
Abstract
Magnetite iron oxide nanoparticles synthesized using the co-precipitation methods were further functionalized with activated carbon. The magnetite-activated carbon nanoparticles were characterized by scanning electron microscopy equipped with energy dispersive X-ray spectroscopy, transmission electron microscopy, X-ray diffraction, Fourier transform infrared spectroscopy, and UV-Vis spectroscopy. X-ray diffraction and Fourier transform infrared confirmed the functionalization of the Fe3O4 nanoparticles with the activated carbon. The X-ray diffraction studies demonstrate that magnetite-activated carbon nanoparticles were indexed into the spinel cubic lattice with a lattice parameter of 0.833 nm and an average particle size of about 14 nm. Various parameters such as dislocation density, microstrain, and surface morphological studies were calculated. However, this work implicated the use of magnetite-activated carbon nanoparticles in antibacterial studies. Further, the antibacterial effect of magnetite-activated carbon nanoparticles was evaluated against three pathogenic bacteria, which showed that the nanoparticles have moderate antibacterial activity against both Gram-positive (Staphylococcus aureus) and Gram-negative (Proteus mirabilis and Pseudomonas aureginosa) pathogenic bacterial strains in the presence of different magnetite-activated carbon nanoparticle concentrations at room temperature.
Keywords

Introduction
During the last decade, there has been an increasing interest in developing and understanding of matter at nanoscale,1,2 as they represent an intermediate dimension between bulk materials and atoms/molecules. 3 Nowadays, the most well-known nanomaterials are the inorganic compounds due to their novel and improved physical, chemical, and biological properties.3–7 Inorganic nanomaterials exhibit very good electric, optical, electronic, magnetic, and biological properties, and are widely use in pharmaceutical, medical, and environmental applications.8–14 Due to the resistance of medicinal nanomaterials toward microorganisms, many researchers have turned toward engineered nanoparticles (NPs) for solving this problem. Recent advancement in the field of nanotechnology has provided attractive method for synthesizing alternative antimicrobial agents and reducing biofilm formation. 15 In the area of antibacterial agents, metal NPs are of a particular interest because they can be synthesized with high surface areas and highly potential active sites. 16 A distinct class of metal oxide with distinctive magnetic properties and superior biocompatibility is found in magnetite iron oxide nanoparticles (MIONs). 17 MIONs with particle sizes less than 100 nm, have been developed for a wide range of biomedical applications. Examples include contrast agents for magnetic resonance imaging (MRI),18–22 hyperthermia agents, 23 and carriers for targeted drug delivery to treat several types of cancer. 24 Specifically, the research group cultured osteoblast (bone-forming cells) with MIONs (at 4.25 mg mL−1 concentration) and found that the cell density was greatly enhanced in the presence of MIONs compared with cells cultured without NPs. 25 The MIONs can also produce antimicrobial effects on different microorganisms such as Staphylococcus aureus, Xanthomonas, Escherichia coli, and Proteus vulgaris bacterial species. 17 Lee et al. 26 stated that zero-valent iron NPs with size of 10–80 nm caused the inactivation of E. coli bacteria. Taylor and Webster 27 also described concentration-dependent bacteria inhibition on Staphylococcus epidermidis when incubated with MIONs concentrations of 100 µg mL−1, 1 and 2 mg mL−1.
In this study, for the first time, we report a simple, low-cost, and environmental friendly method for preparing magnetite iron oxide–activated carbon NPs (Fe3O4@C NPs) via a co-precipitation method. The structural properties of synthesized NPs were characterized by scanning electron microscopy (SEM) equipped with energy dispersive X-ray spectroscopy (EDS), transmission electron microscopy (TEM), X-ray diffraction (XRD), Fourier transform infrared (FTIR), and UV-Vis spectroscopy. In addition, the antibacterial activity of the Fe3O4@C NPs was evaluated against Gram-positive (S. aureus) and Gram-negative (Proteus mirabilis and Pseudomonas aureginosa) bacteria in the presence of different Fe3O4@C NP concentrations.
Results and discussion
Characteristics of the Fe3O4@C NPs
The approach described here is to embed Fe3O4 NPs onto the activated carbon (AC) surface. The SEM image was used in order to investigate the morphology of the synthesized Fe3O4@C NPs. As can be seen in Figure 1, the SEM images show that Fe3O4@C NPs are composed of spherical shaped particles and core–shell particles on the AC surface. The magnetite particle size appeared to depend on the duration over which the ammonia solution was added to the ferric and ferrous salt mixture; a shorter duration (about 5 min) produced smaller particles that “spiked” readily when placed near a magnet. Small magnetite particles are desirable, since they have large surface-to-volume ratios, large reactive surfaces, and strong magnetism. 28 The procedure for the synthesized Fe3O4@C NPs involved coating or mixing the porous AC surface with a magnetic solution. This allowed the small magnetic particles, when formed, to be intimately mixed on the AC surface leading to a desirable structure for the Fe3O4@C NPs (Figure 1).

FESEM images of Fe3O4@C NPs at different resolutions: (a) 30 μm, (b) 50 μm, (c) 1 μm, and (d) 500 nm.
The morphology, structure, and size of the Fe3O4@C NPs were determined by TEM on different scale bars of 40–200 nm (Figure 2(a)–(c)). Figure 2(d) shows the histogram of particle size versus number of particles observed by TEM grid on the scale bar of 100 nm. The particle size distribution is in the range of 4–50 nm. It is clear from the histogram that the mean particle size of Fe3O4@C NPs is 14.66 nm. The particles exhibited core–shell and spherical morphology. In general, magnetite NPs synthesized through co-precipitation tends to aggregate together. As can be seen from Figure 2, a random distribution of Fe3O4@C NPs accrued, and due to the presence of Fe3O4 NPs, magnetization behavior was expected.

(a)–(c) TEM images of the size and morphology of Fe3O4@C NPs with different resolutions of 40–200 nm. (d) Frequency distribution histogram for particle size of Fe3O4@C NPs at 100-nm scale. It is clear from the histogram that the particle size distribution is in the range of 4–50 nm and the mean particle size of the Fe3O4@C NPs is 14.66 nm.
The EDS results shown in Figure 3 qualitatively determined the surface composition of the Fe3O4@C NPs. The EDS spectra showed the strong peaks of Fe, O, and C. This confirms the existence of oxygen and iron in the sample. Therefore, it is assumed that Fe3O4 NPs are coated onto the surface of AC.

EDS results of Fe3O4@C NPs.
The UV-Vis absorption spectra of the Fe3O4@C NPs (Figure 4) shows an absorption band in the region of 300–400 nm (λmax = 300.46 nm) which originates primarily from the absorption and scattering of UV radiation by magnetic NPs, and is in accordance with the previously reported literature.29,30

UV-Vis absorption spectrum for a diluted suspension of Fe3O4@C NPs.
As can be seen in Figure 5, the strong, broad peaks in the FTIR spectrum of the Fe3O4@C NPs at about 560–570 cm−1 are due to the stretching vibrations of Fe–O. The peaks around 3300–3500 cm−1 and 1300–1650 cm−1 have been assigned to the stretching and bending vibrations of the H–O–H bond, respectively, showing the physical absorption of H2O molecules on the surfaces. The spectrum also shows that the H–O–H bending vibration at about 1000–1600 cm−1, typical of the H2O molecule, is less intense. In addition, the second absorption band, between 900 and 1000 cm−1, corresponds to the bending vibration associated with the O–H bond. The O–H in plane and out of plane bonds appear at 1583–1481 cm−1 and 935–838 cm−1, respectively. 31 These first two bands correspond to the hydroxy groups attached by the hydrogen bonds on the iron oxide surface, as well as the water molecules chemically adsorbed to the magnetic particle surfaces. 32

FTIR spectrum of Fe3O4@C NPs.
In addition, the absorption bands at 574 cm−1 correspond to the vibration of tetrahedral, which are indicative of formation of a spinel ferrite structure. The shouldering of the band corresponding to the tetrahedral site is observed for the sample. It is attributed to the Jahn–Teller effect (a geometric distortion of a non-linear molecular system) produced by the Fe2+ ions that causes local deformations in the lattice owing to the non-cubic component of the crystal field potential, and hence leads to the splitting of this band, corresponding to the tetrahedral site. 33 Finally, the band observed at 574 corresponds to Fe3O4. However, after Fe3O4@C NPs formation, there are shifts of notable peaks such as the O–H, C=C, and C–O stretches, indicating that reduction had occurred. This indicates that Fe3O4 NPs synthesized on the AC as a stabilizer.
The XRD analysis of Fe3O4@C NPs is shown in Figure 6. The peaks located at diffraction angles of 2θ = 30.43°, 35.71°, 43.53°, 53.73°, 57.31°, and 62.99° represent the crystalline planes (220), (311), (400), (422), (511), and (440), respectively, which obviously show the crystalline cubic structure of the magnetite phase (Fe3O4) (no. 98-024-0798).34,35 The strongest reflection comes from the (311) plane, which denotes the spinel phase. Also, the spectrum obtained from the XRD profile shows typical peaks at 2θ = 26.6°, 44.68° which are related to AC in the (002) and (100) directions, respectively. 36 The results confirm that Fe3O4 NPs are successfully impregnated onto the AC surface.

XRD of Fe3O4@C NPs powder.
The crystallite size of the nanocrystalline samples was measured from the X-ray line broadening analyses using the Debye–Scherrer formula after accounting for instrumental broadening (equation (1))37,38

where λ is the wavelength of the X-ray used in Å, β is the line broadening at half the maximum intensity (full width at half maximum (FWHM) in radians in the 2θ scale), θ is the Bragg angle, DXRD is the crystallite size in nm. The average of the particle size of Fe3O4@C NPs was found to be 14.9 nm using Debye–Scherrer equation. The lattice parameter (a) and interplanar spacing (dhkl) are determined by Bragg’s law. 14 The values obtained are shown in Table 1. The lattice parameter (a) and interplanar spacing (dhkl) for the Fe3O4@C NPs are lower than the values reported for bulk magnetite JCPDS Card No. (79-0417) (a = 8.394 and d311 = 2.531), but these values are close to some of the values reported for Fe3O4 NPs in the literature.39,40
The values of observed “d,” crystallite size, dislocation density, strain, and h, k, l of Fe3O4@C NPs.

Dislocation density (∂) is calculated with the crystalline size (D) 41

Microstrain (ε) arises due to the lattice misfit, which varies on the deposition conditions and thus it is calculated by the formula (Table 1) 41

As can be seen in Table 1, the dislocation density (∂) for Fe3O4@C NPs (on diffraction angles at 30.43°–62.99° representing the crystalline planes) decreases with an increase in the crystallite size (D). Similarly, the microstrain (ε) increases with a decrease in the crystallite size.
Bactericidal activity of Fe3O4@C NPs
The antibacterial activities of the Fe3O4@C NPs were evaluated against three pathogenic bacteria (Gram-positive and Gram-negative) at a concentration range of 11.50–36.30 mg mL−1 in distilled water (Table 2 and Figure 7). The results of the antibacterial activities of the Fe3O4@C NPs at different concentrations (11.50, 20.10, 23.00, and 36.30 mg mL−1) showed moderate antimicrobial activity against all the pathogenic strains with zones of inhibition ranging from 3.8 to 5.0 cm (Table 2). The zone of the clearance around each well after the incubation period confirms the antimicrobial activity of the synthesized Fe3O4@C NPs.
Comparison of the literature data on the antibacterial activity Fe3O4 NPs at different concentration ranges with the data of Fe3O4@C NPs used in this study.
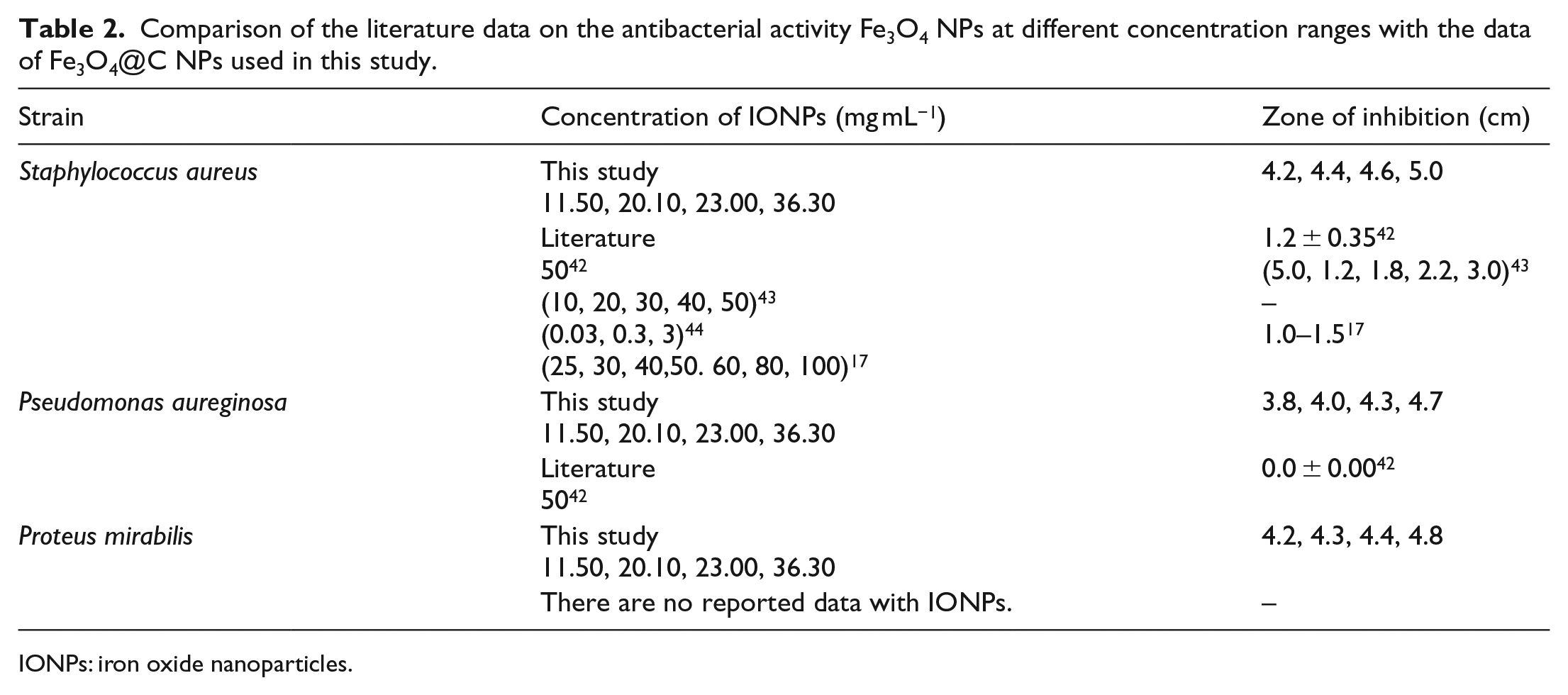
IONPs: iron oxide nanoparticles.

Study of antibacterial activity (zone of inhibition) of Fe3O4@C NPs on different microorganisms (Staphylococcus aureus, Proteus mirabilis, and Pseudomonas aureginosa).
The results also revealed that the microorganisms are sensitive to the test samples at varying concentrations. The antibacterial activities of various concentrations of Fe3O4@C NPs on three bacterial strains are shown in Figure 8. The Fe3O4@C NPs showed antibacterial properties against both Gram-positive and Gram-negative bacterial strains. As the diameter of the zone of inhibition is high, we can conclude that the Fe3O4@C NPs are very effective antibacterial agents. From Table 2 and Figure 8, it is shown that on increasing the concentration of the NPs the antibacterial activity increases. In addition, a comparison of the literature data on the antibacterial activity of Fe3O4 NPs at different concentration ranges along with the results of this study is shown in Table 2. As can be seen, the antibacterial effects of Fe3O4 NPs on Proteus mirabilis have not been reported previously, and for Pseudomonas aureginosa, there is no effect on the zone of inhibition at high concentrations of Fe3O4 NPs. Whereas, in this work, Fe3O4@C NPs present antibacterial activities at different concentration ranges on three bacterial strains.

Effect of the antibacterial activities of the Fe3O4@C NPs at different concentrations on the zones of inhibition of microorganisms.
Conclusion
In this study, Fe3O4@C NPs were synthesized by a facile and rapid co-precipitation method. The prepared Fe3O4@C NPs were characterized by various techniques. The existence of strong signals with the highest percentage of Fe, O, and C in the EDS spectrum together with XRD, TEM, and field emission scanning electron microscope (FESEM) images confirmed the formation of crystalline Fe3O4@C NPs with a crystallite size of about 14 nm. For the synthesized NPs, XRD results demonstrated that the dislocation density and microstrain had decreased with an increase in the crystallite size. In addition, this study highlights the potential application of Fe3O4@C NPs as antibacterial agents against both Gram-positive and Gram-negative bacteria. These NPs can be explored for their topical application in pharmaceutical and biomedical industries.
Experiment
Materials
All chemical reagents used as starting materials were of analytical grade and were used without any further purification. Ferric chloride hexahydrate (FeCl3·6H2O, 98%), ferrous(II) sulfate heptahydrate (FeSO4·7H2O), AC powder, aqueous ammonia (NH4OH; 35%), hydrochloric acid (HCl), and ethanol (C2H5OH) were purchased from SD Fine Chemicals Pvt. Ltd, India. We have used three bacterial species, Gram-positive (Staphylococcus aureus (S. aureus, ATCC25923)) and Gram-negative (Proteus mirabilis (P. mirabilis, ATCC8759)) and (Pseudomonas aureginosa (P. aureginosa, ATCC27853)) obtained from the Department of Biology, Soran University in Kurdistan Regional Government (KRG), Iraq.
Synthesis of Fe3O4@C NPs
Fe3O4@C NPs were synthesized by the co-precipitation method as reported in other articles.45,46 AC was first dispersed in nitric acid (4.0 M HNO3(aq)) at 130 °C for 30 min under stirring to remove the impurities and then washed with distilled water until the filtrate was neutral. The Fe3O4 NPs were prepared by mixing a freshly prepared ferric–ferrous solution consisting of 4.0 mL of FeCl3 (1.0 M FeCl3 in 2.0 M HCl), 1.0 mL of FeSO4 (2.0 M FeSO4 in 2.0 M HCl), and 0.10 g of AC powder with stirring for 3 min at the room temperature. The well-stirred mixture, 25 mL of 1.4 M ammonia aqueous solution was added dropwise over a period of 10 min. During NH3 addition, the suspension became dark brown at pH 6 and then black at pH 11. The precipitate was collected using a magnet and was washed with water several times to remove excess ammonia solution and was finally collected as a powder after being oven-dried at 50 °C. Evidence suggests that the formation involves reduction of the Fe(III) salt into a Fe(II) intermediate on the AC surface as stabilizer 47
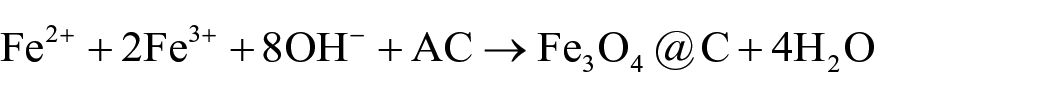
Screening of antibacterial activities
The well-diffusion technique 47 was used for investigating antibacterial effects of Fe3O4@C NPs. About 300 μL of microbe cultures of age 18–24 h were added to Petri plates and nutrient agar was added. Once the medium has solidified, holes were made and each hole was packed with 100 μL (50 mg mL−1) of Fe3O4@C NPs powder in distilled water (different concentrations: 11.50–36.30 mg mL−1 in distilled water). The plates were wrapped in parafilm tape and transferred to an incubator and maintained at 37°C for 24 h. Negative and positive controls were used. The inhibition zones were recorded in centimeters.
Measurement techniques
The synthesized Fe3O4@C NPs were characterized by SEM, TEM, EDS, XRD, FTIR, and UV-Vis spectroscopy. The cross section morphology of the Fe3O4@C NPs was studied by high-resolution SEM (Quanta 450) equipped with EDS (Quantax EDS features the XFlash® 6 detector) at an accelerating voltage of 20 kV. A thin layer of gold was coated on the sample before microscopic analysis. Bright-field TEM images were recorded on a Philips EM 120 TEM at an accelerating voltage of 80 kV. The sample for TEM analysis was cut into slices of a nominal thickness of 100 nm using an ultra-microtome with a diamond knife on a Rechert Ultracut ultra-microtome at ambient temperature. The cut samples were supported on a copper mesh for this analysis. The XRD patterns of the sample were recorded at room temperature on a Philips powder diffractometer type (PW1373 goniometer) using Cu Kα (λ = 1.54060 Å) radiation with a scanning rate of 2° min−1 in the 2θ range from 0° to 80°. Scanning was applied to for the selected diffraction peaks which were carried out in step mode (step size 0.01°, measurement time 0.5 s, accelerating voltage of 45 kV, an emission current of 40 mA) measurement. The absorbance spectrum of the Fe3O4@C NPs suspension after diluting a small aliquot was recorded using UV-Vis spectroscopy (Cary 100, tungsten halogen light sources). The presented results were obtained at room temperature. In addition, Fe3O4@C NPs were investigated by FTIR spectroscopy (IRAffinity-1 Shimadzu Corp. A213750). Dried and powdered NPs were pelleted with potassium bromide (KBr). The spectra were recorded in the wavenumber range of 400–4000 cm−1 and analyzed by subtracting the spectrum of pure KBr.
Footnotes
Acknowledgements
The authors thank Miss Somayeh Khezrian (PhD student) at Tehran University, Mr Shorish Mustafa Abdullah, Karzan Mohammed Khalid (Assistant Lecturer) at Soran University, Mr Karzan Abdulkareem Omar (Assistant Lecturer) at Koya University and Mr Pshtiwan Yusif (Assistant Lecturer) at Salahaddin University for recording the FESEM, FTIR, and UV-Vis spectra, respectively.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.