters of mechanical properties, and the underlying electronic mechanism is also valuable for the design and manufacture of various purpose thermoelectric devices. 27 Furthermore, knowledge of elastic properties is fundamental for hydrogen-storage behavior and for biomaterials suitable as implant components. Therefore, as structural materials, thermoelectric materials, and hydrogen-storage materials, a knowledge of the elastic properties of MgCaSi is important, and hence study of the elastic properties of MgCaSi is both necessary and urgent.
The single-crystal elastic constants are related to the tribology and stiffness, and they are the important parameters of mechanical properties for crystals.
Although experimental investigations on MgCaSi have been carried out,11,17 theoretical research on the mechanical and electronic properties of the ternary MgCaSi phase is rare. Investigations based on density functional theory (DFT) have become a powerful tool in materials science and engineering, which can effectively overcome the limitations of experiments and can predict various properties of materials, and the results are in good agreement with the experimental measurements. So, in this paper, first-principles calculations based on DFT are employed to study the structural, elastic, and electronic properties of the multi-performance ternary MgCaSi phase by comparison with the mother phase Ca2Si. Our goal is to determine the thermodynamic and mechanical stability of the multi-performance ternary MgCaSi phase and to identify mechanical properties for structural applications, and to further probe the features of the electronic structure and chemical bonding to deepen the understanding of the underlying mechanism for important physical and chemical properties as thermoelectric and hydrogen storage materials. Hence, the present investigation not only provides useful references for mechanical properties of the MgCaSi phase and its structural applications, but also is valuable for understanding the thermoelectric properties hydrogen-storage behavior and implant component applications as biomaterials.
Results and discussion
Structure and stability
From its chemical stoichiometry, the ternary MgCaSi compound appears to be associated with the Zintl phases Mg2Si and Ca2Si; it is in fact the complete substitution of Mg for Ca on one of the 4c sites in the Ca2Si structure, which results in a three-dimensional (3D) Mg/Si network forming channels in which the calcium cations reside. Hence, the Mg and Si sites are four-bonded with highly distorted tetrahedral coordination. 9 Finally ternary MgCaSi also possesses an orthogonal structure with the space group Pnma (No. 62), in which the Mg, Ca, and Si atoms are located in three independent 4c sites. The crystal structure is shown in Figure 1.

Crystal structure of (a) Ca2Si and (b) MgCaSi.
Starting from the above crystal structure, the structural optimization was performed by full relaxation of the cell shape and atomic positions. The optimized lattice constants of MgCaSi are a = 7.4669 Å, b = 4.4311 Å, and c = 8.3019 Å, which are in accordance with the available experimental data, a = 7.4752 Å, b = 4.4272 Å, and c = 8.3149 Å, 9 and theoretical results, a = 7.4800 Å, b = 4.4000 Å, and c = 8.2900 Å. 28 The optimized atomic internal positions are tabulated in Table 1, which are also close to the experimental values, showing that the selected calculation parameters are reasonable, and that our calculations are reliable. For comparison, the lattice constants of the mother phase Ca2Si have also been calculated, and the optimal results, a = 7.648 Å, b = 4.818 Å, and c = 9.041 Å, being also in good agreement with experimental, a = 7.691 Å, b = 4.816 Å, and c = 9.035 Å, and theoretical data, a = 7.667 Å, b = 4.799 Å, c = 9.002 Å. This excellent agreement again shows the high accuracy and reliability of the present calculations. Because the atomic radius (1.60 Å) of Mg is 0.37 Å smaller than that of a Ca atom (1.97 Å), the lattice constant of ternary MgCaSi is slightly reduced. The distinct degree of reduction along three principle axes implies a difference in the chemical bonding.
Atomic positions in the unit cell of MgCaSi.

From Whalen et al. 9
To study the phase stability, enthalpy of formation is calculated by the following expression 29

where
Elastic properties
Elastic constants are the measure of the response of a crystal to the externally applied stress. Under small strain, Hooke’s law is valid, so the elastic strain total energy E is a quadratic function of strain. 30 To calculate the elastic constants, a crystal is elastically strained, and the total strain energy is determined. 31 Details of the calculation of elastic constants by the energy-strain method are described elsewhere.32,33 For the orthogonal structure, there are nine independent elastic constants: C11, C22, C33, C44, C55, C66, C12, C13, and C23. 34 The corresponding nine types of elastic strain are required, as listed in Table 2. In this work, five different deformations for each strain, δ = 0, ±0.005, ±0.01, are taken to calculate the elastic constants. The derived nine independent elastic constants for orthorhombic MgCaSi are listed in Table 3. Clearly, the elastic constant C33 of MgCaSi is smaller than C11, and C11 is only very slightly smaller than C22, showing that the compressibility along the c axis is easier than along the a and b axes, while the b axis is the hardest to compress, implying that the atomic bonding along the c axis is weaker than along the a axis and b axis. The shear elastic constants decrease in the order of C44, C66, and C55. For comparison, the elastic constants of the mother phase Ca2Si are also calculated and are listed in Table 3. It can be seen that all the elastic constants Cij increase with formation of the MgCaSi phase due to substitution of Mg for Ca on one of the 4c sites in the Ca2Si structure. This demonstrates that the incompressibility and shear resistance in MgCaSi phase are stronger.
The strains during calculation of the elastic constants of MgCaSi and Ca2Si.

The calculated elastic constants Cij (in GPa) of MgCaSi and Ca2Si.
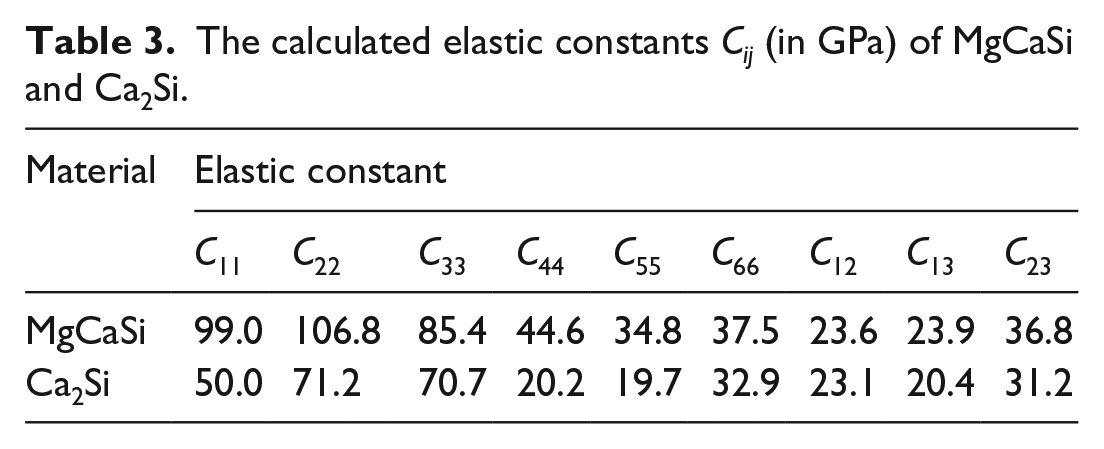
Mechanical stability of crystalline materials requires meeting the condition that the strain energy should be positive against any strain. For an orthorhombic crystal, the requirement of mechanical stability leads to the following restrictions on the elastic constants 35
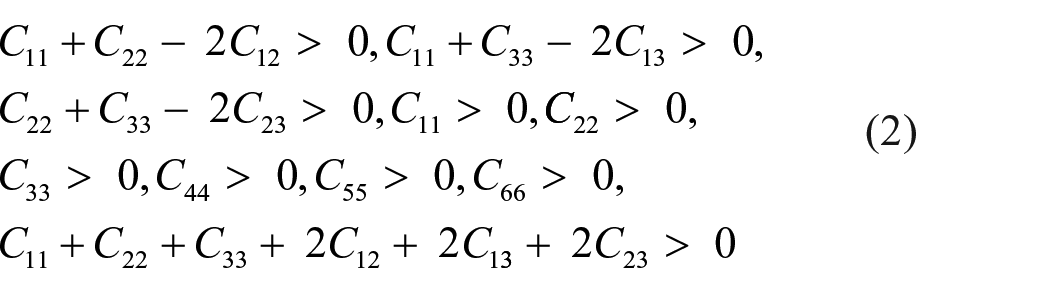
The calculated elastic constants obey the above criteria, so the orthogonal ternary phase MgCaSi is mechanically stable.
Using the single crystal elastic constants, the polycrystalline elastic moduli are further derived using the Voigt–Reuss–Hill (VRH) approximation. 36 The Voigt and Reuss approximations represent the maximum and minimum limits of the polycrystalline elastic moduli, respectively. 37 For the orthorhombic system, the Voigt bounds of B and G are
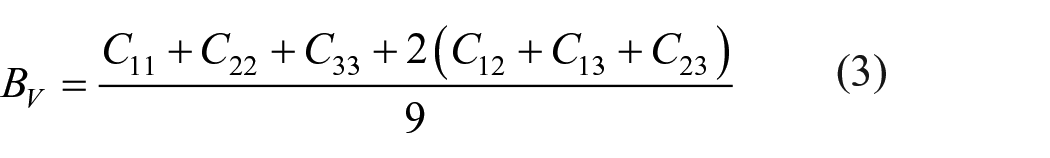

And the Reuss bounds are


where Sij is the elastic compliance constant,
Finally, the VRH mean values of B and G are obtained by


Furthermore, Young’s modulus E and Poisson’s ratio υ can be obtained from the bulk modulus and the shear modulus

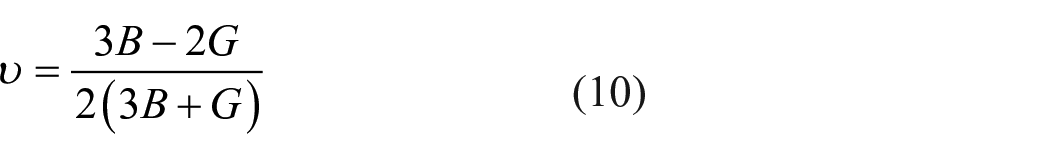
The calculated results are listed in Table 4. The bulk modulus of MgCaSi is larger than that of Ca2Si, showing that the orthogonal ternary MgCaSi phase has strong resistance to volume change because the bulk modulus can be a measurement of resistance to the volume change by applied pressure. Shear modulus is a measure of resistance to reversible shear deformations upon shear stress; it also reflects the strength of directional bonding. The calculated results in Table 4 demonstrate that the shear modulus of MgCaSi is larger than that of Ca2Si, so MgCaSi possesses stronger resistance to reversible shear deformations upon shear stress than the mother phase Ca2Si, and the directional bonding in MgCaSi would also be much stronger. Young’s modulus is essential for technological and engineering applications; it provides an estimate of the stiffness of the solid. The larger the Young’s modulus, the stiffer the material is. 38 From the calculated values, MgCaSi is much stiffer than Ca2Si. Furthermore, Poisson’s ratio is an important quantity, which further reflects the stability of a crystal against shear and which usually ranges from −1 to 0.5. 39 The smaller value corresponds to the stronger stability under the shear stress. In comparison with Ca2Si, MgCaSi is more stable against shear due to the smaller Poisson’s ratio.
The elastic moduli (in GPa) and Poisson’s ratios for MgCaSi and Ca2Si using the Voigt–Reuss–Hill (VRH) approximation.

The ductility of the MgCaSi phase is a crucial quantity of mechanical properties, which can be estimated from the elastic properties. For isotropic polycrystalline materials, Poisson’s ratio is also correlated with the ductility of materials, and it has been employed as a parameter to identify the intrinsic ductility of crystals. A higher Poisson’s ratio implies better ductility of materials. 40 The border line between the ductility and brittleness is about 0.33. 41 As can be seen from Table 4, both MgCaSi and Ca2Si are brittle, and the brittleness of MgCaSi is slightly larger. From the fact that the bulk modulus represents the resistance to fracture, whereas the shear modulus represents the resistance to plastic deformation, Pugh 42 has introduced an empirical ratio G/B to predict the brittle and ductile behavior of materials. A high (low) G/B value is associated with brittleness (ductility). The critical value separating ductility from brittleness is about 0.57. 43 The results in Table 4 show that both MgCaSi and Ca2Si are brittle. Moreover, the brittleness of MgCaSi is slightly higher due to the slightly larger G/B ratio. The Cauchy pressure is also related to the ductility of materials because it implies the angular character of atomic bonding in materials. 44 A positive Cauchy pressure means that the atoms are surrounded by an electron cloud of spherical shape, and the electron distribution is not regional and directional. This implies more metallic bonding, while a negative Cauchy pressure suggests that the distribution of electrons is regional and directional. The more negative the Cauchy pressure, the more directional the bonding. These correlations have been verified for the ductile materials Ni and Al with positive Cauchy pressure due to the typical metallic bonding and for brittle Si semiconductors with negative Cauchy pressure due to directional bonding. 45 The three Cauchy pressure values for the orthogonal structure are computed to be C12 − C66 = −13.70 GPa, C13 − C55 = 10.91 GPa, and C23 − C44 = 7.82 GPa. The negative Cauchy pressure shows the directional bonding and brittle behavior of the orthogonal ternary MgCaSi phase. It is noted that no experimental or theoretical data are available for the elastic properties of MgCaSi, so the present theoretical prediction will provide useful references for further study of MgCaSi in the future.
The elastic anisotropy is important in engineering materials because it is highly correlated with the possibility of inducing microcracks.
46
Furthermore, recent research further shows that the elastic anisotropy has a significant influence on the nanoscale precursor textures in alloys.
47
Therefore, it is crucial in materials science and has been extensively studied. So far several methods have been used to study the elastic anisotropy. The elastic anisotropy in compressibility and shear can be roughly described using two-dimensionless quantities:
48
The percentage of anisotropy in compression AB and shear AG (in %), the universal anisotropy index

It is notable that the above estimate is just considered as the anisotropic level from the individual bulk or shear contribution. In order to determine the extent of the anisotropy considering different contributions from bulk and shear more accurately, a more universal anisotropy index

In this case, AU = 0 defines isotropy, while deviation from zero defines the extent of anisotropy. The calculated AU values in Table 5 also show the elastic anisotropy of both phases. With substitution of Mg for Ca, MgCaSi shows a smaller degree of anisotropy than the Ca2Si phase.
To further reveal the feature of shear anisotropy in detail, the shear anisotropic factor is introduced because it provides a measure of the degree of anisotropy in the bonding between atoms along different directions in crystal planes. 50 For orthorhombic crystals, the shear anisotropy factors between the <011> and <010> directions for the {100} shear plane, those between the <101> and <001> directions for the {010} plane, and those between the <110> and <010> directions for the {001} plane are, respectively, given by Wu et al. 38



The A value of unity corresponds to isotropy, while values smaller or larger than unity indicate the degree of shear anisotropy. The results in Table 5 also show that both MgCaSi and Ca2Si have obvious shear anisotropy. With complete replacement of Ca by Mg on one of the 4c sites in the Ca2Si structure, the shear anisotropy on {100} and {010} seems to be slightly more obvious, while the degree of shear anisotropy on {001} is reduced.
To illustrate the crystallographic direction dependence of elastic properties visually and intuitively, the use of a 3D figure of elastic anisotropy is another effective way. 51 The directional dependence of bulk modulus B, shear modulus G, and Young’s modulus E for a 3D figure can be determined by the following equations 52



where l1, l2, and l3 represent the direction cosines and Sij is the elastic compliance constant that can be obtained through inversion of the elastic constant matrix (

The 3D surface of the directional dependence of (a) bulk, (b) shear, and (c) Young’s moduli of MgCaSi and (d) bulk, (e) shear, and (f) Young’s moduli of Ca2Si.
To further describe the detailed characteristics of elastic anisotropy, the projections on the yz-plane, xz-plane, and yx-plane of the 3D configurations of three elastic modules are accordingly shown in Figure 3. Obviously, these moduli show distinct anisotropy in three planes. For the MgCaSi phase, the bulk modulus on the yz-plane and yx-plane gradually decreases from the y to z and x axes, so the bulk modulus has a maximum along the y-axis and minimums along the z and x axes; it is noted that the value along the x axis is larger than that along the z axis. On the xz-plane, the bulk modulus decreases from the x to z axis. Shear modulus on the yz-plane decreases first and then increases from the y axis to the x axis, reaching a minimum in the 45° angle direction; the value at the y axis is slightly larger than that at the z axis. On the yx-plane, the shear modulus eventually decreases from the y axis to the x axis, dropping to 36.87 GPa. On the xz-plane, the shear modulus increases from the x axis to the z axis, up to 39.89 GPa. The anisotropy of Young’s modulus exhibits approximately the opposite feature to the shear modulus. By comparison, the elastic anisotropy of Ca2Si also exhibits complex features due to the orthogonal structure. It is noted that the projection of the shear modulus on the yz and xz planes is similar. Evidently, with half the Ca atoms of Ca2Si replaced by Mg atoms, the degree of anisotropy is decreased, which is consistent with the above discussion.

The projections of the directional dependence of the (a) bulk, (b) shear, (c) and Young’s moduli of MgCaSi, and the (d) bulk, (e) shear, and (f) Young’s moduli of Ca2Si. The thick lines represent the yz-plane, the short dashed lines are the xz-plane, and the long dashed lines are the yx-plane.
Debye temperature
The Debye temperature is also fundamental parameter in solid materials, which is related to many physical properties, such as melting temperature, thermal expansion coefficient, heat conductivity, and specific heat.53,54 The Debye temperature can also characterize the strength of covalent bonds in solids. 55 The Debye temperature is calculated from the following equation 56

where h represents Planck’s constant, k denotes Boltzmann’s constant, q represents the number of atoms in the molecule, NA refers to Avogadro’s number, ρ denotes the density, M refers to the molecular weight, and υm represents the average sound velocity in the polycrystalline material, which is approximately given by 56

where υs and υl represent the longitudinal and transverse sound velocity and can be obtained from Navier’s equation 57

The calculated density, wave velocities, and Debye temperatures of MgCaSi and Ca2Si are shown in Table 6. The greater elastic moduli of MgCaSi lead to a larger transverse and longitudinal acoustic velocity, and also a higher average sound velocity, so the Debye temperature of MgCaSi is also higher. It should be noted that the Debye temperature depends upon the characteristics of chemical bonds, and higher Debye temperatures correspond to stronger interatomic interactions 55 and stronger bonding strengths.58,59 Therefore, MgCaSi, with a higher Debye temperature, possesses stronger interatomic interactions and chemical bonds than Ca2Si. Moreover, MgCaSi also has better thermal conductivity owing to the higher Debye temperature. 60 The obtained data will be valuable for further theoretical and experimental investigations in the future.

Electronic structure
The electronic properties of MgCaSi are of particular interest. On one hand, MgCaSi may be a semiconducting Zintl phase, like orthorhombic calcium silicide. On the other hand, this MgCaSi phase is considered to span the boundary between charge-balanced semiconducting Zintl phases and fully delocalized intermetallics. 9 Therefore, exploration of the electronic structure for materials falling between these two classifications has been of significant interest. In order to study the electronic properties of the ternary MgCaSi phase and reveal the inherent mechanism for structural stability and mechanical properties from the chemical bonding, the electronic density of states (DOS) for MgCaSi is calculated and displayed in Figure 4(a), in which the Fermi level is set as zero as marked by the vertical lines. For comparison, the calculated DOS of Ca2Si is also shown in Figure 4(b). Clearly, Ca2Si has a narrow band gap, which shows that Ca2Si is semiconducting in nature. The present band gap from generalized gradient approximation (GGA) calculations is 0.34 eV, being close to the other theoretical result of 0.36 eV via the local density approximation (LDA) method. 61 Clearly the theoretical band gap is smaller than the experimental measurement of about 1.90 eV, 62 the deviation originates from the limitations of the pseudopotential and GGA. When half the Ca of Ca2Si is replaced by Mg, the band gap disappears and a pronounced pseudo-gap is formed at the Fermi level (EF). The DOS at the bottom of the pseudo-gap at the Fermi level is close to zero, showing that the MgCaSi phase is intrinsically metallic. Moreover, Ca, Mg, and Si contribute to the states near EF being almost equally; this indicates significant cation–anion orbital mixing (hybridization), 9 which results in closing of the gap at the Fermi level and metallic behavior. The present results are in good agreement with theoretical calculations using the TBLMTO-ASA program package, where crystal parameters are derived from room temperature X-ray single crystal diffraction data. Meanwhile, the Fermi level located at a valley of the pseudo-gap implies pronounced covalence bonding between atoms, so the MgCaSi phase has a strong stability. 63

The total and partial electronic density of the states of (a) MgCaSi and (b) Ca2Si, in which the vertical dotted lines indicate the Fermi levels.
For both, the MgCaSi and Ca2Si phases, the valence band comprises two parts. Figure 4(b) demonstrates that for the Ca2Si phase the valence band in the lower energy region originates mainly from hybridization of the 3s states of Si with the 3p, 3d, and 4s states of Ca. For the valence band in the high energy region near the Fermi level, DOS is mainly composed of the Si 3p states and the Ca 3p, 3d, and 4s states. The present results indicate an obvious covalent bonding between Si−Ca atoms. As shown in Figure 4(a), with the substitution of Mg for Ca, in spite of the similarity of hybridization between Si and Ca atoms, the apparent difference is that in the low energy valence band, the Mg 3s and 2p states hybridize with the Si 3s and Ca 3p, 3d, and 4s states. In the valence band near the Fermi level, the Mg 2p states hybridize strongly with the Si 3p and Ca 3d states, and hybridization of the Mg 3s states with the Si 3p and Ca 3p, 3d, and 4s states can also be observed, implying Mg−Si and Ca−Si covalent bonding in the MgCaSi phase. The Mg−Ca covalent bond is also obvious due to hybridization of the Mg 3s and 2p states with the Ca 3p, 3d, and 4s states in two valence bands. Evidently, with substitution of Ca in Ca2Si by Mg, the newly formed Mg−Si and Mg−Ca covalent bonds are stronger, so the stability of MgCaSi is higher, and the elastic constants and elastic moduli are enhanced.
The charge density distributions were also investigated in order to gain more insight into the features of the bonding. Figure 5(a) and 5(b) displays the calculated contour plots of the charge density distributions on the (010) plane for Ca2Si and MgCaSi. The contour lines are plotted from 0 to 0.05 e Å−3 with 0.005 e Å−3 intervals. High-density regions correspond to the core electron distribution of the Ca and Si atoms, so contributing relatively little to the chemical bonding. 38 From Figure 5(a), one can see clearly that there exists evident overlap of electron densities between Si and Ca atoms in Ca2Si, so the Ca−Si bond possesses a characteristic of covalent bonding. This covalent bonding has somewhat ionicity because the electronegativity of Si is larger than that of Ca. The Si−Si and Ca−Ca bonds are not very strong. With substitution of Mg with Ca on one of the 4c sites in Ca2Si, the charge density of MgCaSi in Figure 5(b) shows that the Si−Si covalent bonds become much stronger due to a large amount of charge overlap between Si atoms. Although an Si−Mg covalent bond is formed, the ionicity of the Si−Mg bond is also obvious. The Ca−Si bond is similar to the Ca−Si bond in Ca2Si. The above features demonstrate that with replacement of Ca by Mg on one of the 4c sites in Ca2Si, the strong Si−Si covalent bond is an important bonding feature of MgCaSi. The calculated charge density distributions and DOS values show that stronger Si−Si, Mg−Si, and Mg−Ca bonds are formed in the ternary MgCaSi phase. Therefore, compared to Ca2Si, the MgCaSi phase has stronger elastic constants and moduli due to the stronger chemical bonding.

The contour plots and charge densities on the (010) plane for (a) Ca2Si and (b) MgCaSi.
Conclusion
The structural, elastic, and electronic properties of multi-performance ternary MgCaSi have been investigated based on DFT within the GGA. The lattice constants of MgCaSi are reduced due to the smaller atomic radii of Mg than that of Ca. Ternary MgCaSi is more thermodynamically stable owing to a more negative enthalpy of formation in comparison with the Ca2Si mother phase; MgCaSi is mechanically stable because the calculated nine independent elastic constants satisfy Born’s criterion. The compressive elastic constant along three principle axes indicates that atomic bonding along the c axis is weaker than that along the a and b axes. The derived bulk, shear, and Young’s moduli increase with the formation of the MgCaSi phase due to replacement of Ca by Mg in the Ca2Si phase, implying that the hardness of MgCaSi has been favorably improved. Compared to the Ca2Si mother phase, the orthogonal ternary MgCaSi phase exhibits smaller elastic anisotropy from several criteria. The calculated Debye temperature of MgCaSi is higher, showing the stronger interatomic interactions, chemical bonds, and higher thermal conductivity. The electronic density of the states and charge density distributions have been further investigated to reveal the bonding features in MgCaSi. The formed stronger Si−Si bond in MgCaSi would play a crucial role for higher stability and increased elastic moduli. These results are expected to be valuable for the further optimization and design of new magnesium alloys with excellent mechanical properties.
Experimental
The DFT calculations were performed with the projector augmented plane wave method, as implemented in the Vienna Ab Initio Simulation Package (VASP) code. 64 The valence electron configurations are 2p63s2 for Mg, 3p64s2 for Ca, and 3s23p2 for Si, respectively. The ion–valence interactions were treated with the projector augmented wave (PAW), 65 and plane-wave cut energy was 400 eV in all calculations. The exchange-correlation functional for electron–electron interactions was treated within the GGA 66 parameterized by Perdew, Barke, and Ernzerhof (PBE). 67 The Brillouin zone integrations were performed using k-point sampling of the Monkhorst–Pack scheme, 62 and 8 × 12 × 8 grids were used for optimization of the geometrical structure of the conventional unit cell and 10 × 14 × 10 grids for calculation of the electronic structure. Structural optimization was performed by full relaxation until the total energy difference was less than 10−4 eV atom−1, the Hellmann–Feynman force on all atoms was less than 10−2 eV Å−1, and the maximum stress was within 0.02 GPa. In the present calculations, the equilibrium lattice parameters were obtained by fitting the calculated total energy–volume curves into the Birch–Murnaghan equation of state (EOS). 68 Elastic stiffness constants were calculated using the energy-strain method, 30 and elastic moduli were derived within the VRH approximation. 36 The numerical accuracy of the DFT-based elastic constants and moduli was set to 0.1 GPa, and the electronic total energy and DOS were calculated using the linear tetrahedron method with the Blöchl correction. 69
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant No. 51461002) and the Key Project of Guangxi Scientific Foundation (Grant No. 2018GXNSFDA281010).