
Abstract
The mechanistic nature of a [3+2] cycloaddition reaction involving zwitterionic species has been investigated, and the changes of electron density related to the O–C and C–C bond formation along the intrinsic reaction coordinate have been characterized. This polar [3+2] cycloaddition reaction, which takes place through a non-concerted two-stage one-step mechanism, proceeds with a moderate Gibbs free activation energy of 21 kcal mol−1. The reaction begins by the creation of a pseudoradical centre at the central carbon, first on the dimethyl acetylenedicarboxylate, and second on the nitrone framework. This immediately favours the formation of the first O–C single bond by donation of some electron density of the oxygen atom lone pairs, which represents the most attractive centre in this cycloaddition reaction.
Keywords
Introduction
The [3+2] cycloaddition (32CA) reactions between a three atom-component (TAC) species and a substituted ethene to afford small heterocyclic compounds 1 attract much attention in bio-medicinal chemistry. 2 However, the TAC compounds are species containing more than four electrons delocalized between three contiguous atoms. Since the introduction of the chemical bonding concept, 3 several theoretical models have been improved to understand structure and chemical reactivity. In this sense, density functional theory (DFT) has proven to be very useful for studying structure and reactivity.4–9 In the present decade, calculations of the quantum mechanical observables along the reaction pathway have led to an understanding of the bond-forming and bond-breaking processes.6,10 For this purpose, the electron localization function (ELF)11,12 method and bonding evolution theory (BET) 13 have emerged to analyze reaction mechanisms.14–19 The ELF is considered as a new tool for analyzing electronic changes in chemical processes and is considered as an appealing procedure that provides a more straightforward relationship between the electron population reorganization and the chemical structure. A number of DFT studies achieved within the molecular electron density theory (MEDT) 20 in 32CA reactions have permitted establishing a valuable classification of this cycloaddition reaction involving TACs (Figure 1); 20 this classification has been permitted the study of reactivity and molecular mechanism.
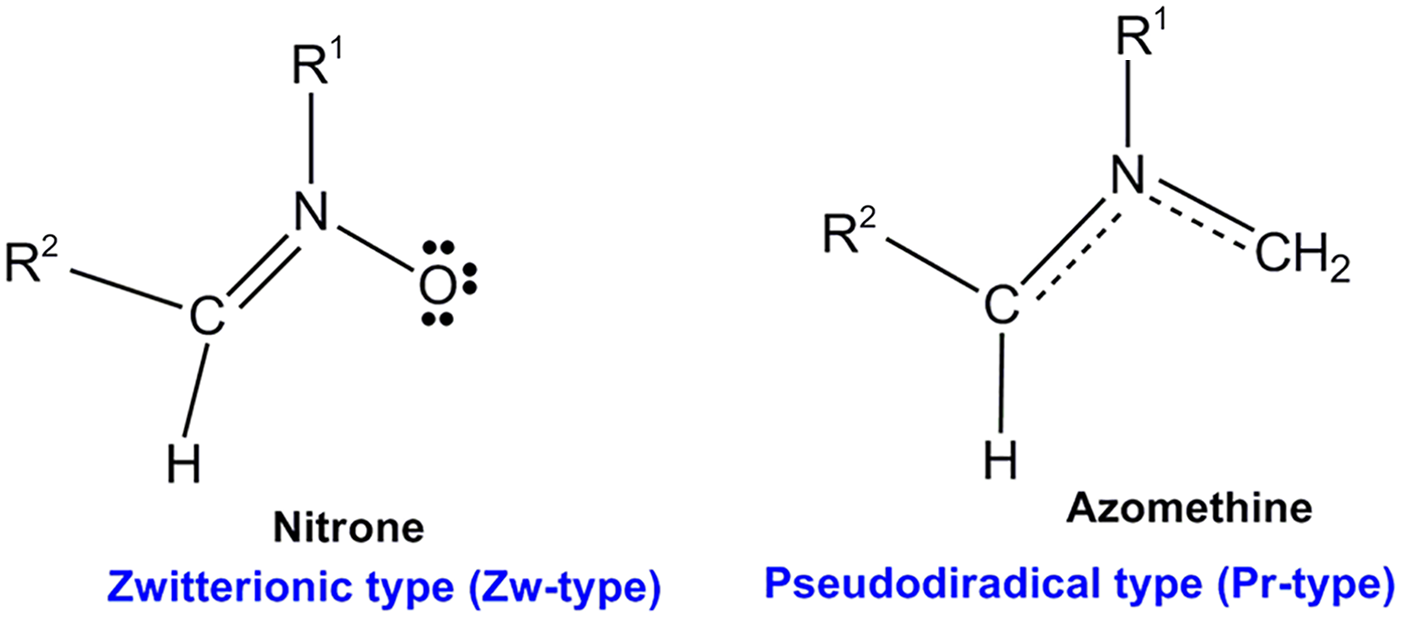
Electronic structure of TACs and proposed reactive zwitterionic and pseudo-diradical types in 32CA reactions.
In this article, the 32CA reaction of a nitrone with electronic-deficient dimethyl acetylenedicarboxylate (DMAD) 21 was studied within the MEDT through DFT calculations at the B3LYP/6-31G+(d,p) computational level. In the course of our studies, we evaluated the reactivity and mechanistic nature of this reaction. Therefore, our primary purpose in this article was the exploration of nitrone reactivity. A quantum chemical topological analysis of the C–C and O–C single bond formation involving zwitterionic-type species along this 32CA reaction was performed in order to characterize the molecular mechanism. Due to the symmetry of DMAD, we can envisage one channel which has a diastereomer OH-trans to the methyl group at the ring junction (Scheme 1).
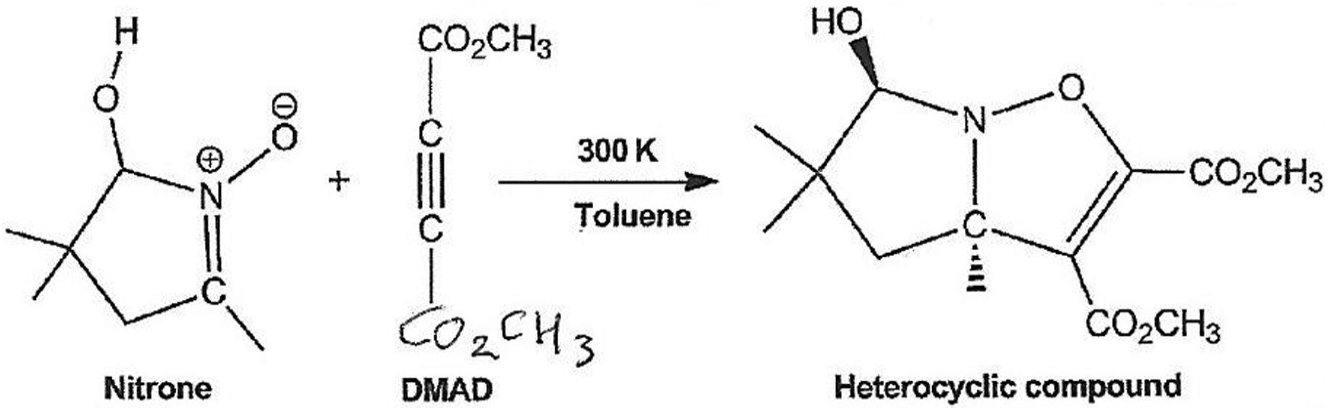
32CA reaction of nitrone with dimethyl acetylenedicarboxylate.
Computational methods
The optimizations of stationary points were performed using the Berny analytical gradient method,
22
and also the intrinsic reaction coordinate (IRC) paths
23
were drawn, in order to ensure the energy profiles linked the transition state (TS) to the two connected minima, using the second-order Gonzalez–Schlegel integration method.
24
The electronic structures of stationary points were analyzed by the natural bond orbital (NBO) method
25
and by ELF topological analysis.
12
The ELF study was carried out with the Multiwfn program
26
using the corresponding mono determinant wave functions of the selected structures. All computations were realized using the B3LYP exchange–correlation functional,27,28 together with the standard 6-31+(d,p) basis set.
29
The calculations were performed with the Gaussian 09 suite of programs,
30
and the charge transfer (CT) was calculated using the natural population analysis.
31
The global electrophilicity index, ω,
32
is given by the following expression, ω = μ
2
/2η, in terms of the electronic chemical potential μ and the chemical hardness η. Both quantities may be approached in terms of the one-electron energies of the frontier molecular orbitals Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), EHOMO and ELUMO, as µ = (EHOMO + ELUMO)/2 and η = (ELUMO – EHOMO), respectively.
33
The nucleophilicity index N
34
based on the HOMO energies is obtained within the Kohn–Sham scheme,
35
and defined as N = EHOMO(Nu) – EHOMO(TCE). The nucleophilicity was referred to tetracyanoethylene (TCE), because it presents the lowest HOMO energy in a large series of molecules. Electrophilic,
Results and discussion
In order to establish the nature of the reaction mechanism and to evaluate the changes of electron density along the IRC of the 32CA reaction between a nitrone and electron deficient DMAD, an analysis of the conceptual density functional theory (CDFT) reactivity indices was performed. This was followed by an examination of the reactivity of the most significant centres, and then the electronic structures associated with the nitrone and DMAD were explored and characterized using an ELF topological analysis. Simultaneously, an ELF bonding analysis along the new bond-formation/breaking pathway to this reaction was completed in order to characterize their electronic structures, and the C–O bond formation and mechanism involving zwitterionic species was investigated.
Analysis of the CDFT reactivity indices of nitrone and DMAD
An analysis of the CDFT reactivity indices of the nitrone with DMAD was performed to predict their reactivity in the 32CA reaction. The global indices, namely, the electronic chemical potential, µ; chemical hardness, η; electrophilicity, ω; and nucleophilicity, N, at the ground state of the reagents involved in this 32CA reaction are given in Table 1.
B3LYP/6-31G+(d,p) electronic chemical potential (µ), chemical hardness (η), electrophilicity (ω) and nucleophilicity (N) of nitrone and DMAD together with HOMO and LUMO energies.
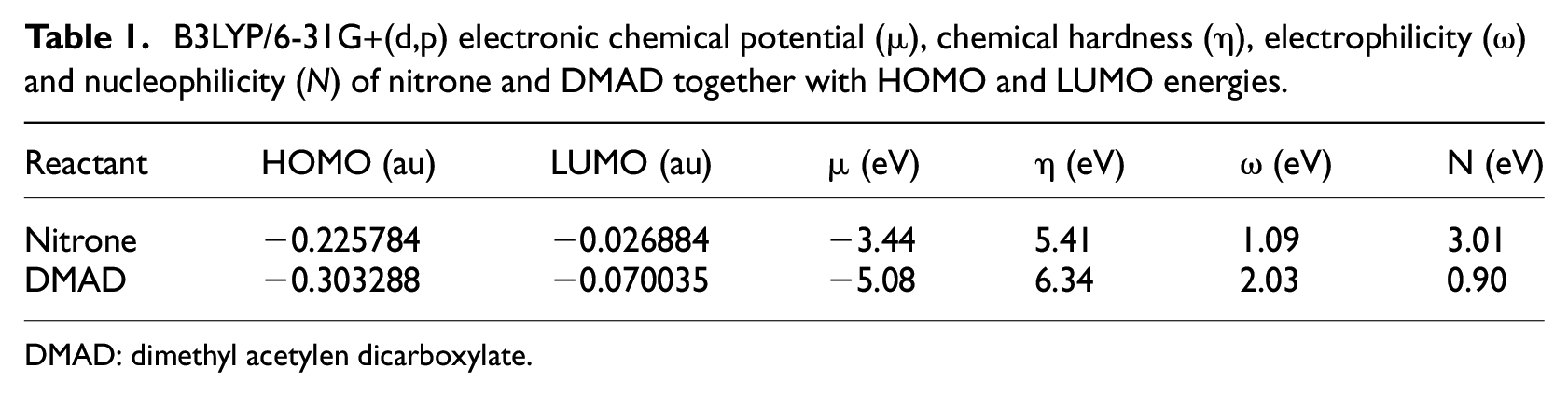
DMAD: dimethyl acetylen dicarboxylate.
The electronic chemical potential of the nitrone µ = –3.44 eV is higher than that of DMAD, µ = –5.08 eV. Thus, the nitrone has a tendency to exchange electron density with DMAD during this 32CA reaction, so the electron flow will take place from the nitrone to DMAD. Besides, DMAD presents a large electrophilicity index ω of 2.03 eV, being classified as a strong electrophile and as a feeble nucleophile, N = 0.90 eV. Thus, the nitrone behaves as a strong nucleophile, with a nucleophilicity index of N = 3.01 eV according to the nucleophilicity scale. However, the electrophilicity of DMAD is larger than that of the nitrone 1.09 eV, indicating strongly that this reaction is broadly polar. Therefore, in this 32CA reaction, the nitrone and DMAD behave as a nucleophile and an electrophile, respectively.
Analysis of the reactivity of the most attractive centres
By using Parr functions, we can investigate with more accuracy the reactivity of each reactive centre, and the nucleophilicity and electropilicity parameter indices were very valuable for exploring the degree of reactivity. The nitrone is a good nucleophile while DMAD is a good electrophile; at this stage, we calculated the nucleophilic and electrophilic Parr functions for the nitrone and DMAD, respectively. The results are given in Figure 2.
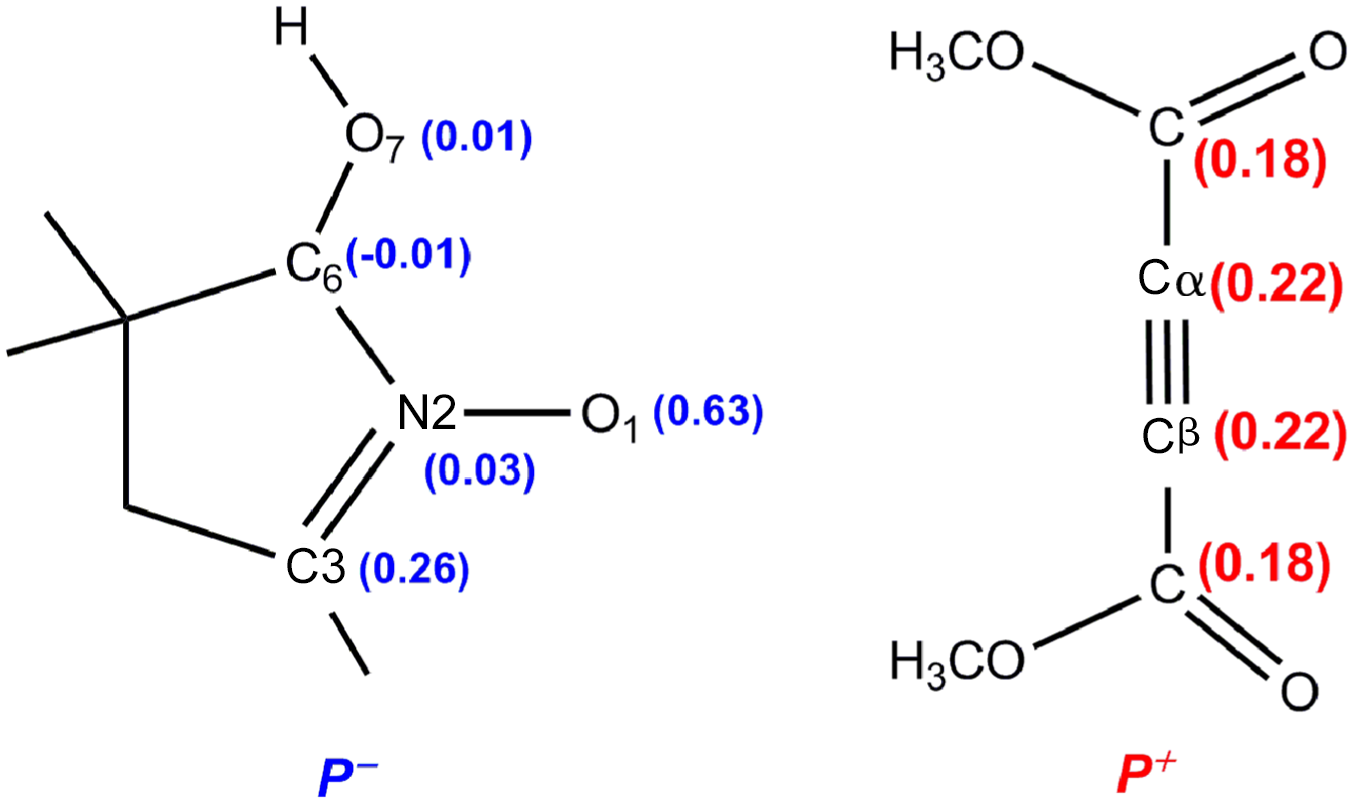
Parr functions of nucleophile (P−/nitrone) and electrophile (P+/DMAD).
Some appealing conclusions can be drawn: O1 is the most nucleophilic atom of the nitrone which has the largest nucleophilic Parr function of 0.63; so such an atom will contribute highly to the reactivity, the presence of three lone pairs of electrons makes the O1 atom very rich electronically. In addition, the C3 atom is the second most powerful nucleophilic centre which has a very significant electron charge and presents 0.26 on the Parr function scale. Moreover, the N2 atom is a feeble nucleophile with Parr function value 0.03 in the TAC sequence (O1–N2–C3). Therefore, the TAC is the most attractive chain of the nitrone. Since the presence of the symmetry axis of DMAD makes the Parr function electrophile values analogous, the Cα = Cβ (0.22), so the Cα and Cβ atoms are equivalent. Subsequently, the O1 atom will react exclusively with one of them.
ELF topological analysis of the electronic structure of nitrone and DMAD
The reactivity of the nitrone can be correlated with its electronic structure. In this sense, the ELF provides a more straightforward connection between the electron density distribution and the chemical structure, in which the ELF divides the electron density of a molecule into basins. Therefore, an ELF topological analysis of the nitrone and DMAD was performed in order to characterize the electronic structure of this TAC sequence. The representation and attractor positions of ELF valence basins, as well as ELF electron populations, arising from the ELF topological analysis are shown in Figure 3. It is interesting to note that the basins are the areas where the chance to find an electronic pair is at a maximum. Therefore, there are monosynaptic, disynaptic and trisynaptic basins. To this end, the monosynaptic basins, labelled V(M), correspond to lone pairs or non-bonding regions, while disynaptic basins, labelled V(M, N), connect the core of two nuclei M and N.
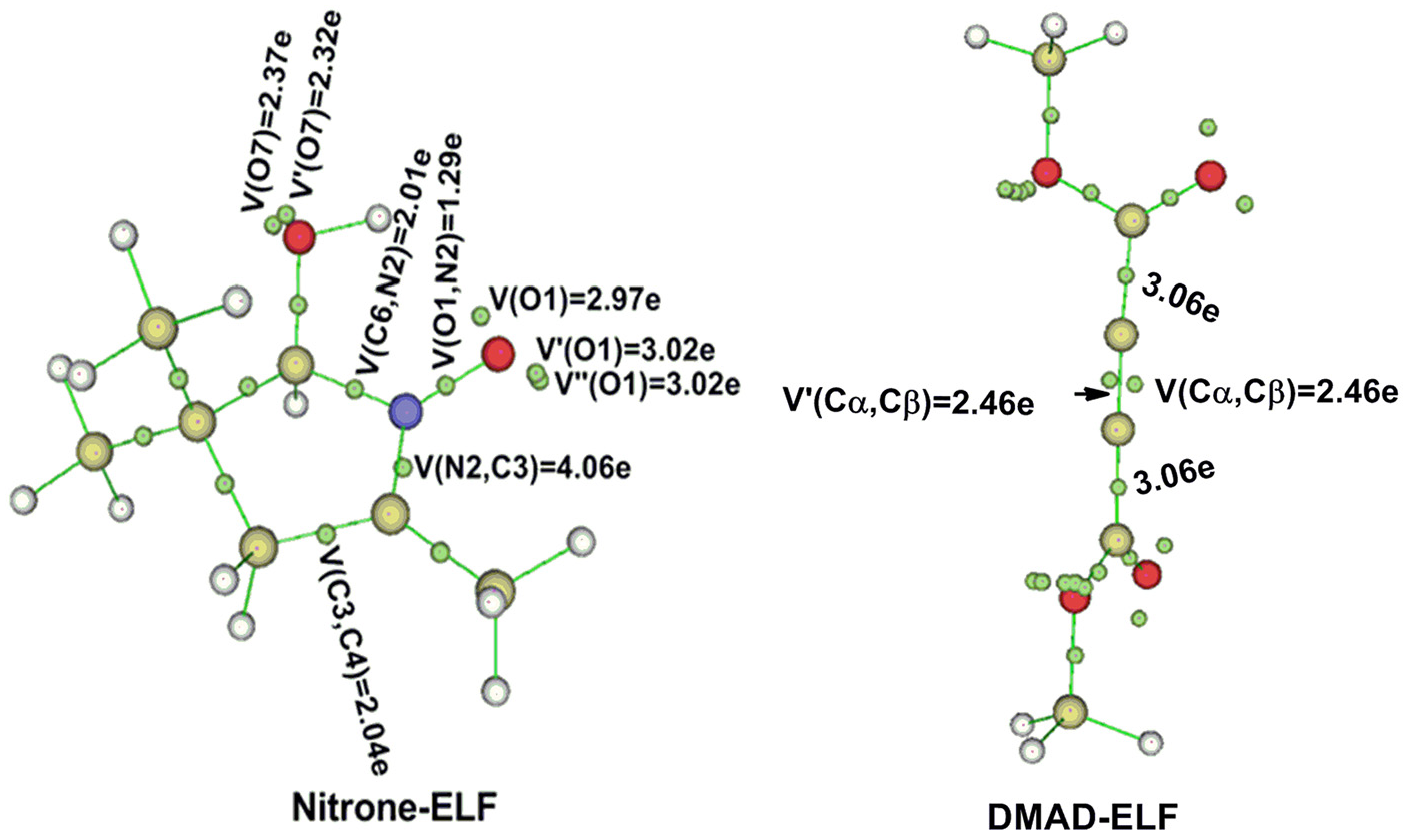
ELF valence basin populations, in e, for nitrone and DMAD.
In this section, we report about ELF topology, i.e. the study permits establishing the electronic structure distribution on this TAC. However, the nitrone presents two analogous V’(O1) and V’’(O1) monosynaptic basins, each with a population of 3.02 e, one monosynaptic basin V(O1) with a population of 2.97 e (total 2 × 3.02 e+2.97 e≈ 9 e), and one V(O1, N2) disynaptic basin with a population of 1.29 e. This behaviour suggests that the O1–N2 bonding region is strongly polarized towards the O1 oxygen atom and a major part of the electron density has been absorbed, i.e. O1 is highly populated electronically. Also, the presence of one V(N2, C3) disynaptic basin integrating 4.06 e indicates that the N2–C3 bonding region has a strong double bond character. Consequently, ELF topology of the nitrone clearly indicates that this TAC is able to participate only in zwitterionic-type 32CA reaction electronic structure (Figure 1), and thus, ELF topological analysis allows establishing of the proposed Lewis structure given in Figure 4. On the contrary, the ELF topology of DMAD shows the presence of two V(Cα,Cβ) and V’(Cα,Cβ) disynaptic basins integrating a total electronic density of 4.92 e ≈ 5 e, indicating strongly that this Cα–Cβ bonding region has a triple bond character. As a result, both reagents have the tendency to exchange electron density between them and provide high reactivity in accordance with the polar reaction.
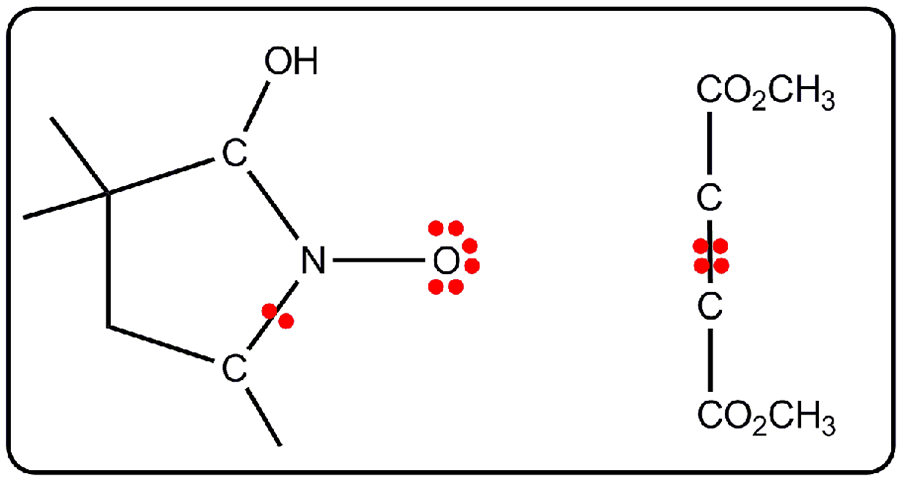
Representation of Lewis structures for nitrone and DMAD obtained from ELF topological analysis in zwitterionic-type species.
Potential energy surface analysis of the stationary points involved in the 32CA reaction between nitrone and DMAD
In this section, we focus on the thermodynamic and kinetic parameters. Accordingly, an analysis of the potential energy surface (PES) was performed in the presence of solvent (toluene) according to the IEF-PCM model at 300 K. However, by reason of the symmetric DMAD, this reaction has one process channel. The results show that this cycloaddition reaction takes place via an asynchronous TS; this result allows us to note that the bond distances are completely different, in which the new bond formed between O1 and C(α or β) has the smallest distance, 1.80 Å instead of 2.52 Å between C3 and C(α or β) (see Figure 5). Subsequently, this 32CA reaction is extensively highly asynchronous. As a consequence, the O1–C(α or β) bond will be formed first along the IRC. On the contrary, the thermodynamic results show that the mechanism is moderately energetic. Thus, we have established that the activation energy is 18.3 kcal mol−1. Likewise, the calculation of the Gibbs free energy of activation ΔG provides a similar outcome, such as we have found, i.e. 20.6 kcal mol−1. Consequently, the activation barrier is feeble to moderate in excellent agreement with the extensive CT in the TS, in which NBO analysis reveals that the CT is about 0.25 e in accordance with a polar reaction, the reaction being exothermic by 12 kcal mol−1 (Figure 6).
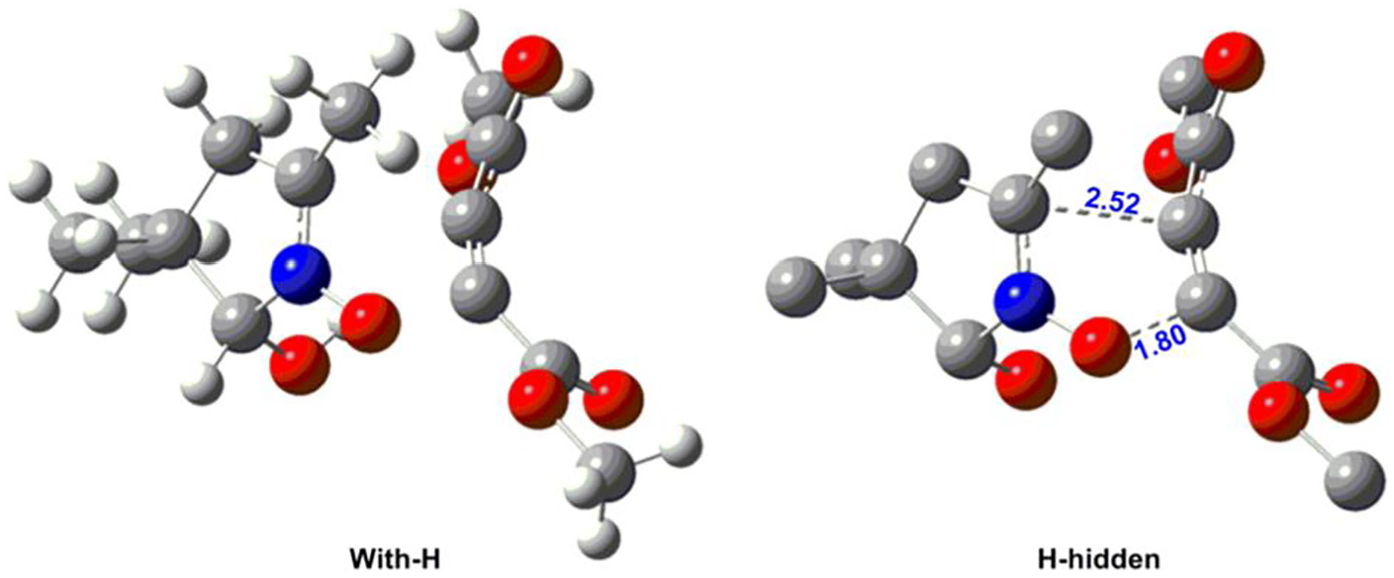
Bond distances, in angstrom (Å), of transition structure in 32CA reaction.
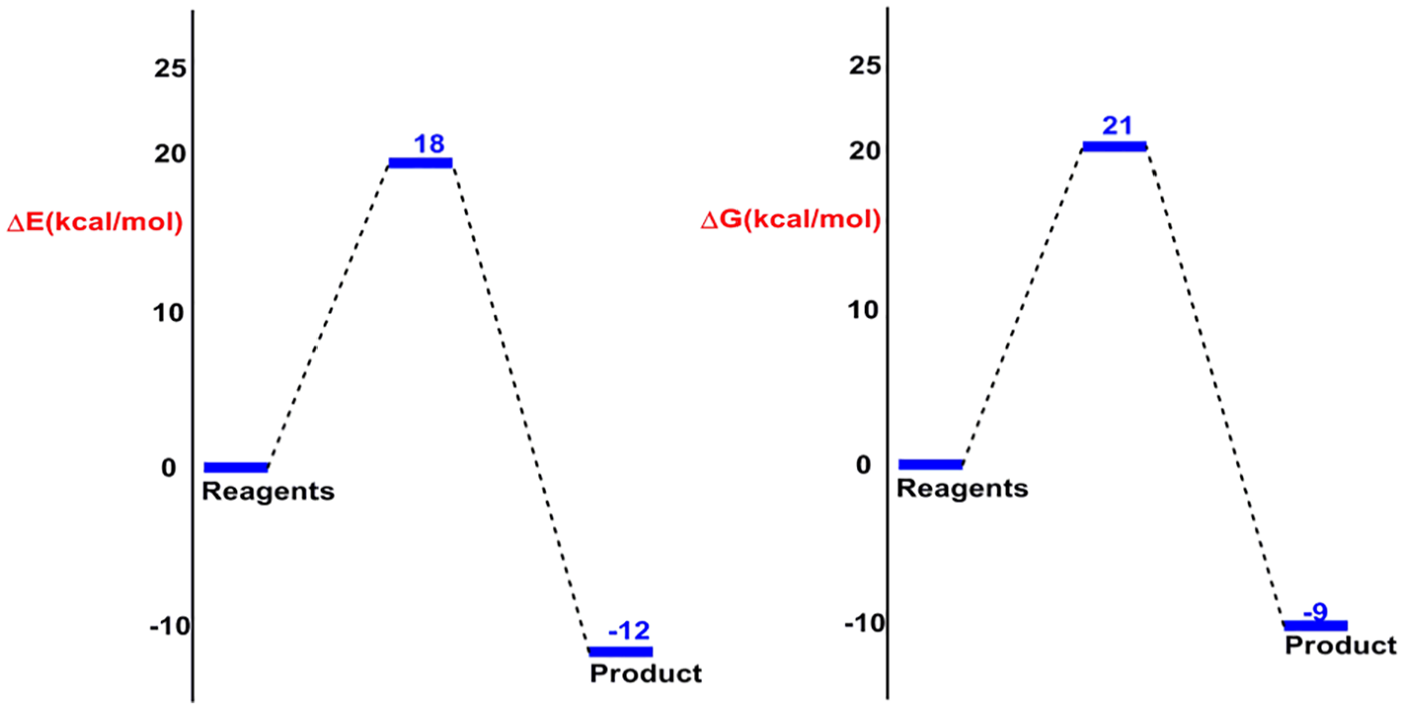
Relative energy and Gibbs free energy profile, in the presence of solvent toluene, of 32CA reaction between nitrone and DMAD.
ELF analysis of the molecular mechanism of the zwitterionic–type in the 32CA reaction between nitrone and DMAD
In previous decades, an ELF study of the bonding changes along the zwitterionic-type 32CA reaction was performed in order to understand the bond formation/breaking processes and the molecular mechanism. 40 BET has been demonstrated to be a very practical methodological device. 41 Several theoretical studies have revealed that the topological investigation of the ELF derived from BET offers a suitable outline for the study of the changes of electron population distribution. This procedural approach is used as a valuable tool to understand the bonding changes along the reaction path, and thus, to establish the nature of the electronic rearrangement as the reaction progresses.
Therefore, an ELF study along the IRC was performed with the aim of characterizing the molecular mechanism of the 32CA reaction involving zwitterionic-type species. The electronic density populations of the most relevant ELF valence basins of chosen structures along the IRC of this 32CA reaction are listed in Table 2, while the attractor positions for the most relevant points associated with the formation of the O1–Cα and C3–Cβ single bonds are shown in Figure 7.
Valence basin populations of the most relevant points calculated from the ELF of the 32CA reaction of nitrone with DMAD, associated with the O1–Cα and C3–Cβ bond formation steps.
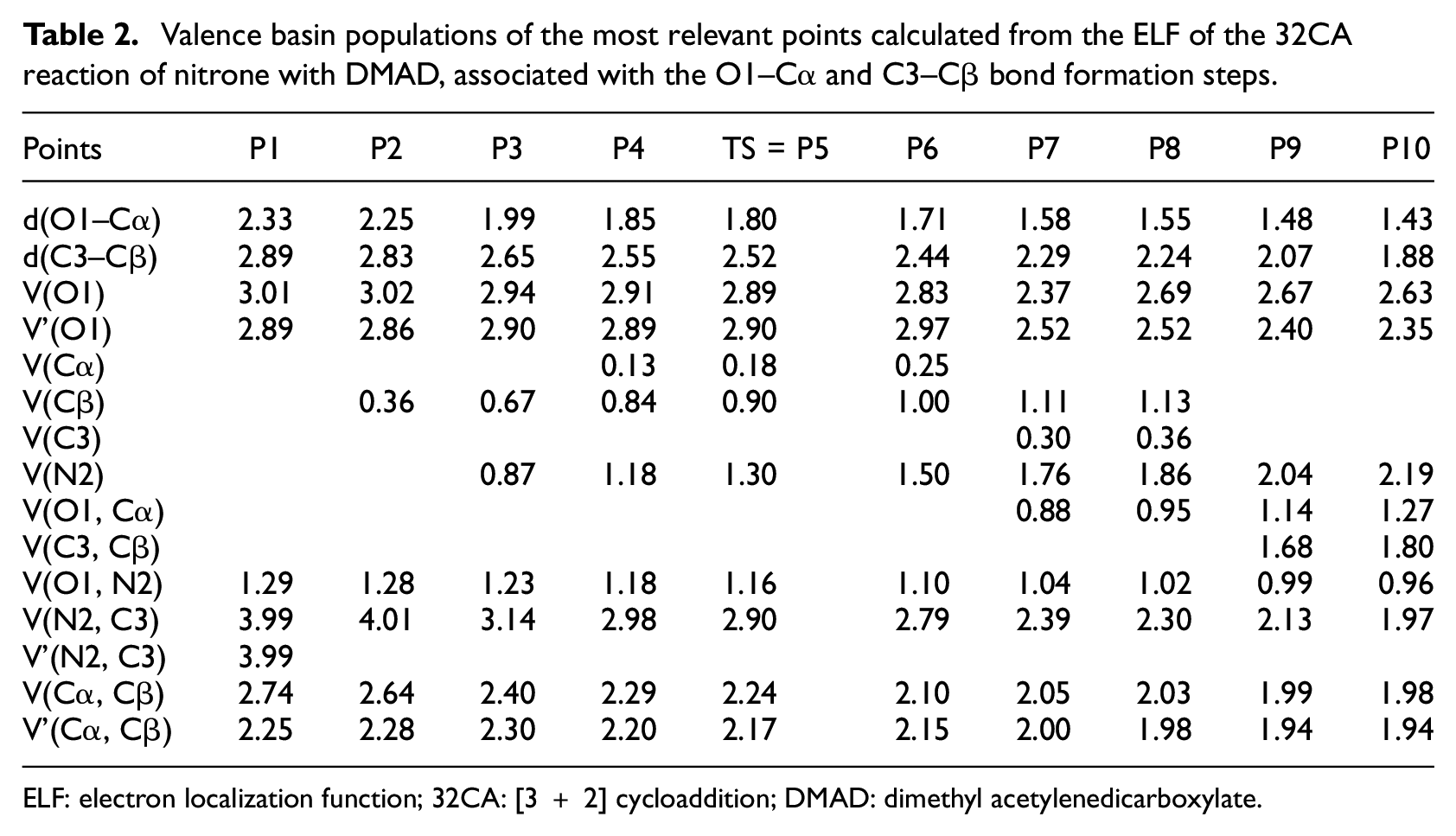
ELF: electron localization function; 32CA: [3 + 2] cycloaddition; DMAD: dimethyl acetylenedicarboxylate.
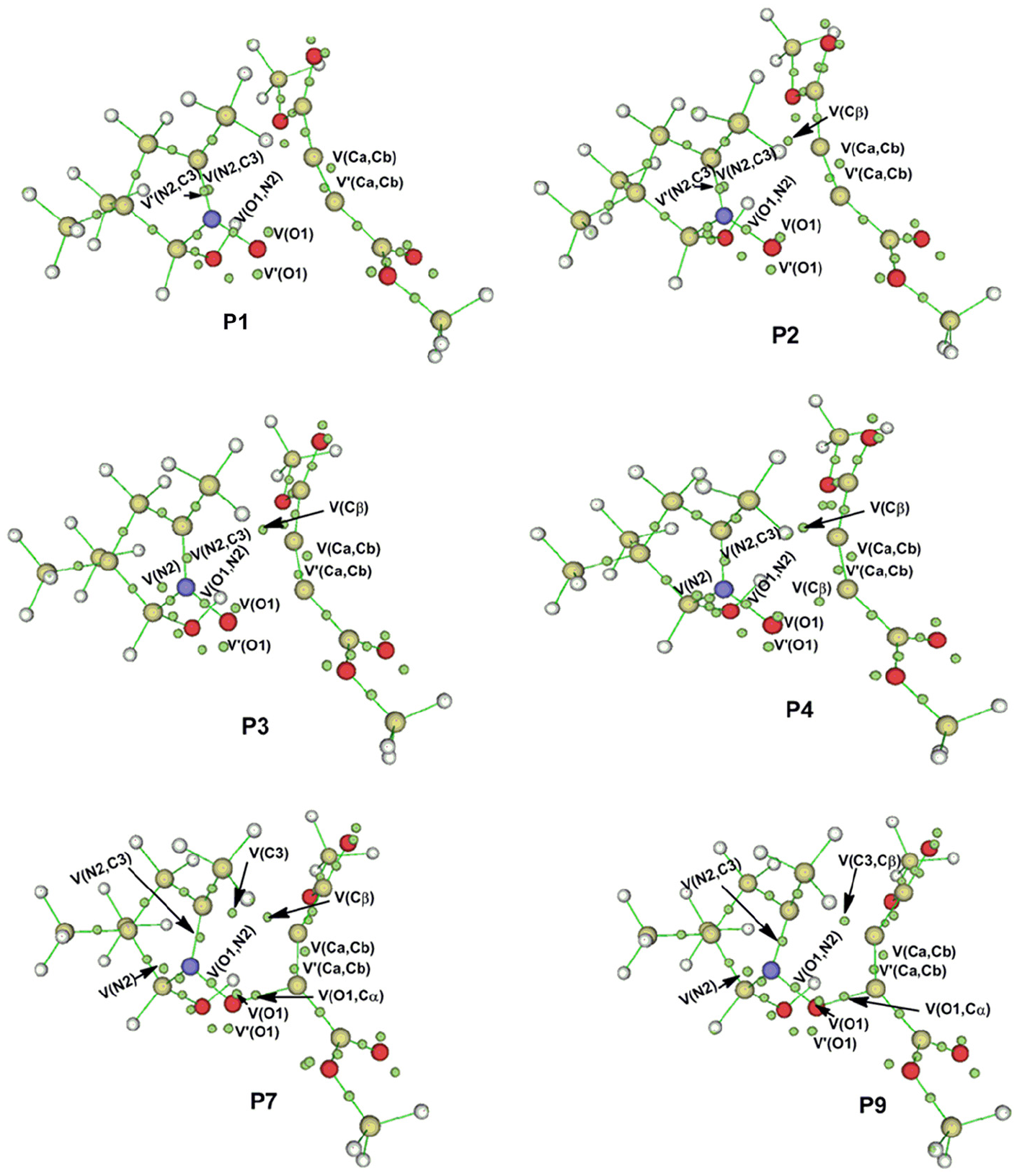
ELF attractors of some relevant points of the IRC related to the formation of the new O–C and C–C single bonds in the 32CA reaction of nitrone with DMAD.
Some appealing conclusions can be drawn from this ELF study: the IRC associated with the 32CA reaction of the nitrone with DMAD is divided into 10 significant differentiated points. A behaviour that clearly indicates that the bonding changes along this one-step mechanism are non-concerted; the formation of the first O1–Cα single bond takes place in the early stage of reaction at an O–C distance of 1.58 Å to an appreciable electronic population of 0.88 e, by the donation of some of the electron density of the O1 oxygen lone pairs to the Cα pseudoradical centre of DMAD (see Table 2 and the V(O1, Cα) disynaptic basin in P7 in Figure 7), as a consequence of the electrophilicity and nucleophlicity scales, in which the O1 oxygen is the most nucleophilic centre of the nitrone and the C (Cα = Cβ) carbon corresponds to the electrophilic centre of DMAD, which demands a symmetric depopulation of the Cα–Cβ bonding region of DMAD (see Parr functions in section ‘Analysis of the reactivity of the most attractive centres’). Note that the electronic population of O1 lone pairs is considerably decreased in P7 due to the donor effect. At this stage, the second C3–Cβ single bond is not yet started. Furthermore, this first stage is characterized by the absence of the monosynaptic basin of lone pair V(N2) in P1 and P2 due principally to their contribution to enriching the N2–C3 double bond region. Moreover, formation of the second C3–Cβ single bond takes place at a C–C distance of 2.07 Å, integrating 1.68 e by the coupling of the pseudo-diradical of the monosynaptic basin of V(C3) and V(Cβ) in point P9, in which the monosynaptic basin V(C3) first appears in P2 with an electronic population of 0.36 e and the monosynaptic basin V(Cβ) created in P7, was e = 0.30 (see Table 2 and the V(C3, Cβ) disynaptic basin in P9 in Figure 5). It is worth noting that the Cβ carbon participates with an appreciable electron density of 1.13 e in the formation of the C3–Cβ single bond. Subsequently, this 32CA reaction follows a two-stage one-step mechanism in which the formation of the second C3–Cβ bond begins when the first O1–Cα single bond is practically already formed. Moreover, the formation of these bonds is totally different and highly asynchronous (O–C and C–C bond distances, see Table 2); this strongly indicates that this reaction has a non-concerted mechanism. The depopulation of Cα–Cβ bonds and the donation of electron density by lone pairs of the O1 atom, which is revealed by this BET/ELF study, can be related to the zwitterionic-type 32CA reaction between the nitrone and electron-deficient DMAD.
A recapitulated picture showing the bonding changes along some selected points involved in this 32CA reaction is given in Figure 8, while a representation of the relative position of the selected points along the IRC with respect to the energy profile along the two-stage one-step mechanism of the 32CA reaction between the nitrone and DMAD is presented in Figure 9.
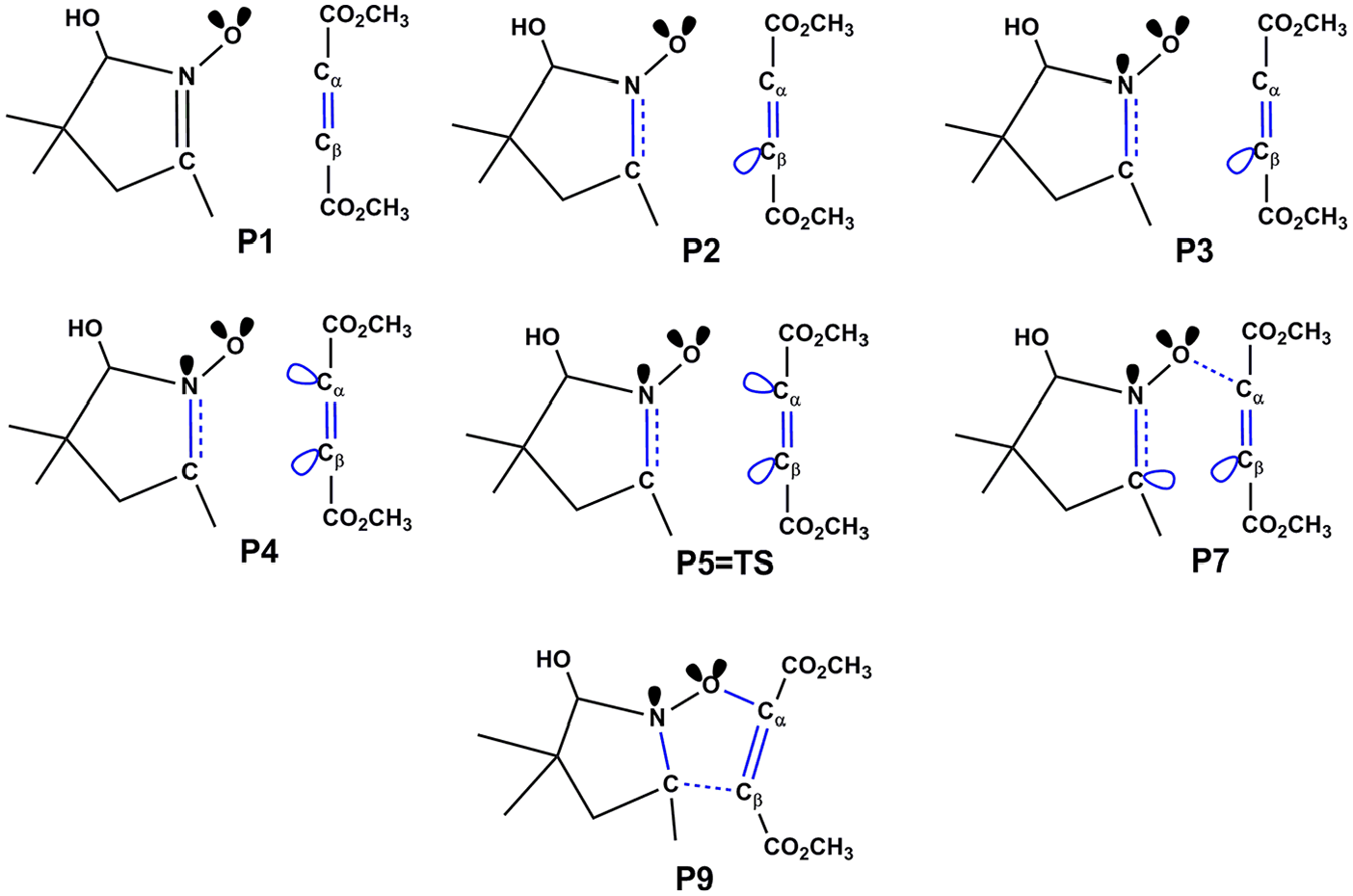
Bonding changes displayed at selected points involved in the 32CA reaction of nitrone with DMAD.
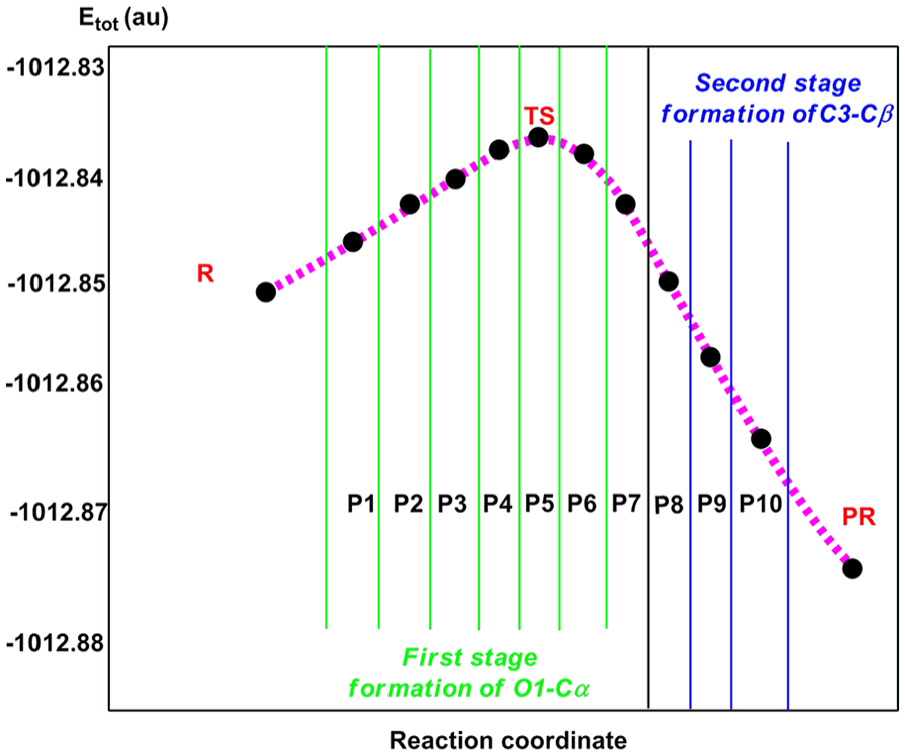
Energy profile, in atomic units (au), along two-stage one-step mechanism associated with the formation of the O1–Cα and C3–Cβ single bonds of the 32CA reaction of nitrone with DMAD.
Conclusion
The 32CA reaction involving zwitterionic species to give heterocyclic compounds has been studied within MEDT at the DFT B3LYP/6-31G+(d,p) computational level in order to explain the mechanism and the C–O bond formation between the electronically-rich and the electronically-deficient atoms. ELF analysis of the nitrone confirmed its electronic structure as a TAC contributing in a zwitterionic-type 32CA reaction. The analysis of CDFT reactivity indices correctly predicts the nucleophilic and electrophilic character of the nitrone and DMAD, respectively, and then, the donor nature of the nitrone. A detailed analysis of the PES corresponding to the reaction reveals that the reaction is moderately energetic, with an exergonic demanding Gibbs free energy, when this thermodynamic study was the performed in the presence of solvent toluene. However, an ELF topological analysis of the stationary points predicted that this 32CA reaction takes place with a two-stage one-step mechanism in which the formation of the second bond will be started once the first is almost completely formed. The formation of the O–C first single bond occurred between the most significant atoms according to the Parr functions, by the donation of some electron density as part of lone pairs associated with the O atom, while the formation of the C–C second bond is a consequence of coupling the pseudoradical centre as a result of the CT and the depopulation of C–C double bonds. Thus, the present computational study confirms the non-concerted nature of the cycloaddition between the nitrone and DMAD.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.