
Abstract
Neuropathic pain affects approximately 7%–10% of the global population, significantly impairing patients’ quality of life and placing a substantial burden on public health systems. Current pharmacological treatments have limited efficacy and are often accompanied by notable side effects, highlighting the urgent need for novel therapeutic targets. Increasing evidence supports the important role of chemokines and their receptors in neuro-immune interactions underlying pain sensitization. Among these pathways, the CXCL12/CXCR4 axis has emerged as an important regulator of both the initiation and maintenance of neuropathic pain. Beyond its canonical function in immune cell trafficking, the CXCL12/CXCR4 axis modulates neuronal excitability, glial activation, synaptic plasticity, and nociceptive sensitization. Notably, this axis is frequently upregulated in both peripheral and central neurons, as well as in multiple glial populations, including astrocytes, microglia, and satellite glial cells, across diverse neuropathic pain models. Importantly, CXCR4 is one of the few chemokine receptors with a clinically approved antagonist, highlighting its unique translational potential. This review systematically summarizes the expression patterns, biological functions, and pain-related mechanisms of the CXCL12/CXCR4 axis in preclinical models of neuropathic pain and discusses current limitations and potential future therapeutic strategies targeting this pathway.
Introduction
Pain is defined as an unpleasant sensory and emotional experience typically associated with actual or potential tissue damage. 1 As a subjective sensation, pain perception is influenced by biological, psychological, and social factors. 2 Acute pain is generally regarded as a protective mechanism, serving as a defense response to harmful stimuli, infections, or secondary injuries.3,4 It typically occurs suddenly and lasts for a short duration. In contrast, chronic pain is characterized by persistent or recurrent pain lasting for more than 3 months. 5 Neuropathic pain (NP) is a common type of chronic pain that arises from lesions or diseases affecting the peripheral nervous system (PNS) or central nervous system (CNS). Classic causes of peripheral NP include painful peripheral neuropathies induced by metabolic diseases (such as diabetes), inflammatory diseases (such as herpes zoster), and traumatic nerve injuries. In contrast, central NP is often caused by conditions such as spinal cord injury, stroke, and multiple sclerosis. 6 The prevalence of NP in the general population is estimated to be approximately 7%–10%, accounting for 20%–25% of all patients with chronic pain, 7 thereby imposing a substantial burden on both individuals and healthcare systems.8,9 Compared with other types of chronic pain, NP is typically more severe and represents a major contributor to the global burden of disease. 10 NP significantly impairs patients’ quality of life and may lead to psychological comorbidities, including anxiety, depression, and sleep disorders.11,12 At present, analgesic drugs remain the cornerstone of pain management; however, their efficacy is often limited, and long-term use is associated with risks of drug dependence and adverse effects. 13 Therefore, there is an urgent need to develop novel analgesic strategies to provide safer and more effective therapeutic options.
Extensive interactions between neurons, immune cells, and glial cells form the fundamental basis for the maintenance of chronic pain. 14 During nociceptive transmission, neurotransmitters and cytokines released by primary sensory neurons act on immune and glial cells. In turn, the activated immune and glial cells promote the release of various inflammatory mediators, including pro-inflammatory cytokines and chemokines, which further sensitize sensory neurons. In chronic pain conditions, chemokines and their receptors function as modulators of neural plasticity and play a critical role in the amplification of nociceptive transmission, making them an emerging focus in pain research. 15 Chemokines, as key signaling molecules, mediate interactions among neurons, glial cells, and immune cells and actively contribute to both peripheral and central sensitization processes. The chemokine CXCL12 and its receptor CXCR4 have been shown to be pathologically upregulated in various NP models, while inhibition of the CXCL12/CXCR4 axis significantly alleviates pain progression. 16 Based on this evidence, further elucidation of the mechanisms underlying the CXCL12/CXCR4 axis may provide novel insights and therapeutic targets for NP.
Background of the CXCL12/CXCR4 axis pathway
Biological characteristics
Chemokines are a large family of highly conserved small proteins that regulate systemic immune responses and cell migration. Based on the positioning and spacing of two conserved N-terminal cysteine residues, chemokines are classified into four subfamilies: CXC, CC, CX3C, and C. CXCL12, also known as stromal cell-derived factor-1 (SDF-1), belongs to the CXC chemokine family and plays critical roles in angiogenesis, cell migration, stem cell maintenance, and neurogenesis. CXCR4, a transmembrane G protein-coupled receptor (GPCR), serves as the principal receptor for CXCL12 and participates in various biological processes, including organogenesis, immune responses, and hematopoiesis.
The intracellular signaling mediated by CXCR4 follows the classical activation mechanism of GPCRs. Upon CXCL12 binding to CXCR4, the receptor couples with heterotrimeric G proteins composed of Gαi, Gβ, and Gγ subunits. This interaction induces the exchange of GDP for GTP on the Gαi subunit, resulting in the dissociation of the G protein complex into a Gαi subunit and a Gβγ dimer. These components then activate several downstream signaling pathways, including the MAPK, PI3K, and PLC pathways. Additionally, the dissociated Gαi subunit can inhibit cyclic AMP (cAMP) production. Beyond G protein-dependent pathways, the CXCL12/CXCR4 interaction can also activate G protein-independent signaling cascades, such as the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway and the β-arrestin pathway. Through these diverse signaling mechanisms, the CXCL12/CXCR4 axis regulates key cellular processes including chemotaxis, cell survival, proliferation, transcription, and gene expression.17,18 The specific downstream signaling events can vary depending on the cell type involved.
Unique features and translational relevance
CXCL12 and CXCR4 are widely expressed in both the PNS and CNS, providing an anatomical basis for their involvement in pain modulation. Knockout of either the CXCL12 or CXCR4 gene results in abnormal nervous system development in animal models, 19 strongly suggesting their essential roles in neural function. In fact, dysregulation of the CXCL12/CXCR4 axis pathway has been implicated in various neurological disorders. 20 The CXCL12/CXCR4 axis participates in multiple key stages of nociceptive processing and coordinates pain signaling at both peripheral and central levels. In the PNS, it modulates the excitability of dorsal root ganglion (DRG) neurons and mediates neuron-glial interactions. 21 At the spinal level, it contributes to the development and maintenance of central sensitization by enhancing neuronal excitability and promoting glial activation.22–24 Through the integration of neuronal activity, glial responses, and inflammatory signaling, the CXCL12/CXCR4 axis may act as a central hub in neuro-immune interactions and contribute to the initiation and persistence of chronic pain.
From a translational perspective, CXCR4 is one of the few chemokine receptors with clinically approved antagonists. The CXCR4 antagonist AMD3100 (plerixafor) has been widely used for hematopoietic stem cell mobilization, supported by clinical data demonstrating a relatively well-characterized safety profile.25–27 This provides a rationale for therapeutic targeting and potential drug repurposing. In contrast, several antagonists targeting other chemokine receptors, such as CCR2, have failed to achieve primary analgesic endpoints in randomized controlled trials involving patients with NP,28,29 indicating that not all chemokine pathways engaged during inflammation and pain possess equivalent therapeutic potential. In this context, CXCR4 antagonists may represent potential therapeutic targets for NP.
Furthermore, the CXCL12/CXCR4 axis is highly conserved across species. The amino acid sequence identity of CXCR4 between humans and mice is approximately 89%, while the major CXCL12 isoforms (α and β) share approximately 92% sequence similarity. 30 In addition, downstream signaling pathways, including Gi protein-mediated PI3K and MAPK cascades, are largely conserved between humans and rodents.31,32 This structural and functional conservation enhances the translational relevance of preclinical findings and provides a basis for the extrapolation of animal data to human pain biology.
Cell-type-specific expression landscape of CXCL12/CXCR4 revealed by single-cell transcriptomics
Although accumulating evidence supports the involvement of CXCL12/CXCR4 axis in NP, its precise cellular localization in the nervous system under physiological conditions remains incompletely defined. Previous studies have reported inconsistent findings regarding the distribution of CXCL12 and CXCR4 in the spinal cord and DRG,33–37 which may be attributed to methodological differences, antibody specificity, and the limited resolution of conventional techniques. Recent advances in single-cell RNA sequencing (scRNA-seq) provide an opportunity to systematically characterize cell-type-specific expression patterns of this signaling axis. To address this issue, we analyzed publicly available scRNA-seq datasets from naïve mouse DRG (GSE298412) and spinal cord (GSE281205) to characterize the physiological distribution of CXCL12 and CXCR4.38,39
After quality control, 16,600 cells from the mouse DRG were retained for downstream analysis. Unsupervised clustering identified 14 distinct clusters, which were further annotated into eight major cell types based on established marker genes: satellite glial cells (SGCs), myelinating Schwann cells (MSCs), non-myelinating Schwann cells (NMSCs), mural cells, fibroblasts, macrophages, endothelial cells, and neurons (Figure 1(a)). In parallel, 24,001 cells from the mouse spinal cord were included after quality control. These cells were grouped into 25 clusters and annotated into 11 major cell types, including microglia, ependymal cells, endothelial cells, oligodendrocytes, pericytes, neurons, dendritic cells, astrocytes, fibroblasts, T cells, and oligodendrocyte precursor cells (OPCs) (Figure 1(e)).

Single-cell RNA sequencing analysis of the DRG and spinal cord from naïve mice: (a, e) t-distributed stochastic neighbor embedding (t-SNE) plots of cells derived from the DRG (a) and spinal cord (e), cell types were manually annotated based on previously reported marker genes,38,39 (b, f) t-SNE feature plots showing the expression patterns of CXCL12 and CXCR4 across cell clusters in the DRG (b) and spinal cord (f), (c, g) dot plots illustrating the expression of CXCL12 and CXCR4 across different cell types in the DRG (c) and spinal cord (g), the color of the dots represents the average expression level, and the size of the dots indicates the percentage of cells expressing the gene, and (d, h) violin plots showing the expression levels of CXCL12 and CXCR4 across different cell types in the DRG (d) and spinal cord (h).
Analysis of gene expression patterns (Figure 1(b)–(d)) revealed that CXCL12 is predominantly expressed in endothelial cells, fibroblasts, and mural cells in the DRG, whereas its expression in SGCs and neurons is minimal. In contrast, CXCR4 is primarily enriched in SGCs and macrophages, with only low expression in neurons. In the spinal cord (Figure 1(f)–(g)), CXCL12 is primarily expressed in endothelial cells, fibroblasts, and pericytes, while showing relatively low expression in astrocytes, neurons, and microglia. Conversely, CXCR4 expression is mainly observed in ependymal cells, dendritic cells, and T cells, with limited expression in astrocytes and microglia, and minimal expression in neurons.
Collectively, these findings provide an overview of the baseline cell-type-specific expression landscape of CXCL12 and CXCR4 under physiological conditions. CXCL12 is predominantly expressed by vascular and stromal cells, whereas CXCR4 is mainly expressed in SGCs and immune-related populations, with minimal neuronal involvement. This baseline provides a critical reference for understanding how pathological upregulation and cellular redistribution of the CXCL12/CXCR4 axis may drive aberrant neuro-immune and neuro-glial interactions in NP.
CXCL12/CXCR4 axis in NP: Evidence from preclinical models
Traumatic nerve injury models
Spared nerve injury (SNI)
The SNI model is established by selectively transecting specific branches of the sciatic nerve, mimicking chronic pain conditions following peripheral nerve injury. In this model, plasma levels of CXCL12 are persistently elevated, and its concentration is positively correlated with pain intensity.40,41 Although direct expression of CXCL12 has not been detected in the injured nerve tissue, the site exhibits a robust release of pro-inflammatory cytokines, chemokines, and adhesion molecules, which may contribute to the increased plasma CXCL12 levels. 40 Studies have demonstrated that a single intravenous injection of exogenous CXCL12 induces mechanical hypersensitivity in naïve mice, whereas repeated injections lead to persistent pain-like behaviors. 40 Furthermore, SNI induces upregulation of CXCL12 and its receptor CXCR4 in both neurons and satellite glial cells within the DRG, and a similar trend has also been reported in the spinal dorsal horn (SDH). Notably, in the spinal cord, CXCL12 is predominantly expressed in neurons and microglia, whereas CXCR4 is mainly found in neurons and astrocytes. 42 These findings suggest that the CXCL12/CXCR4 axis is involved in SNI-induced pain hypersensitivity through coordinated actions in both the PNS and CNS. SNI also leads to sustained upregulation of TNF-α in the DRG and spinal cord. Treatment with the TNF-α synthesis inhibitor thalidomide significantly attenuates pain behaviors and suppresses CXCL12 expression in both regions, 42 implying a key role for inflammatory cytokines in the upregulation of CXCL12. Additionally, SNI activates the ERK pathway in the spinal cord. Inhibition of the upstream MEK pathway effectively alleviates CXCL12-induced mechanical allodynia, highlighting ERK as a critical downstream effector of CXCL12 signaling. 42
Moreover, therapeutic strategies targeting the CXCL12/CXCR4 axis-such as administration of neutralizing antibodies against CXCL12, the CXCR4 antagonist AMD3100, or CXCR4-specific siRNA-have been reported to significantly alleviate SNI-induced pain hypersensitivity.40,42,43
Spinal nerve ligation (SNL)
The SNL model is a classical paradigm for studying peripheral nerve injury-induced NP. In rats subjected to SNL, astrocytes and microglia in the SDH become activated, accompanied by upregulation of the CXCL12/CXCR4 axis and increased expression of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6.44–46 Immunofluorescence analysis reveals that CXCL12 is predominantly expressed in neurons, while its receptor CXCR4 is significantly upregulated in both neurons and astrocytes. Intrathecal injection of CXCL12 induces mechanical hypersensitivity in naïve rats, and co-administration of CXCL12 with fluorocitrate, an astrocyte inhibitor, partially alleviates this effect, indicating that the pro-nociceptive effects of CXCL12 are partially mediated through astrocyte-dependent mechanisms. 45
Furthermore, administration of a neutralizing antibody against CXCL12 significantly reduces pain behaviors in SNL rats by suppressing the activation of both astrocytes and microglia. Similarly, intrathecal delivery of the CXCR4 antagonist AMD3100 has been shown to inhibit glial and neuronal activation and downregulates the expression of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6. 45 In addition, CXCR4 contributes to pain sensitization after SNL by downregulating the inhibitory glycine receptor subunit GlyRα3, thereby impairing inhibitory neurotransmission. 44
Partial sciatic nerve ligation (pSNL)
The pSNL model is another widely used paradigm for studying NP. In this model, both astrocytes and microglia are activated on the ipsilateral side of the spinal cord, and CXCR4 expression is significantly upregulated in the sciatic nerve tissue and spinal glial cells.37,47 The CXCR4 antagonist effectively alleviates pain behaviors in pSNL mice.22,37,47,48 Moreover, various pain-associated molecules, including TNF-α, IL-6, substance P (SP), and calcitonin gene-related peptide (CGRP), are elevated in the ipsilateral spinal cord following pSNL, while AMD3100 treatment reduces the expression of some of these inflammatory mediators, including TNF-α and IL-6. 22 However, some discrepancies have been reported. For example, Luo et al. 48 found that under their experimental conditions, AMD3100 did not alter spinal TNF-α or IL-6 levels but significantly suppressed SP expression. Additionally, AMD3100 was shown to inhibit the expression of phosphorylated JNK1 (p-JNK1), p-p38, and p65 in the spinal cord, while having little effect on the activation of ERK1, ERK2, and JNK2. Notably, microRNAs (miRNAs), as important post-transcriptional regulators, also play a role in modulating CXCR4 expression in the pSNL model. Pan et al. 37 demonstrated that miR-23a targets CXCR4 and regulates the TXNIP/NLRP3 signaling pathway in glial cells, thereby contributing to pain modulation.
Chronic constriction injury (CCI)
The CCI model is a well-established paradigm for investigating the mechanisms of NP. Multiple studies have confirmed that the expression of CXCL12 and its receptor CXCR4 is significantly upregulated in the spinal cord of CCI rats,49–53 where these molecules colocalize with neurons, astrocytes, and microglia. 50 After CCI, the expression of CXCR4 is notably elevated in DRG neurons and satellite glial cells (SGCs). In contrast, CXCL12 expression in the DRG exhibits a biphasic expression pattern: it decreases markedly during the early stage (days 1–3) but partially recovers by day 14. 33 However, another study reported a continuous increase of CXCL12 in the DRG post-CCI. 49 Additionally, serum levels of CXCL12 are also elevated after CCI. 54
The enhanced CXCL12/CXCR4 axis activates downstream pathways such as NLRP3 and RhoA/ROCK2, which in turn promote the release of pro-inflammatory cytokines and exacerbate pain sensitization.24,50,53 Epigenetic regulation also influences CXCR4 expression. For instance, miR-130a-5p, by interacting with the long non-coding RNA MEG3 to form a competing endogenous RNA (ceRNA) network, suppresses CXCL12/CXCR4 expression and inhibits activation of the Rac1/NF-κB pathway. 51 Similarly, miR-185-5p and miR-381 target CXCR4 to reduce neuroinflammation and neuronal apoptosis, thereby alleviating NP.52,53 Furthermore, administration of the CXCR4 antagonist AMD3100 has been shown to alleviate CCI-induced pain hypersensitivity. However, one study reported that although AMD3100 effectively reversed thermal hyperalgesia, its effect on mechanical allodynia was limited, suggesting that the CXCL12/CXCR4 axis may play differential roles in distinct pain modalities. 33
Other traumatic nerve injury models
In the chronic compression of DRG (CCD) model, the expression levels of both CXCL12 and CXCR4 are significantly upregulated in the DRG, with CXCR4 predominantly localized in small- and medium-diameter neurons. 55 CCD also induces marked increases in voltage-gated sodium channels Nav1.8 and Nav1.9 in DRG neurons. These changes can be effectively suppressed by AMD3100 administration, leading to attenuation of NP behaviors. 56
In the spinal cord injury (SCI) model, sustained upregulation of CXCL12 has been observed in the spinal cord. 57 Intravenous administration of a miR-130a-5p mimic significantly downregulates CXCL12 expression and inhibits the activation of its downstream pathways, thereby effectively alleviating SCI-induced NP. 58 However, some studies have reported a decrease in CXCL12 expression within spinal neurons following SCI, 59 suggesting that the increased CXCL12 levels may derive mainly from non-neuronal sources.
In the complete sciatic nerve transection (CSNT) model, the expression of both CXCL12 and CXCR4 in the DRG is also upregulated. Intrathecal injection of AMD3100 suppresses STAT3 expression, while exogenous administration of IL-6 promotes the expression of CXCR4, indicating that the CXCL12/CXCR4 axis may be involved in IL-6/STAT3-mediated neuroinflammatory responses. 60
In the focal demyelination model of the sciatic nerve, CXCR4 expression is elevated in DRG neurons. 61 Moreover, CXCL12 has been shown to induce intracellular calcium increases in these neurons,61,62 suggesting a potential role in sensitization and pain signaling.
Overall, the CXCL12/CXCR4 axis is consistently dysregulated across multiple traumatic nerve injury models. Through modulation of neuron–glia interactions, inflammatory cytokine networks, and epigenetic mechanisms, this pathway appears to contribute to the development and maintenance of NP. Although pharmacological targeting of the CXCL12/CXCR4 axis has shown analgesic effects in preclinical studies, further investigation is warranted to determine its translational potential in clinical settings.
Central neuropathic pain models
Central post-stroke pain (CPSP) is a challenging and treatment-resistant form of NP that typically arises following damage to the thalamus or other central structures after a stroke. In an animal model of CPSP induced by the injection of type IV collagenase into the thalamus, both CXCL12 and CXCR4 are significantly upregulated in the hemorrhagic area, and CXCL12 expression is positively correlated with the severity of pain hypersensitivity. Local blockade of CXCR4 in the thalamus effectively prevents both the development and maintenance of CPSP. 63 Furthermore, even in naïve rats, direct thalamic injection of CXCL12 is sufficient to induce mechanical hypersensitivity, which can be suppressed by the CXCR4 antagonist AMD3100. 63 Notably, thalamic hemorrhage also induces secondary neuroinflammatory responses in distant regions such as the SDH, leading to activation of the spinal CXCL12/CXCR4 axis. Intrathecal administration of AMD3100 significantly alleviates bilateral mechanical hypersensitivity following thalamic hemorrhagic stroke (THS), suggesting that CXCL12/CXCR4 is also involved in the modulation of CPSP via spinal mechanisms. 64 Collectively, these findings indicate that the CXCL12/CXCR4 axis contributes to CPSP through a dual mechanism – local activation in the thalamus and distant regulation within the spinal cord. These findings suggest that targeting this pathway may offer a potential therapeutic approach for CPSP, although further validation is required.
Diabetic neuropathic pain (DNP)
DNP, a common chronic complication of diabetes, primarily results from diabetes-induced damage to the nervous system, especially peripheral neuropathy. Various DNP animal models have been established using high-fat diet (HFD) induction, streptozotocin (STZ) administration, or genetically modified mice (e.g. C57BLKS db/db mice). Across these models, the CXCL12/CXCR4 axis consistently appears to play an important role.65–71 In HFD-induced DNP mice, the CXCL12/CXCR4 pathway is markedly upregulated in DRG neurons, and exposure to CXCL12 elicits a pronounced calcium influx response in these neurons. 67 Knockout of the CB2 gene suppresses the upregulation of CXCR4 and the development of hyperalgesia, suggesting a modulatory role of CB2 in peripheral sensitization. 72 Additionally, studies have shown that Nav1.8-positive DRG neurons are closely associated with mechanical hypersensitivity and small fiber degeneration in DNP mice. Selective deletion of CXCR4 in these neurons reverses both pain behavior and fiber degeneration. 66 In STZ-induced DNP rat models, CXCR4 expression is increased in both the DRG and SDH, with changes in the DRG preceding those in the SDH, indicating that peripheral sensitization may initiate central sensitization. 71 High-mobility group box 1 (HMGB1) indirectly enhances CXCR4-mediated inflammatory responses in both DRG and SDH, whereas administration of the HMGB1 inhibitor glycyrrhizin (GLC) significantly reduces CXCR4 expression and increases pain threshold (i.e. reduces pain sensitivity). 69 These findings suggest that CXCL12/CXCR4 axis contributes to DNP pathogenesis at both peripheral and central levels. Furthermore, CXCL12/CXCR4 axis in the brain is also implicated in DNP. In the anterior cingulate cortex (ACC) of DNP mice, activated microglia upregulate CXCL12/CXCR4, leading to hyperactivity of ACC glutamatergic neurons and exacerbation of pain perception. 68
Therapeutic studies targeting the CXCL12/CXCR4 pathway further support its analgesic potential. Intraperitoneal injection of the CXCR4 antagonist AMD3100 effectively alleviates DNP-associated pain behavior in animal models.65,67,70 However, in advanced stages of DNP, despite sustained high expression of CXCR4, the analgesic efficacy of AMD3100 diminishes, likely due to a cascade amplification of inflammatory mediators (such as CXCL12 and TNF-α) driven by chronic CXCR4 activation. 70 Moreover, whole blood analysis reveals that expression levels of MALAT1 and CXCR4 are significantly correlated with the presence of NP in diabetic patients, suggesting that targeting MALAT1 and CXCR4 may offer a novel therapeutic strategy for DNP. 73
In summary, current evidence suggests a multilayered involvement of the CXCL12/CXCR4 axis in the pathogenesis of DNP, spanning peripheral nerve injury, central sensitization, and neuro-glial network imbalance. Targeted interventions against this pathway have demonstrated analgesic effects in preclinical models; however, their efficacy may vary depending on disease stage, underscoring the need for further investigation to optimize therapeutic strategies.
HIV-associated neuropathic pain (HIV-NP)
HIV-NP is a common complication in people living with HIV, primarily resulting from peripheral nerve injury induced by HIV infection or antiretroviral drugs such as didanosine (ddC). Both mechanisms contribute to sensory deficits and NP. In animal models, HIV gp120 is used to mimic viral injury, while ddC represents antiretroviral drug toxicity. Studies have demonstrated that the CXCL12/CXCR4 axis plays a key role in both gp120- and ddC-induced NP. In both models, TNF-α, CXCL12, and CXCR4 are significantly upregulated in the spinal cord and DRG. Blocking TNF-α significantly alleviates mechanical allodynia and reverses the upregulation of CXCL12/CXCR4 in the spinal cord and DRG.74,75 Similarly, viral vector-mediated overexpression of the anti-inflammatory cytokine IL-10 alleviates pain hypersensitivity and concurrently downregulates spinal cord and DRG levels of p-p38, TNF-α, CXCL12, and CXCR4.76,77 In the gp120 model, SGCs in the DRG exhibit specific and pronounced upregulation of CXCL12. CXCR4 expression appears to be cell type-dependent. Some studies report its upregulation in both neurons and SGCs, 78 while others have observed increased CXCR4 expression restricted to small- and medium-diameter neurons. 79 Administration of the CXCR4 antagonist AMD3100 significantly reduces pain behaviors in both gp120- and ddC-induced models, further supporting the involvement of the CXCL12/CXCR4 axis in HIV-NP.75,78,79 These findings highlight the interplay between neuroinflammatory processes and chemokine signaling in HIV-associated neuropathy and suggest that the CXCL12/CXCR4 pathway may represent a potential therapeutic target. Future studies are needed to further elucidate downstream effectors of this signaling axis to enable more precise analgesic strategies.
Intervertebral disc degeneration-associated low back pain
Intervertebral disc degeneration (IVDD) is a common cause of low back pain, and during disease progression, it can lead to nerve root compression and nerve injury, with some patients exhibiting features of NP. In degenerated intervertebral disc tissues, the expression of CXCL12 and its receptor CXCR4 is significantly upregulated.80–82 Studies have shown that stimulation of degenerated nucleus pulposus cells with CXCL12 induces increased apoptosis and elevated phosphorylation of p65 in the NF-κB signaling pathway, whereas transfection with CXCR4 siRNA can inhibit these changes. 81 Furthermore, CXCL12 promotes CXCR4 expression and upregulates matrix metalloproteinases (MMPs) at both the mRNA and protein levels in cartilage endplate cells, thereby accelerating extracellular matrix (ECM) degradation. 83 Interfering with CXCL12/CXCR4 axis can effectively inhibit the production of pro-inflammatory mediators, delay ECM breakdown and disc degeneration, and alleviate pain symptoms.83–87 Angiogenesis is a hallmark pathological feature of IVDD and is closely correlated with the severity of degeneration. 88 CXCL12 participates in this process via binding to either CXCR4 or CXCR7.89,90 Notably, CXCL12 also plays a positive role in disc regeneration. Its chemotactic signaling with CXCR4 enhances the recruitment and chondrogenic differentiation potential of nucleus pulposus–derived stem cells (NPSCs). 91 In addition, CXCL12 promotes the proliferation of nucleus pulposus cells (NPCs) via cartilage endplate stem cells (CESCs), an effect that is significantly inhibited by AMD3100. 92 Although CXCL12 contributes to ECM degradation, moderate matrix remodeling may facilitate the migration of endogenous stem cells toward degenerated regions, thus promoting tissue repair and regeneration. 83 In summary, the CXCL12/CXCR4 axis plays critical roles in both the pathogenesis and regeneration of IVDD, exerting dual regulatory effects and representing a potential therapeutic target for IVDD-associated low back pain.
Chemotherapy-induced peripheral neuropathy (CIPN)
CIPN represents a significant clinical challenge during cancer treatment, often leading to dysesthesia and abnormal pain sensations. Chemokine signaling has been implicated as an important mediator in CIPN pathogenesis. 93 Studies have shown that intraperitoneal administration of paclitaxel or vincristine activates the STAT3 signaling pathway in SDH neurons. This activation enhances histone acetylation at the CXCL12 gene promoter, thereby increasing CXCL12 mRNA and protein expression. Blocking CXCR4 attenuates mechanical allodynia induced by paclitaxel or vincristine. 94 Similarly, repeated intraperitoneal injections of oxaliplatin activate STAT3 and upregulate CXCL12 expression in the DRG; inhibition of STAT3 expression prevents CXCL12 elevation in the DRG and attenuates oxaliplatin-induced chronic pain. 95 Paclitaxel also activates spinal astrocytes, promoting the release of pro-inflammatory mediators such as TNF-α and CXCL12. Administration of TNF-α or CXCL12 neutralizing antibodies reverses paclitaxel-induced mechanical allodynia. 96 Oxaliplatin treatment also induces elevated serum levels of TNF-α and CXCL12. 97 Bioinformatic analyses further reveal a strong association between CXCR4 expression and the incidence, severity, and susceptibility of CIPN. 98 However, a single intraperitoneal injection of AMD3100 failed to effectively relieve cisplatin- or paclitaxel-induced mechanical or cold hypersensitivity within a short-term (3-hour) window. 99 Collectively, these findings suggest that the CXCL12/CXCR4 axis is involved in the initiation and progression of CIPN. Nonetheless, its precise mechanisms and therapeutic efficacy remain to be fully elucidated, highlighting the need for further studies to develop more reliable therapeutic strategies for CIPN management.
Conclusions and future perspectives
This review systematically summarizes the biological functions, cellular distribution, and roles of the CXCL12/CXCR4 axis in pain signal transmission, with a particular focus on its potential involvement in the initiation and maintenance of NP. Although initially characterized in the context of immune cell trafficking, CXCL12/CXCR4 axis has also been implicated in broader neurobiological processes, including neurodevelopment, neuronal survival, synaptic plasticity, and CNS homeostasis. 100 These broader roles suggest that CXCL12/CXCR4 axis may extend beyond physiological functions to influence pathological processes, including pain modulation. 101 In multiple NP models, upregulation of the CXCL12/CXCR4 axis has been observed in both peripheral and central neurons, as well as in various types of glial cells, including astrocytes and microglia in the CNS and SGCs in the PNS (Table 1). Together, these observations indicate that CXCL12/CXCR4 axis may act as an active contributor to pain processing, rather than merely reflecting injury-associated changes.
Cellular distribution of the CXCL12/CXCR4 axis in preclinical models of neuropathic pain.

CCI: chronic constriction injury; CGRP: calcitonin gene-related peptide; CIPN: chemotherapy-induced peripheral neuropathy; CSNT: complete sciatic nerve transection; DNP: diabetic neuropathic pain; HIV-NP: HIV-associated neuropathic pain; pSNL: partial spinal nerve ligation; SCI: spinal cord injury; SGCs: satellite glial cells; SNI: spared nerve injury; SNL: spinal nerve ligation; SP: substance P.
In recent years, a growing number of preclinical studies have evaluated the effects of CXCR4 antagonists across diverse NP models (summarized in Table 2). Across these studies, CXCR4 blockade has generally been associated with reductions in mechanical allodynia and/or thermal hyperalgesia. Mechanistic investigations suggest that the CXCL12/CXCR4 axis may contribute to the development and maintenance of peripheral and central sensitization through coordinated regulation of neuronal excitability, glial activation, and neuroinflammatory responses (Figure 2). However, the relative contribution of these mechanisms likely varies depending on the injury context and disease stage.
Preclinical evidence for the analgesic effects of CXCR4 antagonists in neuropathic pain.
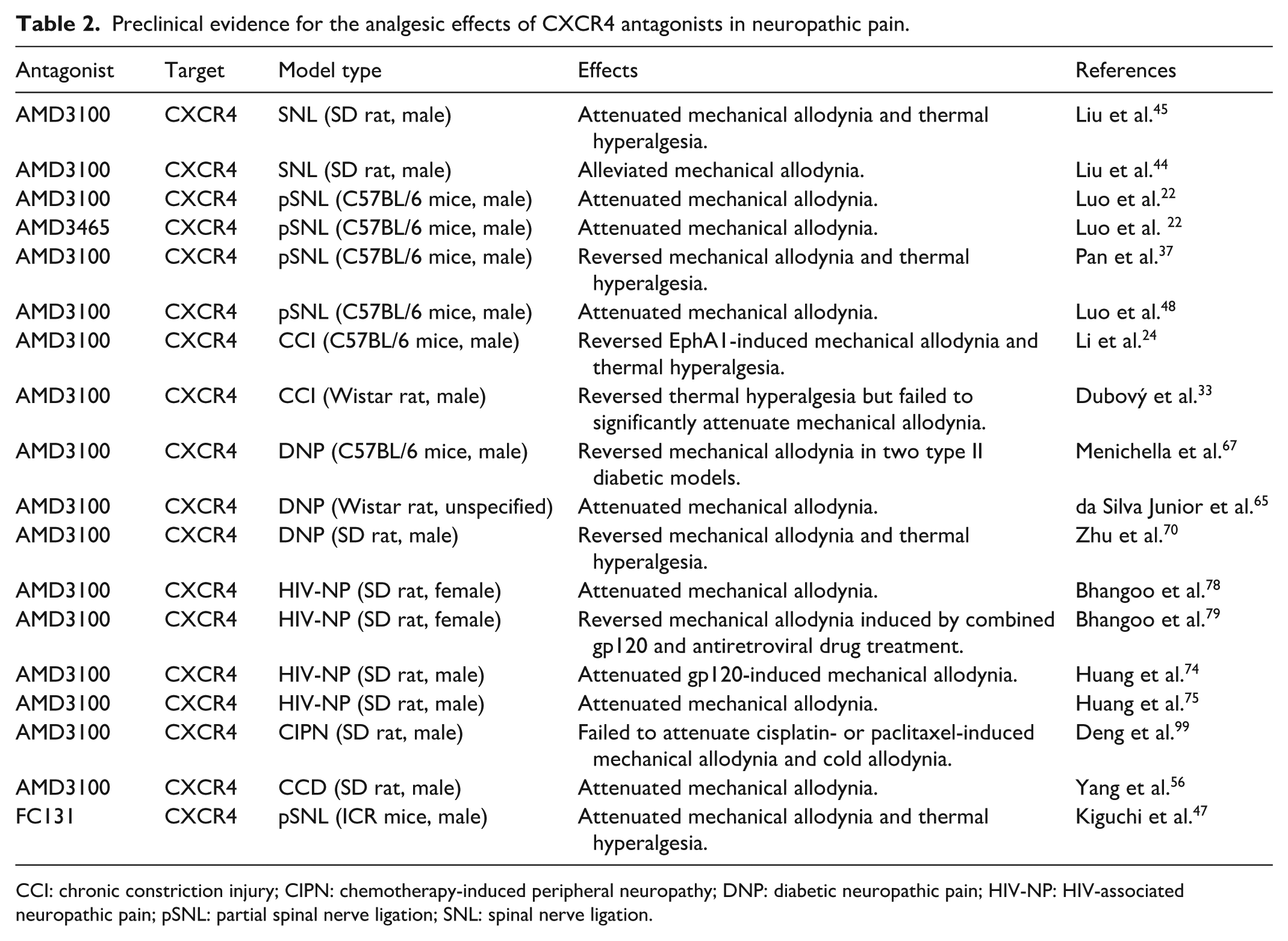
CCI: chronic constriction injury; CIPN: chemotherapy-induced peripheral neuropathy; DNP: diabetic neuropathic pain; HIV-NP: HIV-associated neuropathic pain; pSNL: partial spinal nerve ligation; SNL: spinal nerve ligation.
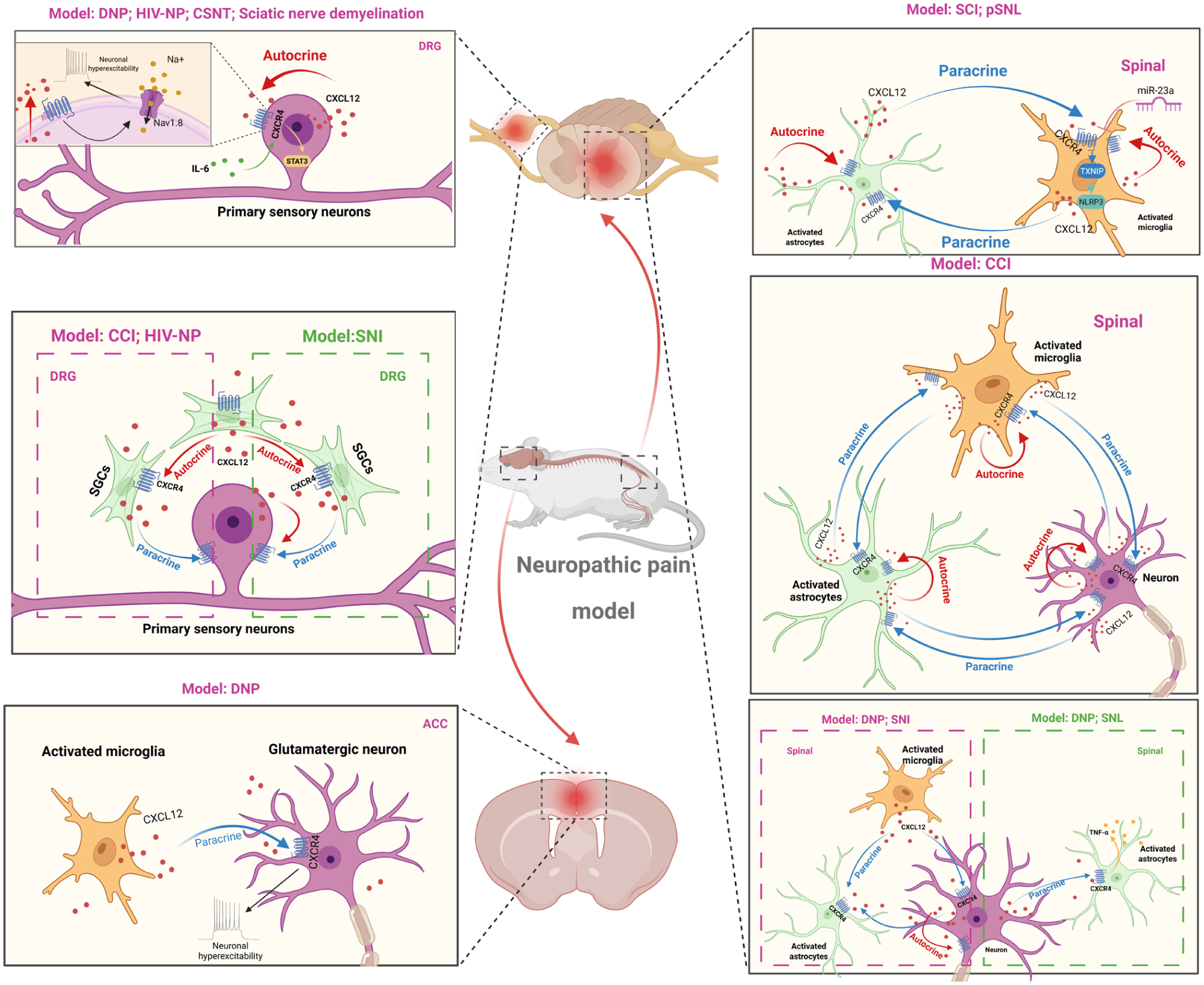
CXCL12/CXCR4 axis-mediated neuron-glia crosstalk in peripheral and central sensitization of neuropathic pain.
Notably, the availability of clinically approved CXCR4 antagonists provides a practical basis for exploring the translational potential of targeting this pathway. AMD3100 (plerixafor), originally developed for hematopoietic stem cell mobilization, has accumulated clinical evidence supporting its pharmacokinetic and safety profiles.25–27 However, despite these advantages, CXCR4-targeted therapies have not yet been systematically evaluated in clinical pain trials, and their efficacy in human NP remains uncertain.
Despite encouraging preclinical findings, several important limitations should be considered. First, current studies on the CXCL12/CXCR4 axis largely rely on global interventions, with limited cell-specific genetic evidence. As complete deficiency of CXCL12 or CXCR4 results in perinatal lethality, the use of conventional knockout models is restricted.102,103 Therefore, the development of cell-specific genetic approaches will be important for clarifying its roles in distinct cellular contexts. Second, it is now recognized that pain circuits exhibit sex-based differences, 104 and the CXCL12/CXCR4 axis may also demonstrate sex-specific patterns in pain modulation. Although women account for the majority of chronic pain patients, preclinical studies have predominantly utilized male animal models. Therefore, inclusion of both sexes in future studies will be important to improve translational relevance. Third, the spatiotemporal dynamics of CXCL12/CXCR4 axis during different stages of NP remain incompletely understood. Given the dynamic activation of glial cells under pathological conditions, 105 further studies are needed to delineate how this axis is regulated across disease progression. Such investigations may help clarify stage-dependent mechanisms underlying pain initiation and maintenance and provide a basis for temporally targeted therapeutic strategies.
In addition, the long-term safety of targeting CXCR4 warrants careful consideration. The CXCL12/CXCR4 axis plays essential roles in immune homeostasis, cardiovascular function, neurogenesis, and tumor biology. Prolonged systemic blockade of this pathway may therefore interfere with immune regulation or tissue repair processes. Although clinical studies in oncology have shown that CXCR4 antagonists are generally well tolerated, with adverse event rates comparable to control treatments, 106 their safety profile in the context of chronic pain, where long-term or repeated administration may be required, remains insufficiently characterized. In particular, the potential effects on immune function, neural plasticity, and central reward circuits require further systematic evaluation. Although no evidence currently suggests that CXCR4 antagonists possess addictive properties, the possible functional overlap between chemokine signaling and dopaminergic or affective regulatory pathways warrants further investigation of long-term neurobehavioral outcomes. 107
Based on current evidence, several directions may help advance the clinical translation of CXCL12/CXCR4-targeted interventions. First, drug repurposing may represent a feasible approach. Leveraging the existing clinical safety data for AMD3100, further exploration of its localized or short-term application in specific NP subtypes may help accelerate translational progress. Second, gene- and RNA-based therapeutic approaches, such as siRNA, shRNA, or CRISPR systems targeting CXCL12 or CXCR4, may provide promising strategies for region- and cell-specific modulation of this signaling axis, potentially preserving analgesic efficacy while minimizing systemic side effects. 108 Third, nanoparticle-based targeted delivery systems may offer an innovative technical avenue for CXCL12/CXCR4 modulation. 109 By directing CXCR4 antagonists to the DRG or spinal cord, such approaches may help enhance local analgesic effects while reducing required doses.
Overall, the CXCL12/CXCR4 axis may represent an important component of neuroimmune signaling in NP. However, its precise cellular contributions, temporal dynamics, and clinical utility remain to be fully clarified. Addressing these gaps will be critical for determining whether targeting this pathway can be effectively translated into clinically meaningful pain therapies.
Footnotes
Author contributions
Ming Li and Xiaoxiao Lu contributed to the literature review and drafting of the manuscript. Luyao Cai and Tao Zhu participated in manuscript revision and editing. Jin Cui supervised the study and revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
Data availability
No new data were generated or analyzed in support of this review. All information discussed is based on previously published literature.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The present project is supported by the Graduate Research Funding Program of Guizhou Province (2024YJSKYJJ351).