
Abstract
N-Acetylcysteine, one of the most prescribed antioxidant drugs, enhances pain threshold in rodents and humans by activating mGlu2 metabotropic glutamate receptors. Here, we assessed the analgesic activity of N-acetylcysteine in the streptozotocin model of painful diabetic neuropathy and examined the effect of N-acetylcysteine on proteins that are involved in mechanisms of nociceptive sensitization. Mice with blood glucose levels ≥250 mg/dl in response to a single intraperitoneal (i.p.) injection of streptozotocin (200 mg/kg) were used for the assessment of mechanical pain thresholds. Systemic treatment with N-acetylcysteine (100 mg/kg, i.p., either single injection or daily injections for seven days) caused analgesia in diabetic mice. N-acetylcysteine-induced analgesia was abrogated by the
Introduction
Diabetic distal symmetrical polyneuropathy represents one of the most common complications of both type-1 and type-2 diabetes mellitus, with about 20% of the patients developing neuropathic pain. 1 The management of painful diabetic neuropathy involves good glycemic control, lifestyle modifications, and the use of analgesic and neuroprotective drugs. 1 According to the European Federation of Neurological Societies (EFNS) guidelines, amytriptiline, duloxetine, venlafaxine, pregabalin, and gabapentine are recommended as first-line drugs for the treatment of painful diabetic neuropathy, whereas opioids are indicated as second-line drugs. 2 Unfortunately, pharmacological treatment of diabetic neuropathy remains suboptimal, with only one-third of patients showing 50% of pain relief. 3 Combination therapy may enhance the efficacy of analgesic agents, as shown for duloxetine and pregabalin in the COMBO-DN study, 4 but at the expenses of troublesome adverse effects. This encourages the search for new analgesic agents, which meet the pharmacodynamic and pharmacokinetic requirements for their use as add-on drugs in the treatment of painful diabetic neuropathy.
Type-2 metabotropic glutamate (mGlu2) receptors are Gi/o-coupled presynaptic receptors, which negatively modulate glutamate release at the synapses between primary afferent fibers and secondary order neurons in the dorsal horns of the spinal cord. 5 Therefore, these receptor are strategically localized to restrain the process of central nociceptive sensitization, which is involved in the pathophysiology of painful diabetic neuropathy and other forms of neuropathic pain. 5 Mice lacking mGlu2 receptors show large increases in nocifensive behavior in the second phase of the formalin test, which reflects the development of central nociceptive sensitization. 6
Recent findings indicate that mGlu2 receptors are expressed by sensory neurons in human dorsal root ganglia, where their activation suppresses prostaglandin-E2-induced sensitization. 7 This suggests that studies on mGlu2 receptors in experimental animal models of chronic pain have translational validity. L-Acetylcarnitine (LAC), a drug used for the treatment of painful diabetic neuropathy,8–11 causes analgesia by inducing the expression of mGlu2 receptors through an epigenetic mechanism.5,12–14 mGlu2 receptors are also targeted by N-acetylcysteine (NAC), a drug that is traditionally used as a mucolytic agent and for the treatment of acetaminophen intoxication and contrast-medium-induced kidney toxicity.15–18
NAC activates the cystine/glutamate membrane exchanger (
We have found that NAC exerts robust analgesic activity in the second phase of the formalin test, and its action was abrogated by genetic deletion or pharmacological blockade of mGlu2 receptors. 32 NAC also caused analgesia in a mouse model of chronic inflammatory pain without the development of tolerance; in contrast, in the chronic constriction injury (CCI) model of neuropathic pain, NAC caused analgesia after a single injection, but not after repeated administrations. 32 This suggests that NAC-induced analgesia is not uniform in different pain models and may be context-dependent.
Here, we examined the analgesic activity of NAC in the streptozotocin (STZ) mouse model of painful diabetic neuropathy extending the study to molecular mechanisms involved in the induction, expression, and maintenance of nociceptive sensitization in the spinal cord.
Materials and methods
Drugs
NAC, sulfasalazine, and STZ were purchased from Sigma Aldrich (St. Louis, MO); (2S)-2-Amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl)propanoic acid (LY341495), pregabalin, erastin, sorafenib, PD0325901, JNJ479655567, capsazepine, memantine, and 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP) were purchased from Tocris Cookson (Avonmouth, Bristol, UK). STZ was dissolved in sodium citrate buffer (0.01 M, pH 4.5). NAC, LY341495, sulfasalazine, and pregabalin were dissolved in saline; erastin, sorafenib, capsazepine, PD0325901, JNJ479655567, and memantine + MTEP were dissolved in saline containing 50% DMSO (vol/vol).
Induction of experimental diabetes in mice and drug treatments
We used two-month-old male C57BL/6 mice (bred in the animal house of IRCCS Neuromed) for the induction of diabetic neuropathy. Mice were kept under control conditions (T = 22°C; humidity = 40%) on a 12-h light-dark cycle with food and water ad libitum. Experiments were performed following the Guidelines for Animal Care and Use of the National Institutes of Health to minimize the number of animals and animal suffering. The experimental protocol was approved by the Ethical Committee of Neuromed Institute (Pozzilli, Italy) and by the Italian Ministry of Health (authorization # 524/2016-PR). Diabetes mellitus was induced by a single injection of STZ (200 mg/kg, intraperitoneal (i.p.)). Blood glucose levels were measured with a glucometer (ACCU-CHEK Active, Roche), according to the manufacturer’s instructions. Mice with random blood glucose levels ≥250 mg/dl 14 days after STZ injection were considered as diabetic and used for the assessment of mechanical pain thresholds. Mice were weighed on a weekly basis. Age-matched, non-diabetic C57BL/6 mice were used as the controls. Groups of 7/10 mice (either diabetic mice or non-diabetic controls) were treated i.p. as follows: single injections of saline, NAC (50 or 100 mg/kg), and pregabalin (30 mg/kg); daily injections of saline, NAC (100 mg/kg) from day 21 to day 28 after STZ administration. Some groups of mice treated with saline or NAC from days 21 to 28 received a single i.p. injection of the mGlu2/3 antagonist, LY341495 (1 mg/kg) or the

NAC-induced analgesia in the STZ model of painful diabetic neuropathy. Blood glucose levels in mice receiving a single injection of saline or STZ (200 mg/kg, i.p.) are shown in (a), where values are means ± S.E.M. of 7–10 mice. *p < 0.05 (Student’s t-test; t = −8.279). Reductions of mechanical pain thresholds in the same mice at 14 and 21 days following STZ injection are shown in (b), where values are also means ± S.E.M. *p < 0.05 (one-way ANOVA for repeated measures + Duncan method; F(1,13) = 77.224). The effect of a single injection of NAC or pregabalin on mechanical pain thresholds in diabetic mice and non-diabetic control mice are shown in (c), where values are means ± S.E.M. of 7–10 mice. *p < 0.05 (one-way ANOVA + Duncan method applied only to the groups of diabetic mice; F(2,18) = 4.562). The effect of repeated administrations of saline or NAC (100 mg/kg, i.p., once a day for 7 days starting from 21 days after STZ injection) on mechanical pain thresholds are shown in (d and e). In (d), all groups of mice were injected once with saline, LY341495, or sulfasalazine at the end of the chronic treatment with saline or NAC (see Methods). Values are means ± S.E.M. of 6–7 mice per group. p < 0.05 versus the control group receiving repeated injections of saline followed by a single injection of saline (*); or versus the group treated with NAC and a single injection of saline (#) (one-way ANOVA + Duncan method; F(5,35) = 3.364). In (e), all groups of mice were injected once with vehicle, erastin, sorafenib, PD0325901, capsazepine, and mementine+MTEP at the end of the chronic treatment with saline or NAC. Values are means ± S.E.M. of 4–10 mice per group. p < 0.05 versus the control group receiving repeated injections of saline followed by a single injection of vehicle (*); or versus the group treated with NAC and a single injection of vehicle (#); or versus the control group receiving repeated injections of saline followed by a single injection of vehicle ($) (one-way ANOVA + Duncan method; F(11,73) = 7.947). Blood glucose levels in mice receiving a single injection of saline or NAC (100 mg/kg, i.p.) are shown in (f), where values are means ± S.E.M. of 9 mice. In (g), four groups of mice were injected once with vehicle or JNJ47965567 (30 mg/kg) at the end of the chronic treatment with saline or NAC. Values are means ± S.E.M. of 7–10 mice per group. p < 0.05 versus all other groups (one-way ANOVA + Duncan method; F(3,28) = 18.643). NAC: N-acetylcysteine; STZ: streptozotocin.
CFA model of chronic inflammatory pain and drug treatment
Inflammatory pain was induced by subcutaneous (s.c.) injections of 20 μl of Complete Freund’s Adjuvant (CFA, Sigma-Aldrich; 1 mg/ml) in the plantar surface of the right hind paw; control mice were injected s.c. with saline in the right hind paw. Groups of six to seven mice were treated i.p. daily for seven days with saline or NAC (25–100 mg/kg). Treatments started 10 min following CFA (or saline) injection. Mechanical pain thresholds were measured 1 h after the last injection. Mice were killed immediately after for protein measurements in the dorsal region of the lumbar spinal cord ipsilateral to CFA injections.
Assessment of mechanical pain thresholds
Mechanical pain thresholds were quantified by measuring the hind paw withdrawal response to von Frey filament stimulation. In brief, mice were placed in a plexiglas box (20 cm high, 9 cm diameter) with a wire grid bottom through which the von Frey filaments (North Coast Medical, Inc., San Jose, CA; bending force range from 0.008 to 3.5 g) were applied by using a modified version of the up-down paradigm. 33 The filaments were applied five times each, in order of increasing forces, and pressed perpendicularly to the plantar surface of the hind paw until they bent. The first filament that evoked at least three responses was assigned as the pain threshold in grams.
Western blot analysis
Western blot analysis of proteins involved in mechanism of action of NAC in the development of nociceptive sensitization was performed in the dorsal regions of the lumbar spinal cord dissected from diabetic or non-diabetic mice treated from days 21 to 28 with saline or NAC and in CFA-injected mice receiving saline or NAC for seven days. In CFA-treated mice, we also examined TRPV1 protein levels in the plantar surface of the hindlimb ipsilateral to CFA injection. Tissue was homogenized on ice with RIPA buffer containing protease and phosphatase inhibitors cocktail tablet (Santa Cruz Biotechnology, Inc., Temecula, CA). Homogenates were centrifuged at 14,000 r/min for 10 min, and an aliquot was used for protein determinations. Equal amounts of proteins (20 µg) from supernatants were separated by 8/10% SDS polyacrilamide gel and transferred on Immuno PVDF membranes (Bio-Rad, Milan, Italy) for 7 min using Trans Blot Turbo System (Bio-Rad, Milan, Italy). Filters were blocked overnight in blocking buffer (TBS, 0.05% Tween-20 and 5% non-fat milk) at 4°C. Membranes were incubated with the following primary antibodies: anti-mGlu2/3 receptors (1:2,000, Sigma-Aldrich, Darmstadt, Germany), anti-pERK1/2/ERK1/2 (1:1000, Cell Signaling, Leiden, The Netherlands), anti-xCT (1:1500, Trans Genic Inc., Kumamoto, Japan), anti-α2δ1 (1:200, Sigma-Aldrich, Darmstadt, Germany), anti-AGS3 (1:500, Santa Cruz Biotechnology Inc., Dallas, TX), anti-P2X7 receptors (1:200, Millipore/Merck KGaA, Darmstadt, Germany), and anti-TRPV1 (1:500, Novus Biologicals, Littleton, CO). Blots were then incubated with corresponding secondary antibodies. The blots were re-probed with anti-β-actin monoclonal antibody (1:50,000 Sigma-Aldrich). Immunostaining was revealed by enhanced chemiluminescence luminosity (Amersham Pharmacia Biotech, Arlington Height, IL) and images captured by ChemiDoc System (BioRad, Berkley, CA).
Statistical analysis
Statistical analysis was carried out by one-way or two-way analysis of variance (ANOVA) followed by Duncan’s method for pain thresholds and for biochemical determinations in diabetic mice and their controls, or by Student’s t-test for blood glucose levels and protein determinations in CFA-injected mice treated with saline or NAC. p values <0.05 were considered as statistically significant.
Results
For the induction of painful diabetic neuropathy, we injected mice with a single dose of STZ (200 mg/kg, i.p.) (see Discussion and References therein). Measurements of blood glucose levels confirmed the induction of type-1 diabetes 14 days after STZ injection (Figure 1(a)). Mechanical pain thresholds were largely reduced at both 14 and 21 days in mice injected with STZ (Figure 1(b)). Diabetic mice received an acute administration of NAC at either 14 or 21 days following STZ injection. A single injection of NAC (100 mg/kg, i.p.) caused analgesia comparable to that induced by pregabalin (30 mg/kg, i.p.) (Figure 1(c)), a first-line drug in the treatment of painful diabetic neuropathy (see Introduction and References therein). NAC was inactive at a dose of 50 mg/kg, i.p. (not shown).
Independent groups of diabetic mice treated daily with saline or NAC from day 21 to day 28 received a single injection of either 1 mg/kg of LY341495 (a preferential orthosteric antagonist of mGlu2 and mGlu3 receptors) or 8 mg/kg of sulfasalazine (an inhibitor of
Additional groups of diabetic mice were treated daily with saline or NAC from day 21 to day 28 and received a single injection of erastin (30 mg/kg) or sorafenib (10 mg/kg) (inhibitors of the
We examined whether NAC treatment could induce adaptive changes in proteins targeted by NAC or in proteins that are involved in mechanisms of nociceptive sensitization in the spinal cord. Seven days of treatment with NAC in diabetic mice did not cause significant changes in protein levels of mGlu2/3 receptors, AGS3, xCT (the catalitic subunit of

Expression of proteins targeted by NAC or involved in mechanisms of nociceptive sensitization in the dorsal region of the lumbar spinal cord of diabetic and non-diabetic mice receiving repeated administrations of NAC. Mice were treated i.p. with either saline or NAC for seven days (100 mg/kg) once a day, starting from 21 days following a single injection of saline or STZ. Densitometric values are expressed as % values obtained in the group of STZ mice treated with saline for seven days (this group is common to two–three different immunoblots used for the analysis). Only one representative blot is shown. Densitometric values are means ± S.E.M. of 2–7 mice. p < 0.05 (two-way ANOVA + Dancan’s method; P2X7: F(1,13) = 11.996 for pathological model (control and diabetic mice) and F(1,13) = 4.717 for drug treatment; P-ERK1: F(1,17) = 13.312 for pathological model (control and diabetic mice) and F(1,17) = 17.514 for drug treatment; and P-ERK2: F(1,16) = 6.652 for pathological model (control and diabetic mice) and F(1,16) = 19.678 for drug treatment)). NAC: N-acetylcysteine.
We extended the study to the CFA model of chronic inflammatory pain, where NAC is known to be effective, 32 to make a head-to-head comparison of protein changes induced in the two models of diabetic and inflammatory pain. Intraplantar injection of CFA (20 µg/µl) caused a significant reduction of mechanical pain thresholds in the ipsilateral hindlimb, which was lasting for at least two weeks. As expected, acute systemic administration of NAC seven days following CFA injection caused analgesia. The action of NAC was dose-dependent, with substantial analgesia being observed at the dose of 100 mg/kg, i.p. (Figure 3(a)). A trend to an increase in the mechanical thresholds was observed at the dose of 50 mg/kg, whereas the dose of 25 mg/kg of NAC was inactive (Figure 3(a)). In another set of experiments, NAC (100 mg/kg, i.p.) was administered daily from days 7 to 14 following CFA injection. This treatment caused the expected increase in mechanical pain thresholds (Figure 3(b); see also Bernabucci et al., 32 ). NAC did not affect mechanical pain thresholds in control mice that did not receive intraplantar injection of CFA (not shown). CFA-treated mice receiving repeated administrations of NAC were used for determination of protein levels in the dorsal regions of the lumbar spinal cord ipsilateral to CFA injection or, only for TRPV1 expression, in the plantar paw surface. NAC treatment in CFA mice did not change protein levels of mGlu2/3 receptors, AGS3, the α2δ subunit of voltage-sensitive Ca2+ channels, or phosphorylated ERK1/2 in the spinal cord, but caused a large reduction in TRPV1 levels in the plantar paw surface (Figure 4).
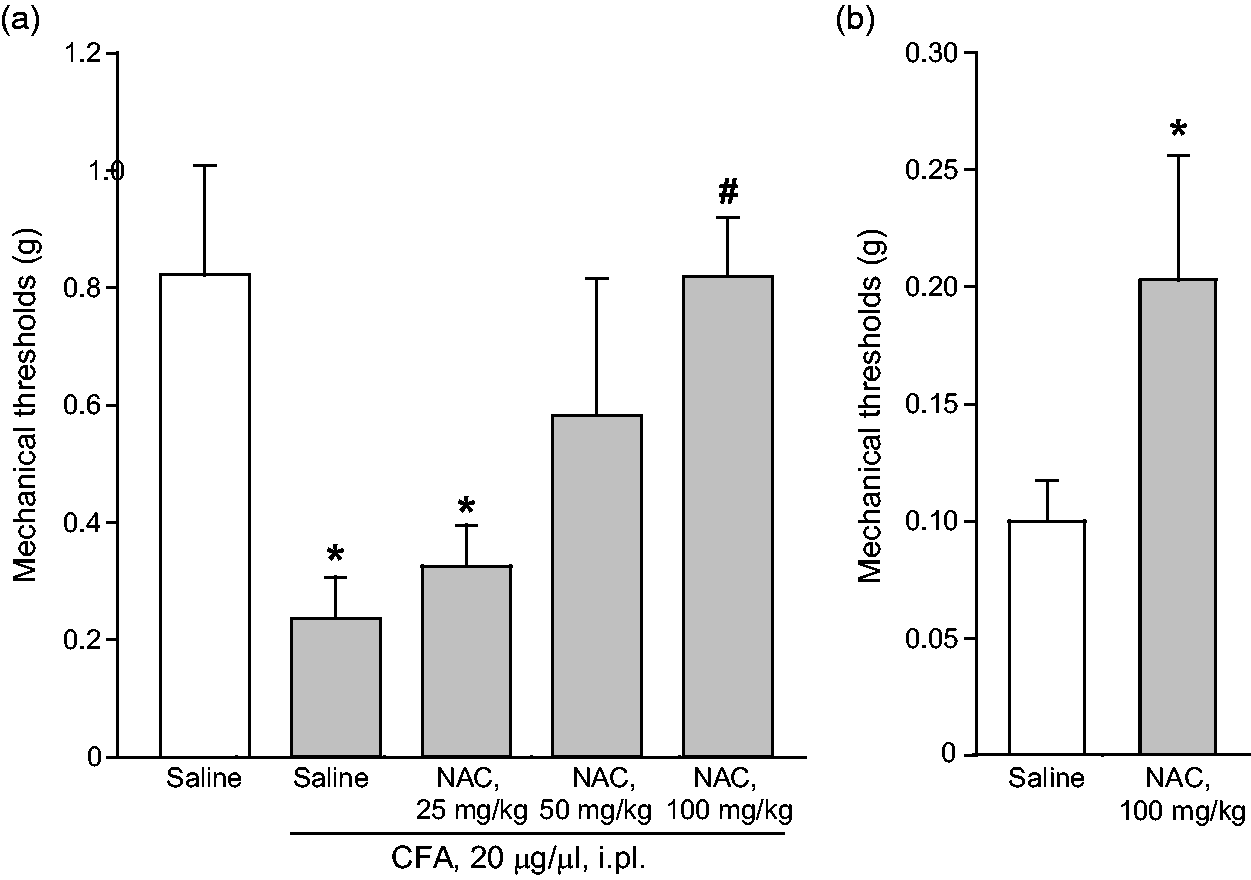
NAC-induced analgesia in the CFA model of chronic inflammatory pain. Mechanical pain thresholds in mice seven days following CFA injection in the plantar paw after acute systemic treatment with saline or NAC (25-100 mg/kg, i.p.) are shown in (a), where values are means ± S.E.M. of 6–7 mice per group. p < 0.05 (one-way ANOVA + Duncan’s method; F(4,28) = 3.834) versus saline treated-mice (*) or versus CFA-injected mice after saline treatment (#). The effect of repeated administrations of saline or NAC (100 mg/kg, i.p., once a day for seven days starting from seven days after CFA injection) on mechanical pain thresholds is shown in (b). Values are means ± S.E.M. of seven mice per group. *p < 0.05 (Student’s t-test; t = −1.841). NAC: N-acetylcysteine; CFA: Complete Freund’s Adjuvant.

Expression of proteins targeted by NAC or involved in mechanisms of nociceptive sensitization in the dorsal region of the ipsilateral lumbar spinal cord or in the plantar paw of CFA-injected mice receiving repeated administrations of NAC. Mice were treated i.p. with either saline or NAC for seven days (100 mg/kg) once a day, starting from seven days following CFA injection in the plantar paw. Densitometric values are means ± S.E.M. of 3–5 mice. *p < 0.05 (Student’s t-test). NAC: N-acetylcysteine.
Discussion
NAC is a safe drug that has been used for decades in the treatment of bronchopulmonary disorders, and liver and kidney intoxication (see Introduction and References therein). NAC causes only mild adverse effects when given chronically for the treatment of bronchitis 34 and is not a substrate, inducer, or inhibitor of cytochrome-P450 or glycoprotein-P. The good pharmacokinetics profile contributes to the widespread use of NAC in intravenous, oral, and nebulizer forms. NAC is also marketed as an over-the-counter product in food stores.35,36 Research interest in NAC has grown substantially in the last years, owing to new insights into its mechanism of action that extend its clinical applications to neurological and psychiatric disorders. For many years, NAC has been considered as a potential source of intracellular reduced glutathione, one of the major cells’ defense against oxidative stress. However, the seminal work of Peter Kalivas and his research group at the University of South Carolina has demonstrated that NAC may influence glutamate homeostasis in the CNS as a result of the activation of the glutamate/cystine membrane antiporter and the induction of the glial glutamate transporter, GLT-1.37–39 The resulting elevation in extrasynaptic glutamate levels enhances the endogenous activation of mGlu2 receptors, which is localized in the preterminal regions of axon terminals, and, therefore, is not accessible to synaptically released glutamate. Through these mechanisms, NAC may correct glutamate deregulation associated with drug addiction. Remarkably, NAC treatment could also restore glutamate homeostasis in addicted patients. 25 These studies demonstrated that the mechanism of action of NAC in the CNS is not limited to its established antioxidant effect and laid the groundwork for an increasing number of clinical trial on NAC in CNS disorders, such as drug addiction, pathological gambling, obsessive-compulsive disorder, schizophrenia, depression, bipolar disorder, and autism. 27
Our interest on NAC in the treatment of pain moved from the comorbidity between some of the above disorders (particularly depression) and chronic pain, and from the evidence that mGlu2 receptors are candidate targets for analgesic drugs in different types of pain. Present findings offer the first demonstration that NAC causes analgesia in an established model of painful diabetic neuropathy. We used the STZ model of type-1 diabetes in C57BL/6 mice, which develop diabetic neuropathy but are relatively resistant to renal damage caused by STZ.40–42 This avoids confounding factors related to the protective effect of repeated administrations of NAC against renal damage caused by oxidative damage.
We found that NAC caused robust analgesia in diabetic mice both after single and repeated injections, suggesting the lack of tolerance to the action of NAC. This is in contrast with findings obtained with the CCI model of neuropathic pain, in which the analgesic activity of NAC was lost after repeated administrations.
32
Why tolerance developed in the CCI model but not in the STZ model of neuropathic pain is unknown. The lack of tolerance confers a translational value to our findings because the management of pain in patients with diabetic neuropathy usually requires long treatment regimens with analgesic drugs. As observed in other pain models,
32
NAC-induced analgesia was abrogated by the
The lack of tolerance to NAC-induced analgesia in the STZ model of diabetic neuropathy gave us the impetus to examine whether repeated administrations of NAC could induce changes in the expression of proteins that are involved in mechanisms of nociceptive sensitization in the first station of the pain neuraxis, i.e., the dorsal horns of the spinal cord. Repeated administrations of NAC did not cause adaptive changes in the machinery of mGlu2 receptor activation, i.e., the receptor itself,
Interestingly, NAC treatment reduced MAP kinase activation and showed a trend to a reduction in P2X7 receptor expression in the dorsal regions of the spinal cord of both control and STZ-treated mice. These adaptive changes might contribute to sustain pain relief in response to repeated administrations of NAC. A large body of evidence indicates that MAP kinase activation in dorsal horn neurons, astrocytes, and microglia is a key event in the development of central nociceptive sensitization, and MAPK inhibitors cause analgesia in models of neuropathic pain, including the STZ model.60–68 To examine whether the observed MAPK inhibition in the spinal cord could contribute to NAC-induced analgesia, we treated diabetic mice with the potent and brain permeant MEK inhibitor, PD0325901, at doses (25 mg/kg, i.p.) that are behaviorally active and inhibit ERK activation in the CNS. 69 PD0325901 caused analgesia in diabetic mice, although the increase in mechanical pain thresholds did not reach statistical significance. PD0325901 occluded NAC-induced analgesia, suggesting that MAPK inhibition lies at the core of the action of NAC. Analgesic drugs, such as dexmetomidine, type-2 cannabinoid receptor agonists, and ketamine alleviate neuropathic pain by restraining MAPK activation in the spinal cord.70–72 In dorsal horn neurons, MAPK phosphorylates and inhibits Kv4.2 channels, which mediate type-A potassium currents and are involved in pain plasticity.73–75 It is possible that NAC treatment in diabetic mice enhances the activity of Kv4.2 channels, therefore reducing excitability of secondary order neurons in the dorsal horns of the spinal cord. This hypothesis warrants further investigation.
P2X receptors are present in neurons, microglia, and immune cells, and their activation contributes to both neuropathic and inflammatory pain. 76 For example, the activation of microglial P2X2, P2X3, P2X4, and P2X7 receptors may maintain pain sensitivity through a mechanism of glial-neuronal interaction, and P2X receptor antagonists attenuate neuropathic pain.77–79 We focused on P2X7 receptors because single nucleotide polymorphisms of these receptors are associated with diabetic neuropathic pain. 80 The reduced expression of P2X7 receptors we have found in the dorsal region of the spinal cord of diabetic mice was unexpected and might represent a compensatory mechanism aimed at restraining the intensity of pain. This reduction may contribute to explain the lack of analgesic activity of the P2X7 receptor antagonist JNJ47965567, 81 which has been reported to cause a modest but significant analgesia in another model of neuropathic pain. 81 NAC treatment caused a trend to a reduction in P2X7 expression in both control and diabetic mice, but, surprisingly, NAC-induced analgesia was largely reduced in mice receiving a single injection of JNJ47965567. This finding is counterintuitive and difficult to explain and suggests that endogenous activation of P2X7 receptors is required for a full analgesic action of NAC. Further studies are needed to elucidate the complex interaction between P2X7 receptor and NAC.
We also examined the interaction between NAC and TRPV1 receptor by using the potent and brain permeant TRPV1 receptor antagonist, capsazepine. 82 We moved from the evidence that the expression of TRPV1 (vanilloid receptor 1) is increased on myelinated primary afferent neurons in STZ model of diabetic neuropathy. 83 Here we did not find changes in TRPV1 protein levels in the spinal cord of diabetic mice. However, we were surprised to find that a single injection of capsazepine caused a robust analgesia in these mice. The action of capsazepine has been extensively investigated in different pain models and data were not homogeneous in different animal species. 84 In mice, capsazepine did not display analgesic activity in the partial sciatic nerve ligation and paclitaxel models of neuropathic pain84,85 and in the CFA model of chronic inflammatory pain.84,86 Our findings indicate that TRPV1 blockade is highly effective in producing acute analgesia in mice developing painful diabetic neuropathy. Interestingly, capsazepine was less than additive with that produced by NAC. This might suggest that NAC-induced analgesia involves the modulation of TRPV1 receptors. Further studies are needed to unravel the nature of this interaction.
To examine whether adaptive changes caused by repeated NAC administrations were dependent on the type of pain, we used the CFA model of chronic inflammatory pain, where we have already shown the analgesic activity of single and repeated administrations of NAC. 32 In this model, NAC had no effect on protein levels (including ERK1/2 phosphorylation) in the dorsal portion of the lumbar spinal cord ipsilateral to CFA injection, but caused a substantial reduction in TRPV1 protein levels in the ipsilateral plantar paw region. TRPV1 receptors form cation-permeable membrane ion channels and are also found in primary sensory neurons and in postsynaptic neurons of the spinal cord.87–89 TRPV1 channels play a key role in the development of inflammatory pain90–92 and are involved in the induction of long-term potentiation in the spinal cord in response to peripheral nociceptive stimulation. 93 Electroacupuncture causes analgesia in the CFA model of inflammatory pain by reducing the expression and function of TRPV1 channels in dorsal root ganglia neurons. 94 Thus, the down-regulation of TRPV1 channels induced by repeated administrations of NAC fits nicely with the analgesic activity of NAC in the CFA model. The different effects of NAC treatment on protein levels in diabetic and CFA mice suggest that adaptive changes induced by NAC are context-dependent and are related to the type of pain.
In conclusion, our findings encourage human studies aimed at establishing whether NAC treatment relieves pain associated with diabetic neuropathy. The translational value of these data is strengthened by the known protective effect of NAC against cardiovascular, renal, dermatological, and CNS complications of diabetes.95–98 Again, NAC is a safe drug with a favorable pharmacokinetic profile and might therefore be combined with conventional analgesic agents in the treatment of painful diabetic neuropathy.
Footnotes
Author Contributions
SN, GM, and PS assessed pain thresholds in STZ-treated mice, CFA-injected mice, and their controls. SN, PS, SM, and AG performed measurements of protein levels. SN, EM, GC, VB, FN, and GB designed experiments and wrote the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Italian Ministry of Health (project code: RF-2011–02352582).