
Abstract
The density functional theory (at the B3LYP level using 6-311++G(2d,2p) basis set) was used for the investigation of the geometry and electronic properties of the carvone. The electronic properties and chemical activity of the titled compound were investigated by means of several theoretical approaches, molecular electrostatic potential surface, natural bond orbital, and frontier molecular orbital analyses. It was established that the oxygen atom in the structure characterized the electrophilic reactivity; the positive regions are localized on the hydrogen atoms, which can be considered as possible sites for nucleophilic attack. A detailed analysis of the intermolecular interactions via Hirshfeld surface analysis and fingerprint plots revealed that the carvone structure is stabilized mainly by the formation of O. . .H/H. . .O hydrogen bonds. However, close contacts were established between C. . .H/H. . .C and H. . .H contacts.
Introduction
Carvone is a monocyclic monoterpene, which exists in two enantiomeric forms—S-(+)- and R-(−)-carvone. It is a colorless to pale yellowish liquid with a characteristic spicy odor of Carum carvi and with warm and sweet taste. They are natural products in the essential oils from dill (Anethum graveolens L.), caraway (Carum carvi L.) (S-(+)-carvone), and spearmint (Mentha spicata L.) (R-(−)-carvone).1,2 The two forms have biological and pharmacological activities: antioxidant, antimicrobial, anticarcinogenic, antimutagenic, antidiabetic, and diuretic activities.3,4
Essential oils containing carvone are used in foodstuffs for the flavoring of various food products and beverages, as well as in perfumery and cosmetics. 2
The carvone is isolated from the oils that contain it in a larger quantity, 5 but for industrial purposes it is synthesized.6–8
From many years, the experimental and theoretical methods are used to investigate the structural and spectroscopic properties of molecules. The infrared, Raman, and vibrational circular dichroism spectra of S-(+)-carvone were calculated and compared to the experimental one with Hartree–Fock (HF) and density functional theory (DFT) methods from the study by Hoffmann. 8 The experimental and theoretical studies of the optical activity of carvone were reported 9 in order to elucidate the potential importance of solvent effects on its optical rotatory dispersion and circular dichroism spectrum. It is found that DFT with B3LYP functional and 6-311G(d,p) basis set gives good results.
The crystal structure of R-(−)-carvone (
Hence, in this work, a theoretical study on the geometry, electronic structure and chemical reactivity of the carvone by DFT calculations using the B3LYP/6-311++G(2d,2p) method was carried out for better understanding the molecular geometry, highest occupied molecular orbital (HOMO)–lowest occupied molecular orbital (LUMO), and electronic excitation. The intermolecular interactions were characterized on the basis of Hirshfeld surface analysis.
Experimental section
The S-(+)- and R-(−)-carvone were purchased from Fluka Chemical.
Fourier-transform infrared spectroscopy
For the identification of S-(+)- and R-(−)-carvone, the Fourier-transform infrared (FTIR) and UV-Vis spectroscopies were used. The infrared spectrum was recorded using a Nicolet iS 50 Thermo Scientific FTIR spectrometer equipped with a DTGS KBr detector (4 сm−1) in the frequency region of 4000–400 сm−1 at scan numbers of 32.
Characterization results for S-(+)-carvone: FTIR (ν, сm−1) 3308.7, 3201.0, 3110.0, 3050.4, 3025.2, 2900.9, 2130.8, 1716.3, 1647.3, 1430.3, 1372.7, 1337.2, 1317.6, 1281.6, 1235.7, 1163.0, 1112.7, 1058.7, 1033.6, 897.2, 664.6, 614.9, 560.1, 518.4, 456.0, and 436.4.
Characterization results for R-(−)-carvone: FTIR (ν, сm−1) 3335.7, 3319.7, 3074.6, 3010.6, 2968.3, 2952.4, 2925.3, 2883.4, 2350.7, 1669.2, 1648.3, 1446.3, 1435.3, 1371.3, 1324.7, 1270.7, 1244.5, 1201.7, 1137.7, 1105.4, 1057.5, 10.47.6, 1031.6, 999.7, 962.7, 903.3, 802.7, 706.7, 664.7, and 611.5.
UV-Vis spectroscopy
UV-Vis spectrum of the sample was recorded in the range from 190 to 500 nm at 298 K using Evolution 300 UV-Vis spectrophotometer equipped with a silicon photodiode detector.
Characterization results for S-(+)-carvone: UV-Vis (λ, nm) for S-(+)-carvone in ethanol: 201 and 234.
Characterization results for R-(−)-carvone: UV-Vis (λ, nm) for R-(−)-carvone in ethanol: 223 and 240.
Computational methods
All quantum chemical calculations of the title compound were performed using the hybrid B3LYP method 11 of DFT with 6-311++G(2d,2p) 12 basis set by means of Gaussian 16. 13 The optimized geometries (converged to 10−8 a.u.) were used in the vibrational frequency calculations to characterize the stationary point as minima. The absence of imaginary frequencies confirms that the structure corresponds to the minimum energy. GaussView program, Version 6 14 was used to visualize the computed outputs.
Natural bond orbital (NBO) analysis was carried out in order to know the charge distribution and the intrinsic property of carvone. The stabilization energy (E(2)) associated with

where qi is the ith donor orbital occupancy,
Molecular electrostatic potential (MEP) was calculated using the B3LYP/6-311++G(2d,2p) level of theory. By this analysis can be visualized the sites or regions of a molecule where an approaching electrophile or nucleophile will be attracted. Moreover, the study of interactions that involve a certain optimum relative orientation of the reactants could also be successfully carried out by means of MEP. 16 The distribution of electrical charge creates an electrostatic potential (V(r)) in the surrounding space. If a molecule has an electron density function ρ(r), then its electrostatic potential at any point r can be calculated by the expression

where ZA is the charge on the nucleus A, located at distance RA. The presented equation is an exact formula for the electrostatic potential due to the set of nuclei (ZA) and the electronic density function ρ(r′).
Frontier molecular orbital (FMO) analysis was realized. The most important parameters in quantum chemistry—HOMO and LUMO—allow the investigation of stability and reactivity of the molecules. 17 A smaller energy difference between them would determine a larger reactivity between two molecules. The HOMO acts as an electron donor and the LUMO acts as an electron acceptor. 18
The reactivity parameters (chemical hardness, chemical potential, softness, electrophilicity, and nucleophilicity) are calculated with natural population charges obtained from the B3LYP/6-311++G(2d,2p) method. Using the restricted method, the energy calculations of the N-electron species were done, while the energies of the N − 1 and N + 1 electronic species were calculated by the open shell-restricted method using the optimized geometry of the N-electron species. By using Fukui function (f(r)), the reactivity of a chemical system can be measured as follows 19

where ρ(r) is the electron density at each point r, N—electrons, and v(r)—potential acting on an electron due to all present nuclei. Yang and Mortier 20 proposed three different condensed forms of the Fukui function for an atom k in a molecule, based on atomic charges of N, N+ 1, and N − 1 electron systems



where qk is the atomic charge, evaluated from the natural population analysis (NPA) by the NBO method, in the neutral (N), anionic (N + 1), and cationic (N − 1) chemical species. To predict the reactivity of molecule, Yang and Parr 21 introduced local softness s(r), which is obtained by Fukui function f(r) and global softness S

For investigation and visualization of intermolecular interactions in R-(−)-carvone, Hirshfeld surface analysis was used.22,23 Using the program CrystalExplorer,
24
Hirshfeld surfaces and fingerprint plots were generated. The crystallographic information file (.cif) was retrieved from the Cambridge Crystallographic Data Center as supplementary publication no. CCDC 131923.
25
In the field of this analysis, a given molecule is defined by a set of points in three-dimensional (3D) space where the contribution to the electron density from the molecule of interest is equal to the contribution from all other molecules.
26
The intermolecular interactions are obtained by mapping normalized contact distance (
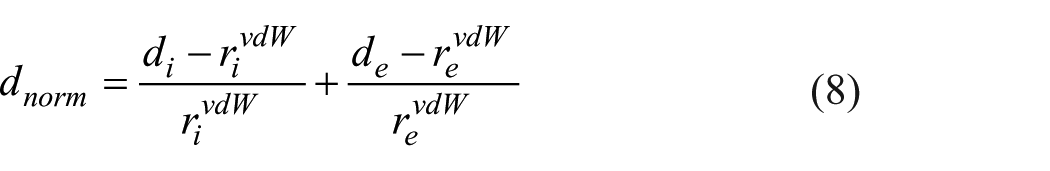
where
Results and discussion
Structural properties
The optimized geometrical parameters—bond lengths and bond angles for R-(−)-carvone and S-(+)-carvone—were computed by the DFT using the B3LYP method with 6-311++G(2d,2p) basis set. The experimental (X-ray diffraction (XRD)) crystal data obtained by Sane et al. 10 are added for comparison. Results showed that the theoretically calculated values correlate well with obtained experimentally (Figure S1 and Table S1). The calculated energy values, dipole moments, polarizability, and hyperpolarizability of R-(−)-carvone and S-(+)-carvone with B3LYP/6-311++G(2d,2p) are presented in Table S2. It can be seen that the energies of the two enantiomeric forms are the same. There are small differences in the values for dipole moment, polarizability, and hyperpolarizability. Dipole moment in a molecule is an important property, which is used to study the intermolecular interactions. As shown in Table S2, the title compound has larger dipole moments (3.5610 D for S-(+)-carvone and 3.5609 D for R-(−)-carvone), and the stronger dipole moments in the cases of title compound are mainly attributed to an overall imbalance in the charge from one side of a molecule to the other. The calculated polarizability values are equal to 18.2485 × 10−24 esu (123.13433 a.u.) for S-(+)-carvone and 18.2484 × 10−24 esu (123.13367 a.u.) for R-(−)-carvone. The S-(+)-carvone is slightly softer and has more polarizability than R-(−)-carvone.
Figure 1 illustrates the optimized molecular structure and atom numbering of S-(+)-carvone (

Optimized molecular structures and atom numbering of S-(+)-carvone.
It is visible from the data presented in Table S1 that the double bonds C1=C2 and C8=C9 are characterized with lengths of 1.342 and 1.332 Å, respectively. The double bond C1=C2 is longer than the C8=C9 because the π C6–O24 orbital is involved in the conjugation with the antibonding σ* C1–C2 orbital. On the contrary, slightly higher values were calculated as lengths for the single C3–C4, C4–C5, and C5–C6 bonds (1.539, 1.538, and 1.520 Å, respectively) in comparison with other single C–C bonds. This may be attributed to the conjugation of π C8–C9 with the antibonding σ* C3–C4 and σ* C4–C5orbitals (1), on the contrary (2) it is known that the length of a given bond strongly depends on the hybridization state of the atoms formed that bond and it increases in the following order: sp3 < sp2 < sp.29,30 For this reason, an NBO analysis was used to confirm the differences between the bond lengths in the title compound.
It was presented earlier that the distance (1.342 Å) between the atoms C1 and C2 is slightly longer than that observed for the C8=C9 (1.332 Å) interaction (Table S1). According to NBO analysis, the σ component of the double C1=C2 bond (1.980e) possessed a contribution of 50.83% sp1.64(C1) and 49.17% sp1.55(C2), whereas the π component (1.862e) was expressed via 52.34% p(C1) and 47.66% p(C2) (Table 1). The data obtained for the C8=C9 bond are as follows: σ component (1.984e) was composed of 51.37% sp1.63(C8) and 48.63% sp1.43(C9), and π component (1.957e) was composed of 48.38% p(C8) and 51.62% p(C9). It is visible that occupancy of the π bond for C1=C2 is lower in comparison with the π bond for C8=C9. The absence of a difference between the lengths of single C3–C4 (1.539 Å) and C4–C5 (1.538 Å) bonds (Table S1) is confirmed by NBO analysis: a contribution of 49.88% sp2.5(C3) and 50.12% sp2.92(C4) characterized the σ C3–C4 orbital (1.967e) and values of 50.35% sp2.89(C4) and 49.65% sp2.49(C5) were calculated as contributions in the C4–C5 orbital (1.967e). These findings completely coincide with the difference between the lengths of single C2–C3 (1.501 Å) and C1–C7 (1.502 Å) bonds.
Occupancy of NBOs and hybrids of S-(+)-carvone by the B3LYP method with 6-311++G(2d,2p) basis set.

NBO: natural bond orbital; AO: atomic orbital; LP: lone electron pair.
Hybrids on atoms.
Percentage contribution of AOs in NBO hybrid.
Electronic properties and chemical reactivity
The chemical reactivity of a given molecule depends on its electronic structure, that is why the MEP surface of the S-(+)-carvone was investigated (Figure 2).
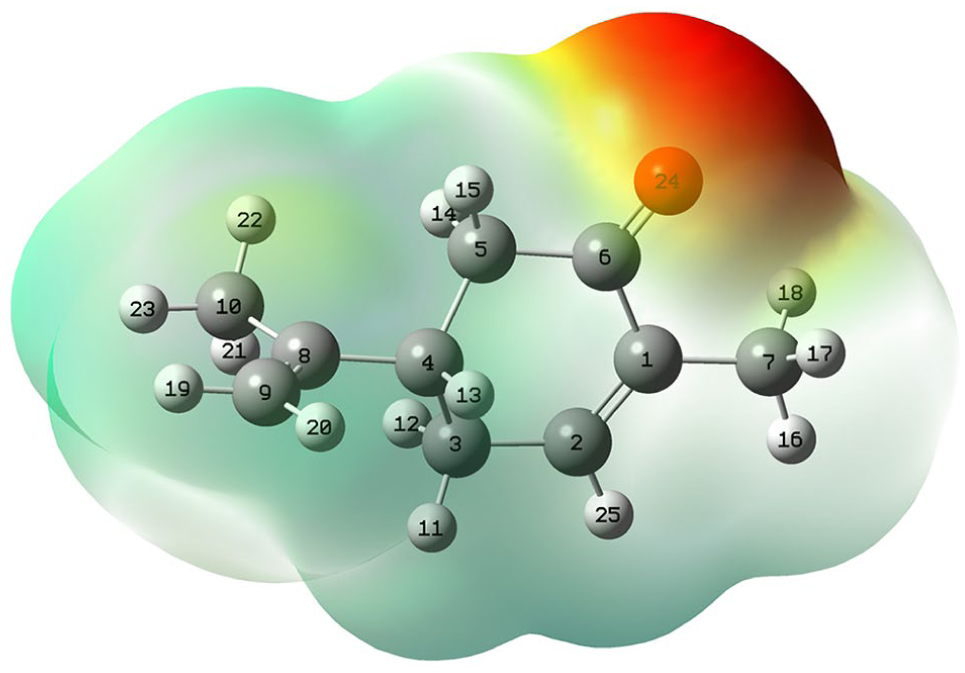
The total electron density mapped with electrostatic potential surface of S-(+)-carvone.
The theoretical calculations showed that the MEP of S-(+)-carvone varies from −0.054 to +0.017 a.u. The electron-rich and electron-deficient sites of the molecule are represented by different colors. The red color corresponds to the regions localized the most negative charge, whereas the blue color denotes the most positive ones. The molecular electron density surface is used to identify the electrophilic and nucleophilic reactive sites and to study interactions between molecules and ions or solvents and biological properties of molecules.31,32 The negative potential region in the studied molecule is found around the polar oxygen atom, which shows high activity of this site. The positive regions are localized on the hydrogen atoms, which can be considered as possible sites for nucleophilic attack.
In addition, the atomic charges of the neutral, cationic and anionic species of S-(+)-carvone were established by NBO using the B3LYP method with 6-311++G(2d,2p) basis set and are presented in Table 2.
NPA atomic charges and bond orders in S-(+)-carvone.

NPA, natural population analysis.
It is visible that the most positive charge of +0.557 was detected on the carbon atom (C6) linked with the oxygen one, followed by the charge value of +0.228 located on the H15 atom. These data propose that the nucleophilic reactivity of the titled molecule depends in a greater extent on the C6 atom rather than H15 one. The calculated NPA charges showed that a significant amount of negative charge is located on the carbon atoms C10 (−0.602), followed by the negative charge of −0.589 on the carbon atom C7 and −0.568 on the oxygen atom O24, which assumed the existence of a charge compensation effect between the oxygen atom and C6 one. The latter was confirmed by means of MEP surface analysis, where it is clearly exposed that O24 is located in the area with most negative charge. Hence, we state that the electrophilic reactivity of S-(+)-carvone depends mainly on the oxygen atom.
The carbon atoms 1 and 8 have a similar bonding environment, but NBO gives different charges, −0.120 and +0.001, respectively. The latter is linked with lowest amount of electron density (+0.557) on the carbon 6 atom bonded to the oxygen (electronegative atom). According to the NPA analysis (Table 2), values of +0.557, −0.099, and −0.589 were calculated as charges located on the C6, C2, and C7 atoms, respectively. On the contrary, the C4, C9, and C10 atoms were characterized with charges of −0.232, −0.377, and −0.602, respectively. Consequently, a stronger charge compensation effect between C4, C9, C10, and C8 atoms with respect to those between C6, C2, C7, and C1 was assumed. As a result, a positive charge exists on the C8 atom.
A useful method for investigating the chemical reactivity of a given molecule is the analysis of the FMOs. 33 The FMOs and their negative (green) and positive (red) regions obtained by the B3LYP method with 6-311++G(2d,2p) basis set for the S-(+)-carvone are shown in Figure 3(a) and (b), respectively.

Frontier molecular orbitals of S-(+)-carvone: HOMO (a) and LUMO (b).
The HOMO is with the contribution: ψHOMO = 16.8%
The local reactivity descriptors such as Fukui function and local softness are related to the selectivity of the specific site in a molecule, whereas chemical potential, hardness, and softness are the global properties related to the chemical reactivity. The Fukui’s indices provide information about the molecule’s tendency to lose or receive an electron and predicting which atom in the molecule would be more susceptible to an electrophilic or nucleophilic attack.20,40 The condensed reactivity parameters for the title compound were calculated by NPA. Morell et al. 35 proposed new dual descriptors for chemical reactivity: Δfk and Δsk. They are defined as follows


If ∆fk (Δsk) is positive, then the site is electrophilic (atom k), and if ∆fk (Δsk) is negative, then the site is nucleophilic. For the titled compound, O24 acts as an electrophilic site and it is prone to nucleophilic attack, whereas C6, C2, C1, and all the hydrogen atoms are nucleophilic sites and are prone to electrophilic attack. The condensed Fukui functions and local softness and dual descriptors for S-(+)-carvone are given in Table 3.
The condensed Fukui functions, local softness, and dual descriptors (Δfk, Δsk) for S-(+)-carvone.

For studying intramolecular bonding, charge transfer, and conjugative interactions, NBO analysis was provided. When there is more intensive interaction between electron donors (occupied (lone pair) orbitals) and electron acceptors (unoccupied (antibonding) orbitals), the second-order stabilization energy (E(2)) value is larger, it corresponds to a stable donor–acceptor interaction. These interactions and their E(2) values for carvone are presented in Table 4.
Second-order perturbation theory analysis of Fock matrix in NBO basis.

NBO: natural bond orbital; LP: lone electron pair.
ED—electron delocalization (occupation number); E(2)—energy of hyperconjugative interactions (stabilization energy), kcal mol−1; E(j)− E(i)—energy difference between donor and acceptor i and j NBO orbitals, a.u.; and F(i, j)—Fock matrix elements between i and j NBO orbitals, a.u.
Results showed (Table 3) a strong hyperconjugative interaction of the π electrons of C1–C2 to the π* C6–O24 with a value of 20.97 kcal mol−1 as stabilization energy, which decreases the occupied orbital population (1.862e) and increases the electron density of the acceptor orbital (0.144e). In the same way, slightly higher stabilization energy of 29.60 kcal mol−1 was observed for the hyperconjugative interaction π* C6–O24 → π* C1–C2. The values of 3.29 and 2.89 kcal mol−1 were calculated as stabilization energies for the interactions π C8–C9 → σ* C3–C4 and π C8–C9 → σ* C4–C5, respectively. Comparing the stabilization energies, it is clear that the double bond C1=C2 is longer than the double bond C8=C9. The latter was in accordance with the bond order value (1.875) calculated for C8=C9, which is higher in comparison with bond order (1.727) for C1=C2. A considerable electron density distribution from the lone electron pair LP(2) O24 to the antibonding σ* C5–C6 and σ* C1–C6 orbitals was established. In detail, E(2) values of 20.18 and 19.27 kcal mol−1 characterized the LP(2) O24 → σ* C5–C6 and LP(2) O24 → σ* C1–C6 interactions, respectively. This increases the electron density of the acceptor orbitals (0.058e and 0.070e) and weakens the nonbonding LP(2) O24 (1.895e) orbital. The latter was in accordance with the order value of 1.082 calculated for the C1–C6 bond, which is slightly higher in comparison with bond order of C5–C6 (1.041). Weaker stabilization energies were obtained for LP(1) O24 → σ* C1–C6 and LP(1) O24 → σ* C5–C6 interactions (expressed with E(2) of 1.86 and 1.55 kcal mol−1, respectively).
Intermolecular interactions
The Hirshfeld surface and associated two-dimensional (2D) fingerprint plots were used to examine the interactions in the crystal structure of R-(−)-carvone.
41
The Hirshfeld surface is not simply a function of the molecular geometry but is only defined within the crystal.
27
The Hirshfeld surface shows the result of the interaction between different atomic sizes and intermolecular contacts in the crystal.
28
The blue regions are longer than the van der Waal distance of the interacting atoms, whereas the red regions show the region of the molecule contacts that are shorter than the van der Waal’s distances, the white regions are those where the distances are comparable to the distances of Van der Waal’s. Hirshfeld surface of R-(−)-carvone mapped with

Hirshfeld surface for R-(−)-carvone mapped with
The mapping
The complementarity between molecules in the crystal packing structure is a feature of Hirshfeld surface analysis namely Shape index.42,43 The areas of intermolecular complementarity have an identical pattern but opposite colors. The blue patches represent convex regions which are indication for ring atoms of the molecule inside the surface, the red ones represent concave regions related to atoms of the π-stacked molecule above them (Figure 4(b)).

Hirshfeld surface for R-(−)-carvone mapped with dnorm and neighboring molecules associated with close contacts are shown along with distances between the atoms involved.
The 2D fingerprint plots identify each type of intermolecular contact and enable the analysis of very small differences in these patterns and represent a new way of summarizing the major intermolecular contacts of an entire crystal structure. 44 Figure 6 shows the associated fingerprint plots of the four R-(−)-carvone molecules showing the contributions of different intermolecular interactions on the Hirshfeld surface.

Two-dimensional fingerprint plots with dnorm view of the contacts in the R-(−)-carvone.
It is visible (Figure 6(b)) that the highest contribution of total Hirshfeld surface is attributed to H. . .H contacts with 76.6% and appear in the middle of the scattered points in the two-dimensional fingerprint map with a single broad peak at di = de = 1.2 Å. In detail, the spike characterized the O. . .H interactions with a contribution of 7.4%. The calculated distances between nearest atoms present inside (O) and outside (H) the surface were approximately equal to 1.28 and 0.95 Å, respectively. The H. . .O interactions with a contribution of 6.7% of the total surface were detected to form a spike with values of 0.95 and 1.28 Å as di and de, respectively (Figure 6(c)). It was stated that the presence of O. . .H/H. . .O interactions are a result of hydrogen bond formation, which ensures the stability of the crystal framework. The C. . .H/H. . .C interactions with contribution of 7.4% are other significant contacts for the investigated compound (Figure 6(d)). The C. . .H interactions were exhibited by a spike with di ≈ 1.5 Å and de ≈ 1.2 Å. These types of interactions involve 4% of the total surface in the bottom right region of the fingerprint plot. The H. . .C interactions represented by spike with di ≈ 1.3 Å and de ≈ 1.6 Å cover 3.4% of the total surface in the top left region of the fingerprint plot. Figure 6(e) shows that the C. . .O/O. . .C interactions
The enrichment ratio (E) was determined in order to confirm both the nature and contribution of the bonds established in R-(−)-carvone. The enrichment ratio is defined as the ratio between the proportion of actual contacts in the crystal and the theoretical proportion of random contacts, derived from the Hirshfeld surface analysis. 45 The E value is larger than one for a pair of elements with a higher propensity to form contacts. The E value is lower than one for pairs with a lower propensity to form contacts. 41 It is visible from Table 5 that the 87.35% of the molecular surface is generated by H atoms, followed by the O atoms with surface contribution of 7.75%, while C atoms complement the total molecular surface with contribution of 4.90%.
Hirshfeld contact surfaces and enrichment ratios for R-(−)-carvone.

The C. . .C and C. . .O interactions are the most enriched contacts (
A study of Hirshfeld surface carried out on R-(−)-carvone gives the following measurement: molecular volume, 236.88 Å3; surface area, 231.17 Å2; globularity, 0.801; and asphericity, 0.114.
Conclusion
Analyses on the molecular geometry, chemical bonds nature, electronic properties, and chemical activity of the carvone were described using theoretical calculations (expressed by MEP surface, FMO, and NBO analyses at the B3LYP/6-311++G(2d,2p) level). The MEP surface and FMO theory showed that the nucleophilic activity of the carvone was connected with the low electron density on the hydrogen atoms. In contrary, a large electronegative potential was detected on the oxygen atom. Using FMO analysis, values of −6.813 and −1.648 eV were related to HOMO and LUMO orbitals, respectively. As a result, the chemical hardness and softness, electronegativity, chemical potential, and electrophilicity index were calculated as 2.583, 0.387, 4.231, −4.231, and 3.465 eV, respectively. The intermolecular interactions were examined via Hirshfeld surface analysis. It was established that
The theoretical calculations will be used to interpret the difference in antimicrobial and antioxidant activity of essential oils containing the two forms of carvone, which are the subject of our next research.
Supplemental Material
Supplementary_information – Supplemental material for Electronic structure, reactivity, and Hirshfeld surface analysis of carvone
Supplemental material, Supplementary_information for Electronic structure, reactivity, and Hirshfeld surface analysis of carvone by Rumyana Yankova, Milen Dimov, Krasimira Dobreva and Albena Stoyanova in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.