
Abstract
Background: For many years, studies have shown that the results of clinical trials are often published or reported selectively with a statistically significant bias in favour of positive trial results. Trial registration as a precondition for publication had only limited effects on current practice. Results of trials which were approved by research ethics committees (RECs) are often published only partially, with a substantial time lag or not at all. This study examined existing procedures of RECs in the European Union to monitor and prevent incomplete registration of trials and selective reporting of trial results. It further investigated opinions of REC members about the need to update current legislation on this matter. Methods: Web-based survey on members of RECs in 22 European countries. Results: Over 90 percent of respondents agreed that the incomplete publication of trial results had a strong or somewhat negative impact on public health and on healthcare professionals’ trust in the validity of clinical research, yet only 30 percent reported that their REC had some (often unsystematic) mechanism in place to check that findings of approved studies are published in some form. Less than 10 percent stated that their REC has further specific procedures in place to prevent or minimize selective reporting of study results. Respondents stated variously that their REC did not have the resources to follow up on this matter. Conclusions: The existing legislation to regulate trial registration and encourage complete publication of trial results leaves room for improvement. REC members welcome guidelines to adequately address both problems. The new Regulation EU No. 536/2014 as well as the FDA Amendment Act from 2007 require the reporting of summary results within 1 year after study end. As recent reviews demonstrated, without any systematic approach to monitor the adherence to these regulations, publication rates remain rather low.
Introduction
For many years, studies have shown that the results of clinical trials are often published selectively, with a statistically significant bias in favour of positive trial results (Chan, 2008; Song et al., 2000; Turner et al., 2008). Selective reporting occurs where primary end points are not published or are reported in a distorted or modified form or when the selection of published results favours positive findings and leaves out negative findings (Chan et al., 2004; Mathieu et al., 2009).
One suggested response to the challenge is the registration of all clinical trials as a precondition for publication (Drazen and Zarin, 2007; Strech, 2012). This also reflects an explicit demand of the Declaration of Helsinki, which states in §35: ‘Every research study involving human subjects must be registered in a publicly accessible database before recruitment of the first subject’ (World Medical Association, 2013a). Because registration does not itself improve an unbiased dissemination of findings, the Declaration of Helsinki adds in §36: ‘Researchers have a duty to make publicly available the results of their research on human subjects and are accountable for the completeness and accuracy of their reports. All parties should adhere to accepted guidelines for ethical reporting. Negative and inconclusive as well as positive results must be published or otherwise made publicly available’. However, studies still demonstrate that in many instances the results of trials which were approved by Research Ethics Committees (RECs) are published only partially or not at all (Blumle et al., 2014; Kasenda et al., 2014a, b; Schmucker et al., 2014).
To inform discussions and policy decisions about the role that RECs can play in reducing the selective reporting of results of clinical trials in the future, we conducted a survey as part of the OPEN project. OPEN is an interdisciplinary initiative, funded by the European Commission, that brings together academics and key stakeholders from across Europe who aim to develop evidence-informed recommendations and strategies which focus on overcoming the failure to publish negative research findings (OPEN, 2012; Littmann et al., 2013; Wager et al., 2013).
Here, we report findings from an OPEN work package that aimed to survey members of RECs across Europe, and collect data on current procedures that are supposed to ensure complete registration of trials and subsequent publication of research findings. We also asked respondents about their personal views and attitudes towards the issues of trial registration and the effects of biased publication.
Methods
Study sample and design
Our study aimed to collect responses from around 20 European countries, in order to provide a broad overview of national and region-specific approaches, viewpoints and attitudes. To this end, we assembled a database of RECs for the EU-27 countries plus Norway and Switzerland.
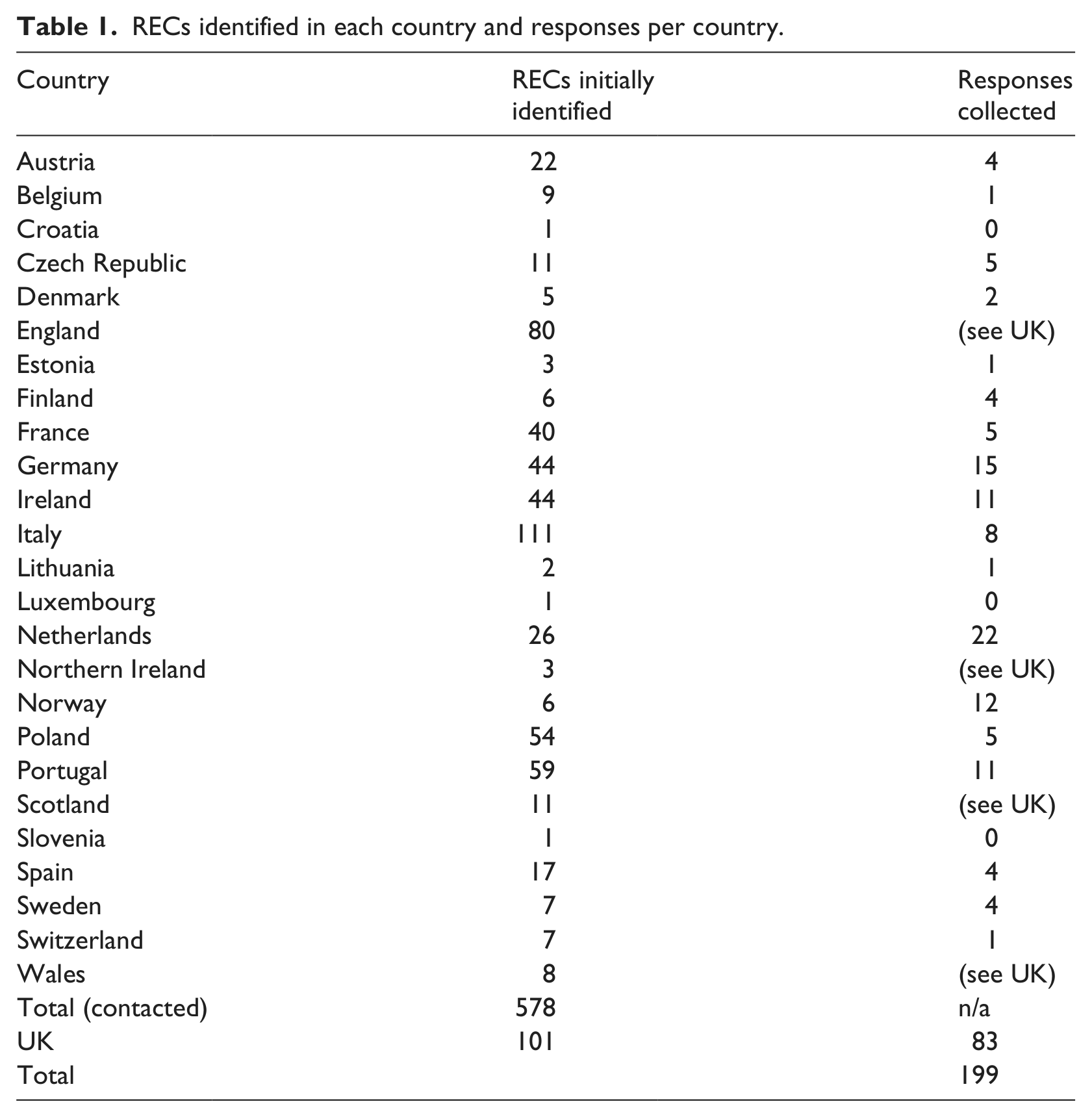
Overall, we contacted 578 RECs in 22 countries (see Table 1). For the remaining countries, contact information was not available online, or could not be identified. Where possible, we relied on the previously compiled national contact information for RECs published by the European Network of Research Ethics Committees (EUREC), and contacted spokespersons for each country individually to request the latest available set of contact information for the specific country (Lanzerath, 2013). If no contact person was named, or contact could not be established, RECs were identified with the help of OPEN project partners in their respective home country, or alternatively through online research. Where possible, we contacted REC chairpersons individually. If no contact information for the chairperson was available, we wrote to the REC’s administrator instead. In cases where neither chairperson nor administrator could be identified, the REC’s email address for general inquiries was used.
RECs identified in each country and responses per country.
We contacted all RECs for which contact details could be established (n = 578) by email, describing the OPEN project, inviting REC members to participate, and providing a link to the online survey. All RECs that were included in our database were contacted three times: with an initial introductory email followed by two reminders sent in three-week intervals. The survey itself contained a brief introduction of the OPEN project and its goals.
The design of this survey did not allow for calculating a reliable response rate, because we needed to accept that more than one person from the same REC completed the survey. We did not collect IP addresses from respondents or ask them to identify their REC by name. Therefore, it was not possible to verify how many persons per REC did complete the survey. This limitation of our study was unavoidable because the pre-test had shown that contacted REC members had a strong preference for a high level of anonymity. Furthermore, the available contact data did not always allow for the identification of individual contact persons at each REC. Therefore, we accepted that the survey was completed by or passed on to more than one REC member. Thus, in the case of Norway, for instance, the total number of responses (n = 12) was greater than the number of RECs that had been contacted (n = 6).
To address this limitation we distinguished two categories of our survey data: ‘policy data’ (e.g. procedures for encouraging registration of all approved studies) and ‘attitudes data’ (e.g. attitudes about the impact of incomplete publication of study findings on healthcare professionals’ trust in the validity of clinical research). Furthermore, we distinguished two categories of survey respondents: ‘REC-chairs’ and ‘other REC members’. For the ‘policy data’ the responses from ‘REC chairs’ were considered the most valid. For the ‘attitudes data’ the responses from both ‘REC chairs’ and ‘other REC members’ were considered relevant.
Development of survey instrument
The survey design and formulation of its items was informed by reviewing the existing literature on incomplete publication of trial results, and by discussions with experts of the OPEN project. The survey was first tested in five cognitive telephone interviews with REC members from different EU member countries, who had different professional backgrounds and nationalities. Subsequently, the survey was adjusted to improve clarity, internal consistency, and accessibility for non-native English speakers. Three weeks before the general roll-out of the survey, we randomly selected 20 potential respondents from most countries that were eventually surveyed for a pre-test, to verify that the email send-out and response via web-link worked smoothly, and to gauge an initial response rate. Survey items were presented in multiple-choice format, as Likert-scales or as open-ended questions. All answers were collected via a dedicated online platform. To guarantee anonymity of respondents, we did not record respondents’ IP addresses, and did not ask respondents to identify their REC beyond the country in which it was based.
Domains of the survey
Characteristics of respondent
We collected basic socio-demographic data about age, gender and professional background of respondents. Owing to the fact that contact details for REC chairpersons were only available in 181 cases, we asked respondents to identify their position on the REC. Furthermore, to get a better idea about the activity of the REC and the experience of the respondent, we requested information about the number of study protocols reviewed per year by the REC, an estimate of the total number of study protocols reviewed by the respondent in their role as REC member, and the number of years the respondent had been serving on the REC.
Trial registration
We asked multiple questions about requirements of the respondent’s REC to register different types of clinical study prior to granting ethics approval. Furthermore, we asked if the REC informed applicants about the requirements of most journals to register trials, and finally in an open-ended question asked if the respondents felt that there were any conditions under which the registration of trials was not necessary.
Incomplete or selective publication of trial results
We first introduced the term ‘selective publication’ as the failure to publish primary end points of the study, or the distortion or modification of trial results that may lead to a biased reporting of positive and negative study outcomes. We then asked respondents Likert-style questions about the impact that selective reporting may have on public health, the trust of healthcare professionals and the public in the validity of clinical research findings, and the willingness of the public to participate in clinical trials. We also asked respondents to identify any circumstances which in their view would merit the selective publication of clinical trial results.
REC procedures to address incomplete registration/selective publication
In the final section, we posed multiple response questions about the need for additional guidelines or legislation to address the issues of incomplete registration and selective publication, and inquired about procedures that their commission may already have in place to deal with either. Furthermore, we used a Likert-style response format to find out more about survey participants’ views on potential obstructions that might make complete trial registration difficult in their respective country. Finally, we asked open-ended questions about proposals on how RECs could help to reduce publication bias and incomplete trial registration in the future, and whether or not respondents felt that the survey had overlooked any areas of concern with regard to these issues.
Statistical analysis
We calculated frequency data by standard descriptive statistics. If a respondent did not answer on a specific survey item or answered that this item was not applicable to his work, we did not count this respondent for the respective frequency analysis. Absolute numbers and frequency data presented in the Results section only reflect the item-specific full sample of responses.
Ethics statement
The study was not reviewed and approved by an institutional review board (ethics committee) before the study began because, according to German regulations for RECs and medical professional law, no ethics approval is necessary for socio-empirical research that does not involve patients. In our web-based survey we only contacted members of research ethics committees.
Data safety and informed consent in this study were secured as follows. The survey template explained the study rationale, the study design and the later reporting of anonymized data to each survey participant, and asked whether they were willing to participate in this interview research project. Consent was then considered to be implied when participants filled out the survey instrument. Beside gender, age, profession and experience with RECs, no further personal data were gathered. All personal data and interview data are stored on computers protected by the security policy of Hannover Medical School.
Results
590 RECs were initially identified, and after excluding those RECs for which no contact information were available (n = 12), 578 were contacted (see Table 1). Based on the email responses, we excluded another 54 RECs after the initial contact. Reasons for exclusion were inactive (3) or invalid (40) email addresses, ethics committees which did not deal with clinical trials (4), retirement of the contact person from the REC with no mentioning of a successor (5), discontinuation of the REC (1), and in one instance a REC which had not yet commenced its work.
We collected 199 valid responses (three responses were excluded from consideration, as the respondent had started the online survey but not answered any of the questions), and in one instance a respondent asked for his answers to be removed after completing the survey. Of the respondents to the survey, 25 percent (n = 50) were ‘REC chairs’ and 75 percent (n = 149) identified themselves as ‘other REC members’ (including researchers, physicians, lay members, REC administrators or substitute members).
The professional background of respondents was diverse, which was reflected by the wide range of responses. The largest single group was made up by physicians (39% of respondents, n = 78) followed by epidemiologists (6.5%, n = 13), nurses (6.5%, n = 13) and lawyers (5%, n = 10). The majority of respondents were employed in a hospital or healthcare institution, either at a teaching hospital (56%, n = 107) or a non-academic hospital (21%, n = 41). Other employers reported were government institutions (9%, n = 18), and a similar proportion of respondents continued to work for their REC after retiring from their profession.
Most of the survey respondents had substantial experience in their role. The average time of service on a REC for all respondents was 8.6 years, and more than half of all respondents (56%, n = 112) estimated that they had reviewed in excess of 150 applications since joining their REC. Respondents were on average 54 years old, and the gender split was fairly even: 51 percent male and 49 percent female.
Not all 199 respondents responded to each survey item. The presentation of frequency data for specific survey questions was calculated based on the question-specific number of responses.
RECs’ control of trial registration
Overall, 79 percent (n = 31/39) of REC chairs reported that their REC regularly checks whether or not trials which fall under the EU Directive 2001/20/EC have applied for a EudraCT number. 1 It is notable that only 7 percent (n = 3/39) of REC chairs but 33 percent (n = 46/140) of other REC members responded that they did not know whether checks were performed.
Less often, RECs demand the registration of other types of clinical trials that are not covered by the Directive 2001/20/EC. For non-interventional trials (e.g. observational studies on authorized medicines), 47 percent (n = 17/41) of REC chairs responded that they demand trial registration. Furthermore, 60 percent (n = 25/42), 36 percent (n = 16/45) and 27 percent (n = 9/33) of REC chairs answered that they demand trial registration for clinical trials on medical devices, surgical procedures and psychotherapeutic procedures.
When asked about whether or not respondents’ RECs routinely inform principal investigators about the fact that, according to policy agreed on by the International Committee of Medical Journal Editors (ICMJE), results cannot be published without the previous registration of the trial, 41 percent (n = 17/41) of REC chairs answered that this information was passed on to applicants.
Although the majority of all respondents (81%, n = 138/170) thought that clinical trials should always be registered, a smaller group (12%, n = 21/170) felt that under certain circumstances registration might not be necessary. Respondents’ views varied widely on what such circumstances might be, although some common themes emerged. A number of respondents (n = 6) stated that in cases where intellectual property rights were threatened by registration, exemptions could be made. Others expressed the opinion that research in very early stages or pilot studies did not have to be registered (n = 5). Some respondents also listed types of studies that they felt did not require registration, namely non-interventional trials (n = 4) and research for educational purposes (n = 3). It should be noted, however, that neither of these types of trials currently require registration, which means that the opinions of respondents who named them are otherwise consistent with the requirement to register interventional trials. A final reason that was given for foregoing registration was the protection of patient confidentiality, although this was only provided by two respondents.
Incomplete or selective reporting
Respondents were generally concerned about the effect of incomplete and selective reporting of trial results and the negative impact this might have. 94 percent (n = 153/162) of respondents agreed that the incomplete publication of trial results had a ‘strong negative impact’ (61%, n = 99) or ‘somewhat negative impact’ (33%, n = 54) on public health. Even more respondents (97%, n = 157/162) answered that a ‘strong’ (61%, n = 99) or ‘somewhat’ (36%, n = 58) negative impact exists on healthcare professionals’ trust in the validity of clinical research. Furthermore, 91 percent (n = 147/161) of respondents agreed that incomplete publication of results had either a strong or somewhat negative impact on public trust in evidence-based medicine.
It was therefore not surprising that 97 percent (n = 152/157) of respondents stated that there were no circumstances under which the incomplete or selective publication of trial results was acceptable. Five (3%) respondents (one REC chair and four other REC members) thought that selective reporting was acceptable when it is ‘in the public interest’, ‘for reasons of limited space’, ‘if related to a patent request’, ‘if the primary outcomes turn out to be unimportant and other secondary outcomes are more important to report’ or ‘if specifically asked for during the peer review of submitted publications’.
RECs’ ways of dealing with incomplete registration/publication
The final section of the survey asked participants about any specific procedures that their REC employed to encourage registration and to prevent or minimize selective reporting of study results. When asked about procedures of their REC to encourage registration of trials, only 27 percent (n = 11/41) of REC chairs and 37 percent (n = 39/105) of other REC members stated that such procedures existed. When asked to specify what these procedures consisted of, it emerged that in many cases, RECs merely advised that registration was important and desirable but did not follow up whether or not this advice was followed. Respondents stated variously that their REC did not have the resources to follow up on this matter. A notable exception that was repeatedly mentioned was the use of a standardized form as part of the UK’s Integrated Research Application System (IRAS), which specifically asks for registration details (NHS Health Research Authority, 2013).
Similarly, only 30 percent (n = 12/40) of REC chairs and 30 percent (n = 32/107) of other REC members reported that their REC has some mechanism in place to check that findings of approved studies are published in some form. Additional qualitative commentaries highlighted, however, that such mechanisms often lacked a systematic approach, or that there was insufficient funding available to carry out this task.
Even fewer respondents (8%, n = 3/39 of REC chairs; 9%, n = 10/108 of other REC members) stated that their REC had further specific procedures in place to prevent or minimize selective reporting of study results. None of the respondents gave examples of measures their REC employed to reduce selective publication.
A majority of respondents (85%, n = 34/40 of REC chairs; 89%, n = 99/113 of other REC members) stated that the work of their REC to improve complete registration and unbiased publication of studies would benefit from new (or modified) national/international laws or guidance that explicitly demanded complete registration and unbiased publication of clinical studies.
Respondents were also asked about barriers in their respective countries that complicated trial registration and the complete publication of trial results. 77 percent (n = 30/39) of REC chairs and 82 percent (n = 89/109) of other REC members agreed that the ‘legal protection of commercial interests’ creates a problem for complete publication and registration of all trials. Other widely shared concerns were ‘practical difficulties in overseeing ongoing research by RECs’ (77%, n = 30/39 of REC chairs; 64%, n = 68/106 of other REC members) and ‘conflicts of interest of principal investigator’ (53%, n = 20/38 of REC chairs; 51%, n = 54/106 of other REC members). A smaller proportion (37%, n = 14/38 of REC chairs; 28%, n = 30/108 of other REC members) found that ‘ethical difficulties [due to] the unjustified intrusion into the researcher’s independence’ pose a barrier for complete registration and publication of reviewed trials.
Discussion
In this survey of members of RECs in 20 European countries, we found widespread support for the need to improve complete registration and publication/dissemination of approved trials. However, our survey results suggest that only a minority of European RECs undertake specific efforts to address a lack of or incomplete registration or publication of trials. Currently, only a minority of the surveyed RECs systematically check whether trials are registered and even fewer monitor the publication of results for the trials they previously approved. Furthermore, the evidence we collected suggests that whether or not RECs check for registration partly depends on the type of trial in question. Trials which are not covered by the EU directive 2001/20/EC are less likely to be checked for registration.
Our findings also suggest that the discrepancy between current practice and the strong support for registration and complete publication of trial results may be owing to a lack of available resources in RECs. In part, however, it may also be because in many instances the role which RECs can play in addressing both issues has not been clearly defined. This is also reflected by the fact that a majority of respondents were in favour of better or more detailed guidelines to regulate the role of RECs in improving standards of registration and publication of trials.
Generalizability of results and limitations of study
Our survey findings provide the first overview on the status quo of European RECs’ policies and attitudes towards complete registration and publication of approved studies. As described in the Methods section, the sampling of European RECs as well as the evaluation of response rates were limited for technical reasons and because of REC members’ preferences for a high level of anonymity. Owing to the heterogenic group of respondents, the relatively small sample size per country and the uneven distribution of responses across the 20 countries included in the survey, the generalizability of the survey findings for individual European countries is limited. Low response rates in some countries may be partly due to the fact that the survey was only sent out in English, which may have presented a barrier for REC members with limited English language skills.
Although not representative for all European RECs, the results we have collected still offer a unique insight into the attitudes and views of a substantial number of experienced REC members, and are the best available evidence regarding current European practices concerning the monitoring of trial registration and incomplete publication.
Conclusion
Overall, the findings of our survey show that many REC members are concerned about the effect that incomplete registration of clinical trials and biased reporting of trial findings may have, not only on clinical research but also on public health and the public’s trust in clinical research. Judging from our results, the existing legislation to regulate trial registration and encourage complete publication of trial results leaves room for improvement, and many of the REC members who responded to our survey would welcome more specific guidelines to adequately address both problems. Additionally, our results show that REC members are worried about increasing workload that a greater focus on monitoring would lead to and which they feel they cannot meet with existing resources. This may be overcome by standardizing and streamlining the application process for clinical trials, as has been the case in some countries, most notably in the UK and the Netherlands.
So far, EU-wide policies have not focused on the issue of monitoring the publication of trial results. The European Commission’s Directive 2001/20/EC that was recently revised into the Regulation EU No. 536/2014 states in Article 37(4): ‘Irrespective of the outcome of a clinical trial, within one year from the end of a clinical trial in all Member States concerned, the sponsor shall submit to the EU database a summary of the results of the clinical trial’ (European Union, 2014). This new database (neither EudraCT nor Eudravigilance) still has to be developed by the EMA. Furthermore, Article 29(6) specifies necessary content for informed consent forms, including that: ‘The subject shall be informed that the summary of the results of the clinical trial and a summary presented in terms understandable to a layperson will be made available in the EU database … irrespective of the outcome of the clinical trial, and, to the extent possible, when the summaries become available’ (European Union, 2014). The annexes IV and V specify the content of both the expert and layperson summary.
However, so far there appears to be no systematic approach to monitor the adherence to this regulation. Anderson et al. demonstrated that 13 percent of eligible trials (n = 1790) had reported summary results to ClinicalTrials.gov within the 1 year time period as it is required in the USA by the FDA Amendment Act from the year 2007 (Anderson et al., 2015). Within 5 years of a trial’s completion 38 percent reported results. The role RECs could play to monitor and increase timely publication rates has not yet been specified in either the US or the EU regulations.
The recent revision of the Declaration of Helsinki (DoH) includes a supplement in the section that covers the REC’s right to monitor ongoing studies: ‘At the end of the study, the investigators must submit a final report to the committee containing a summary of the study’s findings and conclusions’ (World Medical Association, 2013b). REC policies built on the DoH content might consider forcing the submission of summary results as soon as the study’s end is indicated in the respective register.
In May 2013, the NHS Health Research Authority announced a plan of action to increase transparency in health research. Particularly, this plan aims to ‘strengthen the Research Ethics Committee (REC) review of researcher intentions to make findings, data and tissue available by including this assessment in the current pilot of the ethics officer before REC review’ (Health Research Authority, 2013).
Sufficient resources should be made available for allowing RECs or other more centralized institutions to follow up actively on approved trials with regard to registration and publication. This should apply to all trials and not only drug or medical device trials. Principal investigators and sponsors could be further encouraged to adhere to complete registration and publication if RECs require further monitoring measures/safeguards for trials protocols from sponsors or principal investigators who demonstrated inadequate registration or publication of results for previously approved trials.
Footnotes
Acknowledgements
The authors would like to thank Ana Marusic, Joost Kleijnen, Gerd Antes and Joerg Meerpohl for comments and suggestions.
Authors’ contributions
DS has made substantial contributions to the conception, analysis and interpretation of data, and to drafting the article for important intellectual content. JL has made substantial contributions to the conception and acquisition of data and to drafting the article. DS and JL approved the final version to be submitted.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 285453.